


Abstract
In the battle against viruses, antiviral peptides (AVPs) had demonstrated the immense potential. Presently, more than 15 peptide-based drugs are in various stages of clinical trials. Emerging and re-emerging viruses further emphasize the efforts to accelerate antiviral drug discovery efforts. Despite, huge importance of the field, no dedicated AVP resource is available. In the present study, we have collected 1245 peptides which were experimentally checked for antiviral activity targeting important human viruses like influenza, HIV, HCV and SARS, etc. After removing redundant peptides, 1056 peptides were divided into 951 training and 105 validation data sets. We have exploited various peptides sequence features, i.e. motifs and alignment followed by amino acid composition and physicochemical properties during 5-fold cross validation using Support Vector Machine. Physiochemical properties-based model achieved maximum 85% accuracy and 0.70 Matthew’s Correlation Coefficient (MCC). Performance of this model on the experimental validation data set showed 86% accuracy and 0.71 MCC which is far better than the general antimicrobial peptides prediction methods. Therefore, AVPpred—the first web server for predicting the highly effective AVPs would certainly be helpful to researchers working on peptide-based antiviral development. The web server is freely available at http://crdd.osdd.net/servers/avppred.
INTRODUCTION
There has been a considerable focus per se on the antiviral research from the past decade due to the limited availability of therapeutic molecules for many viral infections (1). Apart from existing drugs and vaccines, there is a need to explore new antiviral candidates to control pathogenic re-emerging and resistant viruses (2). Antiviral peptides (AVPs) are a potential alternative strategy in this context (3).
AVP were experimentally proven in blocking virus attachment or entry into host cells or inhibiting viral replication. In other words, these may interfere with the key steps of pathogenic human viruses (4). These inhibitory peptides could then be used as starting point for the design of more active molecules targeting the generic steps involved in virus attachment, fusion and replication, etc. These peptides may sometimes be preferable because of their relatively low molecular weight, lesser toxicity, rapid elimination from the host, lesser side effects and also cost effective synthesis nowadays (5).
Many workers have reported highly efficient peptides against human viruses, e.g. influenza (6–8), HIV (9,10), WNV (11), HCV (12), Rabies (4), HSV (13) and RSV (14), etc. Synthetic analogues of several naturally occurring antimicrobial peptides have been made in an attempt to identify important structural features contributing to the antiviral activity (15). Currently, 15 antimicrobial peptides have entered clinical trials (16). The importance of antimicrobial peptides has been further reviewed (17–20).
Despite immense potential of this field, there is no virus specific AVP prediction algorithm available. Although some general antimicrobial peptide prediction servers exist like CAMP (21), APD2 (22), AntiBP2 (23) and Wang et al. (24). At the same time, presently there is no dedicated database pertaining to AVPs. As far as AVP sequences are concerned, collectively less than 200 AVPs are provided in the functional antimicrobial peptide database APD2 (22) and CAMP (21). In the present study, we have developed the first AVP prediction method based on the collected peptides which were experimentally proven for antiviral activity.
MATERIALS AND METHODS
Algorithm development
Data collection
We have screened several hundred research articles and patents indexed in Pubmed and Patent Lens, respectively, to retrieve AVP sequences. From more than 80 relevant articles; we have extracted 1245 peptide sequences with a reported antiviral activity against human viruses like HIV, HCV, SARS and Influenza, etc. About 150 AVP of our collection were also present in the existing antimicrobial databases (21,22). Nearly 91% of the collected peptides were from natural source and remaining have synthetic source. Identical peptides were removed to finally have 604 highly effective and 452 least or non-effective AVPs which were used in the training T544p+407n (544 positive and 407 negative) and validation V60p+45n (60 positive and 45 negative) data sets, respectively. Simultaneously, we have made another training T544p+544n* and validation V60p+60n* data sets where non-experimental negative peptides [as employed in earlier antimicrobial peptide prediction method (23)] were used in place of experimentally verified negative data set.
Peptide features
General antimicrobial peptide prediction methods exploited several peptide sequence features in the past. We have also utilized the most important features like amino acid composition and physicochemical properties for SVM model development and also used sequence alignment technique. We have included another important sequence motif feature which was not exploited earlier in the AMP predictions.
Motif search
Motif-based approach has been used in past for protein and peptide identification (25,26). However this approach was not exploited for finding antimicrobial peptides so we searched for the conserved motifs in the AVP using MEME/MAST (27,28), where MEME is used to discover motifs and MAST is used to search these motifs in peptides. Correct prediction of MEME/MAST depends on E-values. If at given E-value MAST searches any motif then this peptide is predicted as AVP and if not found then it is predicted to be non-AVP. Firstly we extracted 20 motifs (as provided in the Supplementary Table S1) from AVP using MEME and searched these motifs in antiviral and non-AVPs using MAST at various E-values and best results were found at E-value 10.
Sequence alignment
The sequence segments with high identity are inclined to share the structure and function. This method has been widely used in the past for proteins and peptide prediction, like in signal peptide prediction using BLASTP (29). Similarly Wang et al. (24) used BLASTP for prediction of antimicrobial peptide. We have also implemented BLASTP algorithm for prediction of AVPs. Briefly we created two databases; antiviral database having 544 and Non-Antiviral database having 407 peptides. Both databases were then formatted into BLAST readable format using ‘formatdb’. Optimized parameters were used as reported earlier (24). Each query sequence was matched against antiviral and non-AVP databases using BLASTP program. If the most significant hit is found in the antiviral database then peptide is predicted to be AVP else non-AVP if hit is found in the non-antiviral database.
Amino acid composition
Amino acid composition is the fraction of each amino acid in a peptide. The fraction of all 20 natural amino acids was calculated using the following equation:
Physicochemical properties
Antimicrobial peptide properties cannot be defined by amino acid sequence pattern alone, various combination of structural and physicochemical features like secondary structure, overall charge, size, residue composition, hydrophobicity and amphiphilic character are responsible for overall activity of AMP (30). Torrent et al. (31) has used physicochemical properties for the analysis and prediction of antimicrobial peptides and have shown good results. We tried all 544 physicochemical properties available in AAINDEX database (32) individually, to look into the importance of each property in antiviral activity of the peptides. Model of 544 physicochemical properties was not implemented due to its very large (10 880) vector size. Finally selected 25 best performing physicochemical properties were used in developing AVPphysico model as provided in the Supplementary Table S2.
Implementation
The Support Vector Machine (SVM) approach has been used extensively in the areas of pattern recognition and classification (26). SVM is a learning algorithm which, upon training with a set of positively and negatively labelled samples, produces a classifier that can then be used to identify the correct label for unlabelled samples. The best performance was obtained using the RBF kernel parameters. For the present study, we used the freely downloadable SVMlight package.
Validation
In the present study, 5-fold cross validation technique has been adopted to evaluate the performance of the various SVM modules constructed. In this technique, the data set was partitioned randomly into five equally sized sets. The training and testing was carried out five times, each time using one distinct set for testing and reaming four sets for training. The following equations were often used in literatures to reflect the prediction quality:
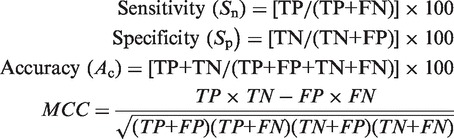 |
where Sn reflects the sensitivity, Sp the specificity, Ac the accuracy and MCC, the Mathew’s correlation coefficient; while TP represents the true positive, TN, the true negative; FP, the false positive and FN, the false negative. Sn, Sp and Ac stand for the success rates of prediction on positive, negative and overall data sets, respectively. MCC is used to evaluate the performance of the predictor when the positive and negative samples in the data set are out-of-balance. Its value ranges from −1 to 1 and a larger MCC means a better prediction.
RESULTS
Performance evaluation during 5-fold cross validation
During 5-fold cross validation, all four AVP prediction models were evaluated, namely AVPmotif based on 20 AVP motifs; AVPalign based on sequence alignment; AVPcompo based on amino acid composition and AVPphysico based on 25 physico-chemical properties. All models were made on data set T544p+407n. Similarly four new models (suffixed by #) as above were developed on another data set T544p+544n* having non-experimental peptides. Performances of all these models are shown in the Table 1.
Table 1.
Performance of AVPpred models during 5-fold cross validation
| Data set | Model | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|---|
| T544p+407n | AVPmotif | 72.3 | 82.2 | 76.8 | 0.54 |
| AVPalign | 88.3 | 81.0 | 85.0 | 0.70 | |
| AVPcompo | 80.7 | 87.0 | 83.4 | 0.67 | |
| AVPphysico | 82.2 | 88.2 | 85.0 | 0.70 | |
| T544p+544n* | AVPmotif# | 72.3 | 88.4 | 80.4 | 0.62 |
| AVPalign# | 86.0 | 92.7 | 89.3 | 0.79 | |
| AVPcompo# | 84.2 | 96.1 | 90.2 | 0.81 | |
| AVPphysico# | 89.7 | 90.3 | 90.0 | 0.80 |
Training data set T544p+407n is consisting of experimental AVP, while training data set T544p+544n* was containing non-experimental peptides.
#AVP models developed using T544p+544n* data set.
Performances of all the models were very good. AVPmotif model based on antiviral motifs also performed well; however, we achieved maximal 85% accuracy and 0.70 correlation on T544p+407n experimental data set by AVPphysico model using sequence physicochemical features. AVPalign model also performed equally well. On the other T544p+544n* data set, amino acid composition feature-based model AVPcompo# showed best performance with 90% accuracy and 0.81 correlation. When comparing the results on both the data sets, it is observed that all four models developed on T544p+544n* data set performed slightly better than the models developed using T544p+407n complete experimental data set during 5-fold cross validation. This could be attributed to the fact that T544p+544n* data set contain non-experimental peptides in the negative data set. These negative peptides were generated from the non-secretary proteins which have least chance of being positive and enhancing the model accuracy. We have also checked the performance of a hybrid model by combination of all four methods during 5-fold cross validation and on independent validation data set. However, performance of the hybrid model is almost similar to that of best individual method.
Performance evaluation on independent data set
Since 5-fold cross validation evaluation is not considered sufficient, therefore, we have also evaluated the performance of our models on the validation/independent data set. This data was not included anywhere in the training or testing. There were two validation data sets: (i) V60p+45n having complete experimental verified 105 peptides and (ii) V60p+60n* data sets with non-experimental negative peptides (see material and method). Each of these independent data set was evaluated using four models developed using T544p+407n and T544p+544n* data sets, respectively. Detailed results are shown in Table 2.
Table 2.
Performance of AVPpred models on validation/independent data sets V60p+45n and V60p+60n*
| Data set | Model | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|---|
| V60p+45n | AVPmotif | 70.0 | 77.8 | 73.3 | 0.47 |
| AVPalign | 81.7 | 82.2 | 81.9 | 0.63 | |
| AVPcompo | 83.3 | 88.9 | 85.7 | 0.72 | |
| AVPphysico | 88.3 | 82.2 | 85.7 | 0.71 | |
| AVPmotif# | 70.0 | 81.7 | 75.8 | 0.52 | |
| AVPalign# | 83.3 | 48.9 | 68.6 | 0.35 | |
| AVPcompo# | 83.3 | 62.2 | 74.3 | 0.47 | |
| AVPphysico# | 93.3 | 48.9 | 74.3 | 0.48 | |
| V60p+60n* | AVPmotif | 70.0 | 81.7 | 75.8 | 0.52 |
| AVPalign | 81.7 | 78.3 | 80.0 | 0.60 | |
| AVPcompo | 83.3 | 88.3 | 85.8 | 0.72 | |
| AVPphysico | 88.3 | 65.0 | 76.7 | 0.55 | |
| AVPmotif# | 70.0 | 83.4 | 76.7 | 0.58 | |
| AVPalign# | 83.3 | 95.0 | 89.2 | 0.79 | |
| AVPcompo# | 83.3 | 98.3 | 90.8 | 0.83 | |
| AVPphysico# | 93.3 | 91.7 | 92.5 | 0.85 |
V60p+60n* was containing non-experimental peptides.
#AVP models developed using T544p+544n* data set.
On the V60p+45n independent data set both AVPcompo and AVPphysico method achieved maximum accuracy of 86% and 0.71 MCC while on the same data set AVPphysico# model showed 74% accuracy and 0.48 MCC. Similarly on V60p+60n* independent data set AVPcompo model performed best with 86% accuracy and 0.72 MCC while AVPphysico# also outperformed other models with 93% accuracy and 0.85 MCC.
On both the independent data set, performance of the AVP models developed on T544p+407n data set were quite good and similar. At the same time, results also revealed that all models except AVPmotif which were developed on the experimental T544p+407n data set outperformed the models developed on the T544p+544n* data set having non-experimental peptides on complete experimental data. This suggests that it may be better to include experimental verified negative peptides data also for the antimicrobial peptides prediction.
Comparison with existing antimicrobial peptide prediction algorithms
It is not appropriate to compare the AVPpred with any other method since none such method available. Still in the context of antimicrobial peptide prediction algorithm we made the comparison with latest AMP prediction methods of Wang et al. (24) and Thomas et al. (21). Results are shown in the Table 3.
Table 3.
Comparison of AVPpred models with recent antimicrobial peptide prediction methods on independent data sets V60p+45n, V60p+60n* and T604p+452n
| Data set | Model | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|---|
| V60p+45n | Wang et al. (24) | 61.7 | 80.0 | 69.5 | 0.42 |
| Thomas et al. (21) | 40.0 | 77.3 | 55.8 | 0.18 | |
| V60p+60n* | Wang et al. (24) | 61.7 | 90.0 | 75.8 | 0.54 |
| Thomas et al. (21) | 40.0 | 86.7 | 63.3 | 0.30 | |
| T604p+452n | Wang et al. (24) | 56.1 | 75.4 | 64.4 | 0.32 |
| Thomas et al. (21) | 37.0 | 76.8 | 54.1 | 0.15 |
T604p+452n data set is consists of both experimental training and validation data sets.
Results showed that on the V60p+45n complete experimental validation data set, best models of Wang et al. (24) method could achieve only 70% accuracy and 0.42 MCC despite the fact that 15 of these positive peptides were also included in the Wang et al. (24) training model while model of Thomas et al. (21) performed rather poorly with 56% accuracy and 0.18 MCC. On the other side, our both AVPcompo and AVPphysico models based on complete experimental data performed far better with accuracy of 86% and 0.71 MCC on the same independent data set. Even overall performance of general AMP prediction method on the whole T604p+452n experimental data is not good. Similarly on the V60p+60n* independent data set, our AVPphysico# model outperformed the Wang et al. (24) model with 93% accuracy and 0.85 MCC. This is due to the fact that our method is specific for the AVPs than the existing general antimicrobial peptide prediction methods are mostly suitable for bacteria. It further suggests that AVP may have some unique features which our AVPpred algorithm best able to incorporate and thus outperformed other AMP prediction in case of AVPs.
Webserver
AVPpred web server is freely accessible via the URL http://crdd.osdd.net/servers/avppred. On the ‘Home’ page a general description of the server is provided. Documentation regarding usage and algorithm are available under ‘Help’ and ‘Algorithm’ pages. A flowchart depicting the workflow of AVPpred web server is shown in Figure 1. Data sets used in model development are also displayed in separate pages. A general outline of the server is displayed in Figure 2.
Figure 1.

AVPpred workflow. Flowchart of the AVP model development.
Figure 2.

Web server overview. Outline of AVPpred web server and functionality.
Input
To use this prediction model, an online web server available via the URL http://crdd.osdd.net/servers/avppred has been provided. On the submit page user may paste single or multiple peptide sequence(s) in FASTA format in the provided text-box or upload a FASTA file from the system. User can remove the entries in the text-box by using the clear button. An ‘Example link’ has been provided to help the users. If user has a protein sequence, then, an option for making peptide fragments of desired length and overlapping residues is provided. User can paste these peptide fragments in the ‘Predict AVP’ form.
Output
Results of the prediction are shown in a tabular as well as graphical layout. In first three columns of the result Table; Sequence No, Peptide Sequence Name and Length are given. In fourth column BLAST option is provided to search similar peptide sequences in CAMP and AMP antimicrobial peptide databases and also in Uniprot. By clicking the properties column, some relevant properties of the peptide like charge, polarity, amino acid composition, hydrophobicity, preference for alpha/beta strand, etc. are be displayed. In next columns prediction of Models 1–4 corresponding to motif search, sequence alignment, amino acid composition and physicochemical properties are shown. Eighth and ninth columns display the numerical value of SVM score. This is displayed in colour code as red (high), yellow (medium) and green (low). The graph displays the specificity of the peptides corresponding to the ‘composition’ and ‘physicochemical’ models. Also users can download the prediction results as a tab delimited text file.
Tools
Users may BLAST their peptide for similarity against APD2, CAMP and Uniprot. The result shows distribution of hits, their score, E-value and alignment with sequences with significant similarity will be displayed. Also user can predict AVPs from other databases (CAMP and APD2). Further users can check some relevant physicochemical properties like charge, polarity, amino acid composition, hydrophobicity, preference for alpha/beta strand, for their peptides. Also the collection of positive and negative data sets has been provided on the server along with their sequence, length, target virus and references. In the figures below is shown the development of prediction models (Figure 1) and general outline of the web server (Figure 2).
Server implementation
The server AVPpred is implemented on Red Hat Linux and Apache (2.2.17) in back-end and front-end of web interface is implemented with PHP (5.2.14).
CONCLUSION
AVPpred is the first algorithm for prediction of highly effective AVPs based on experimentally validated positive and negative data sets. Four prediction models; the composition-based, physico-chemical properties and sequence alignment-based were implemented in the web server to make comprehensive predictions. This web server would be helpful for researchers working for the development of peptide-based antiviral therapeutics.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2.
FUNDING
Council of Scientific and Industrial Research (CSIR); Department of Biotechnology, Government of India and CSIR-IMTECH [Communication No. 013/2012]. Funding for open access charge: CSIR-Institute of Microbial Technology, Sector 39A, Chandigarh.
Conflict of interest statement. None declared.
REFERENCES
- 1.Thakur N, Qureshi A, Kumar M. VIRsiRNAdb: a curated database of experimentally validated viral siRNA/shRNA. Nucleic Acids Res. 2012;40:D230–D236. doi: 10.1093/nar/gkr1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sadanand S. Vaccination: the present and the future. Yale J. Biol. Med. 2011;84:353–359. [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang H, Xu Y, Li L, Weng L, Wang Q, Zhang S, Jia B, Hu H, He Y, Jacob Y, et al. Inhibition of influenza virus replication by constrained peptides targeting nucleoprotein. Antivir. Chem. Chemother. 2011;22:119–130. doi: 10.3851/IMP1902. [DOI] [PubMed] [Google Scholar]
- 4.Real E, Rain JC, Battaglia V, Jallet C, Perrin P, Tordo N, Chrisment P, D’Alayer J, Legrain P, Jacob Y. Antiviral drug discovery strategy using combinatorial libraries of structurally constrained peptides. J. Virol. 2004;78:7410–7417. doi: 10.1128/JVI.78.14.7410-7417.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castel G, Chteoui M, Heyd B, Tordo N. Phage display of combinatorial peptide libraries: application to antiviral research. Molecules. 2011;16:3499–3518. doi: 10.3390/molecules16053499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones JC, Settles EW, Brandt CR, Schultz-Cherry S. Identification of the minimal active sequence of an anti-influenza virus peptide. Antimicrob. Agents Chemother. 2011;55:1810–1813. doi: 10.1128/AAC.01428-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drummond JE, Shaw EE, Antonello JM, Green T, Page GJ, Motley CO, Wilson KA, Finnefrock AC, Liang X, Casimiro DR. Design and optimization of a multiplex anti-influenza peptide immunoassay. J. Immunol. Methods. 2008;334:11–20. doi: 10.1016/j.jim.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 8.Wunderlich K, Juozapaitis M, Ranadheera C, Kessler U, Martin A, Eisel J, Beutling U, Frank R, Schwemmle M. Identification of high-affinity PB1-derived peptides with enhanced affinity to the PA protein of influenza A virus polymerase. Antimicrob. Agents Chemother. 2011;55:696–702. doi: 10.1128/AAC.01419-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl Acad. Sci. USA. 2007;104:12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Narumi T, Komoriya M, Hashimoto C, Wu H, Nomura W, Suzuki S, Tanaka T, Chiba J, Yamamoto N, Murakami T, et al. Conjugation of cell-penetrating peptides leads to identification of anti-HIV peptides from matrix proteins. Bioorg. Med. Chem. 2012;20:1468–1474. doi: 10.1016/j.bmc.2011.12.055. [DOI] [PubMed] [Google Scholar]
- 11.Bai F, Town T, Pradhan D, Cox J, Ashish, Ledizet M, Anderson JF, Flavell RA, Krueger JK, Koski RA, et al. Antiviral peptides targeting the west nile virus envelope protein. J. Virol. 2007;81:2047–2055. doi: 10.1128/JVI.01840-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Portal-Nunez S, Gonzalez-Navarro CJ, Garcia-Delgado M, Vizmanos JL, Lasarte JJ, Borras-Cuesta F. Peptide inhibitors of hepatitis C virus NS3 protease. Antivir. Chem. Chemother. 2003;14:225–233. doi: 10.1177/095632020301400501. [DOI] [PubMed] [Google Scholar]
- 13.Akkarawongsa R, Pocaro NE, Case G, Kolb AW, Brandt CR. Multiple peptides homologous to herpes simplex virus type 1 glycoprotein B inhibit viral infection. Antimicrob. Agents Chemother. 2009;53:987–996. doi: 10.1128/AAC.00793-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambert DM, Barney S, Lambert AL, Guthrie K, Medinas R, Davis DE, Bucy T, Erickson J, Merutka G, Petteway SR., Jr Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl Acad. Sci. USA. 1996;93:2186–2191. doi: 10.1073/pnas.93.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jenssen H, Hamill P, Hancock RE. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006;19:491–511. doi: 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fjell CD, Hiss JA, Hancock RE, Schneider G. Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discov. 2012;11:37–51. doi: 10.1038/nrd3591. [DOI] [PubMed] [Google Scholar]
- 17.Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 18.Kavanagh K, Dowd S. Histatins: antimicrobial peptides with therapeutic potential. J. Pharm. Pharmacol. 2004;56:285–289. doi: 10.1211/0022357022971. [DOI] [PubMed] [Google Scholar]
- 19.Popovic S, Urban E, Lukic M, Conlon JM. Peptides with antimicrobial and anti-inflammatory activities that have therapeutic potential for treatment of acne vulgaris. Peptides. 2012;34:275–282. doi: 10.1016/j.peptides.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 20.Peters BM, Shirtliff ME, Jabra-Rizk MA. Antimicrobial peptides: primeval molecules or future drugs? PLoS Pathog. 2010;6:e1001067. doi: 10.1371/journal.ppat.1001067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas S, Karnik S, Barai RS, Jayaraman VK, Idicula-Thomas S. CAMP: a useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2010;38:D774–D780. doi: 10.1093/nar/gkp1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang G, Li X, Wang Z. APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009;37:D933–D937. doi: 10.1093/nar/gkn823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lata S, Mishra NK, Raghava GP. AntiBP2: improved version of antibacterial peptide prediction. BMC Bioinformatics. 2010;11(Suppl. 1):S19. doi: 10.1186/1471-2105-11-S1-S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang P, Hu L, Liu G, Jiang N, Chen X, Xu J, Zheng W, Li L, Tan M, Chen Z, et al. Prediction of antimicrobial peptides based on sequence alignment and feature selection methods. PLoS One. 2011;6:e18476. doi: 10.1371/journal.pone.0018476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saha S, Raghava GP. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;34:W202–W209. doi: 10.1093/nar/gkl343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rashid M, Saha S, Raghava GP. Support Vector Machine-based method for predicting subcellular localization of mycobacterial proteins using evolutionary information and motifs. BMC Bioinformatics. 2007;8:337. doi: 10.1186/1471-2105-8-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 29.Frank K, Sippl MJ. High-performance signal peptide prediction based on sequence alignment techniques. Bioinformatics. 2008;24:2172–2176. doi: 10.1093/bioinformatics/btn422. [DOI] [PubMed] [Google Scholar]
- 30.Svenson J, Karstad R, Flaten GE, Brandsdal BO, Brandl M, Svendsen JS. Altered activity and physicochemical properties of short cationic antimicrobial peptides by incorporation of arginine analogues. Mol. Pharm. 2009;6:996–1005. doi: 10.1021/mp900057k. [DOI] [PubMed] [Google Scholar]
- 31.Torrent M, Andreu D, Nogues VM, Boix E. Connecting peptide physicochemical and antimicrobial properties by a rational prediction model. PLoS One. 2011;6:e16968. doi: 10.1371/journal.pone.0016968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawashima S, Kanehisa M. AAindex: amino acid index database. Nucleic Acids Res. 2000;28:374. doi: 10.1093/nar/28.1.374. [DOI] [PMC free article] [PubMed] [Google Scholar]