

Abstract
Oxidant stress and compromised microcirculation underlie renal pathophysiology in septic acute kidney injury (AKI). Holthoff et al. report that resveratrol ameliorates these coupled abnormalities. They did not establish the primacy of either defect in septic AKI. However, tubule mitochondrial defects were recently reported to be involved in septic AKI pathogenesis, and resveratrol targets PGC-1α and respiratory enzymes. Together, these findings open new avenues for research into long-unresolved issues in the pathophysiology of septic AKI.
Renal failure during gram negative sepsis can be profound, difficult to treat and fatal (1, 2). It occurs in a well known setting of endotoxemia, endotoxin binding to endothelium and leukocytes, production and release of cytokines and a systemic “cytokine storm” accompanied by decreased peripheral vascular resistance and hypotension – septic shock. In light of the dire systemic disturbance, a puzzling aspect of “acute kidney injury” (AKI) in sepsis is the paucity of structural renal damage despite severely impaired function (2, 3). Although glomerular filtration rate (GFR) can decrease during septic AKI as a consequence of generalized circulatory collapse and attendant reduction of renal blood flow (RBF) (1), kidney specific factors have important roles as well, and may dominate. Renal events that determine septic AKI have been examined in animal models (3, 4). When the macrocirculation fails, RBF and GFR decrease, but these hallmarks of AKI are also evident in normotensive models of murine endotoxemia (1, 3, 5). Moreover, persistent azotemia develops in sheep injected with live E.Coli despite increased RBF (3). Along these lines, early volume replacement can restore GFR in some models, whereas in other situations, it only allows AKI to be expressed, by delaying death due to systemic sepsis (3). Thus, kidney injury seems likely during sepsis. Nevertheless, as stated earlier, overt structural damage of the kidney is uncommon in septic AKI (2) and the best documented lesion is subtle vacuolation of tubule epithelium (3), a pathology that has not received rigorous investigative attention. On the other hand, we know that renal damage in experimental septic AKI is potentially reversible, at least as inferred from the benefits afforded by early interventions that restore renal function. Such interventions include volume replacement, renal denervation, free radical scavengers and anti-inflammatory therapies (1, 3, 5). Although these modalities could work entirely through their effects on systemic hemodynamics or the renal circulation, equally plausible arguments could be mounted for the primacy of a tubular lesion that in murine and rat models leads to reflex renal vasoconstriction. In the latter case, the effects of early beneficial interventions could conceivably be directed at preventing further renal damage by purchasing time for damaged tubules to recover. To us, it is a conundrum: does decreased RBF – the result of systemic cytokine storm and hemodynamic collapse and/or a renal circulatory abnormality – cause tubule pathology in the form of non-specific reversible damage? Or, is there a specific tubule lesion that is produced by cytokines or endotoxin? Both TNFα and LPS have direct proinflammatory effects on tubules (4, 6), and LPS directly induces TNFα expression in tubules (7), an effect that synergizes with other stresses to promote the tubular production of toxic cytokines (8). Moreover, endotoxic AKI was shown to require TNFα receptors in kidneys (9), although it was not demonstrated if tubule or endothelial receptors or both, played the critical roles. AKI was attended by subtle tubule vacuolation and modestly increased apoptosis in tubules. Arguably, these findings could weigh in favor of a primary tubule lesion that is produced by LPS/TNFα.
In this issue of the journal, Holthoff et al clarify some aspects of the renal vascular pathophysiology of sepsis (10). They report that intraperitoneal injections of the plant polyphenol resveratrol rapidly correct the decrease of RBF and to a lesser extent the reduction of GFR that occur during septic shock induced by cecal ligation and puncture (CLP). Septic shock produced by CLP was accompanied by hypotension; nevertheless, improvements of RBF and GFR took place without alterations of mean arterial pressure (MAP) or heart rate. This is an important observation because it suggests that the actions of resveratrol were targeted at the kidney (although it has multiple systemic effects), and therefore implies that septic AKI developed as a consequence of kidney specific pathophysiology. Treatment with resveratrol led to amelioration of tubule oxidant stress, improvement of blood flow along capillaries adjacent to stressed tubules, diminution of protein nitration in tubules, less azotemia, and prevention of tubule pathology characterized by brush border loss with focal vacuolation and sloughing of tubule cells. These benefits were associated with increased survival.
In previous studies cited in the bibliography, research by this group of investigators examined the relationships between tubule oxidant stress and renal microcirculation in murine and rat models of septic AKI induced by endotoxin and CLP. They used fluorescence intravital video microscopy of kidneys to monitor blood flow and oxidation of dihydrorhodamine (DHR) in tubule epithelium to demonstrate close coupling of tubule oxidant stress and sluggish blood flow along immediately adjacent capillaries. Notably, oxidized rhodamine fluorescence colocalized with areas of tubules with cytoplasmic vacuolation observed in vivo (11). Tubule cell vacuolation is the most consistent pathology of septic AKI (3), and these findings suggest that oxidant stress is involved in its pathogenesis. Furthermore, this work showed that inducible nitric oxide synthase (iNOS) becomes increased and nitro-tyrosine protein adducts are formed in tubules during endotoxic or CLP AKI, and that selective iNOS inhibition by L-N6-(1-Iminoethyl)lysine (L-NIL) abolishes tubule oxidant stress and corrects the microcirculatory abnormality (11). Although some of this earlier work was handicapped by omission of blood pressure and RBF measurements, these investigators did show that RBF is lowered without decrease of MAP during septic AKI in rat pups. As in murine models, reduction of RBF was reflected by compromised circulation in capillaries immediately adjacent to tubules with DHR fluorescence, suggesting a close relationship between tubule stress and sluggish microcirculation (12).
Holthoff et al believe that resveratrol protected against septic AKI by decreasing the activity of reactive nitrogen species. This is supported by their previous work showing that resveratrol scavenges peroxynitrite and that iNOS inhibition is also protective (11, 13, 14). Furthermore, it is known that interventions that scavenge superoxide also protect (5), consistent with known requirement of both superoxide and nitric oxide to produce the major agent that nitrates proteins – peroxynitrite. Interestingly, resveratrol scavenges diverse free radicals including superoxide. These properties of resveratrol neatly fall into place with efficient protection against tubule oxidant stress and protein nitration afforded by the compound. Holthoff et al do not take a position regarding the primacy of tubular vs. microcirculatory abnormalities that they document. Sluggish capillary blood flow could lead to tubulointerstitial hypoxia and secondary tubule cell effects. However, tubule pathology could also occur primarily, a contention that would be supported by tubule selective induction of TNFα by LPS (7). The relative importance of hemodynamic as opposed to parenchymal cell dysfunction in the pathophysiology of sepsis has long been debated for other tissues with substantial evidence for parenchymal cellular metabolism as the target in many cases (15). Consistent with the latter possibility in the kidney, sluggish capillary flow that is closely coupled to tubule stress can also be explained by reflex vasoconstriction at the single nephron level, such as that documented for single tubules with oil blocks (16). The nature of this reflex is obscure but similar phenomena probably contribute to the renal vasoconstriction seen during diverse renal injuries – a mechanism designed to prevent fluid loss – termed “acute renal success” (17). Clearly more studies are needed to investigate whether an endothelial pathology that is sufficiently severe to obstruct peritubular capillary blood flow occurs during septic AKI. Barring such a demonstration, the findings of Holthoff et al are better explained by functional vasoconstriction.
The case for a tubule pathology that determines the course of septic AKI was strengthened in other recent work. Tran et al (18) attribute and relate lipopolysaccharide (LPS) induced AKI to mitochondrial dysfunction in the kidney, accompanied by diminished expression of PPARγ coactivator-1α (PGC-1α), a regulator of mitochondrial biogenesis, and of genes coding for proteins required for oxidative phosphorylation. To a limited extent, they verify these findings in the CLP model of AKI. They demonstrate that although RBF is reduced in endotoxic AKI, tissue oxygenation remains intact, suggesting that kidney oxygen consumption is decreased. Enzyme histochemistry showed that cytochrome c-oxidase and NADH dehydrogenase functions in mitochondria become profoundly reduced. The latter findings are consistent with prior observations in other tissues (15). Using mice with global as well as proximal tubule deletion of PGC-1α and cultured cell models, Tran et al offer provocative data implying that PGC-1α is required to restore mitochondrial functional integrity during recovery from endotoxic insults. Furthermore, it would appear that TNFα can suppress PGC-1α expression and oxygen consumption, and that overexpression of PGC-1α corrects the mitochondrial defect. Importantly, Tran et al demonstrate profound mitochondrial dysfunction in tubule cells that displayed little pathology by light and electron microscopy beyond occasional cells with diffuse mitochondrial swelling and only focal and subtle mitochondrial changes in the vast majority of cells. The authors are cautious not to invoke a primary role for PGC-1α suppression in causing the observed changes, speculate that mitochondrial defects could have resulted from the actions of reactive oxygen and nitrogen species, and urge further studies to investigate the relationships between mitochondrial pathology and PGC-1α. It is also conceivable that some forms of suppression of mitochondrial function during sepsis in tubule and other cells, including endothelial cells, are adaptive (15, 19). These studies emphasize the value of further exploring how LPS, or one or more of the cytokines involved in septic shock, could produce the described changes as a consequence of their direct actions on tubules. Multiple mechanisms have been proposed in nonrenal cells (15) and others are possible. The findings of Tran et al are novel, need confirmation, and validation to demonstrate their importance in determining tubule dysfunction and clinical outcome. That they are irrelevant downstream events appears unlikely. The actions of resveratrol in septic AKI reported by Holthoff et al (10) were quite rapid, and consistent with its role as a scavenger of oxidant species. However, resveratrol has prominent and important actions on PGC-1α, mitochondrial biogenesis and cellular antioxidant enzymes related to its effects on sirtuins (20), mechanisms that should be investigated. Together, the findings of Holthoff et al in this issue of Kidney International (10) and the report by Tran et al in the Journal of Clinical Investigation (18) provide a wealth of interesting and intriguing new data that will enable investigators to formulate new hypotheses and more intensively pursue research in a field that heretofore has not received the attention it deserves – the tubule pathology of septic AKI.
Figure 1. Renal pathophysiology in septic AKI as suggested by the findings of Holthoff et al10 and Tan et al.18.
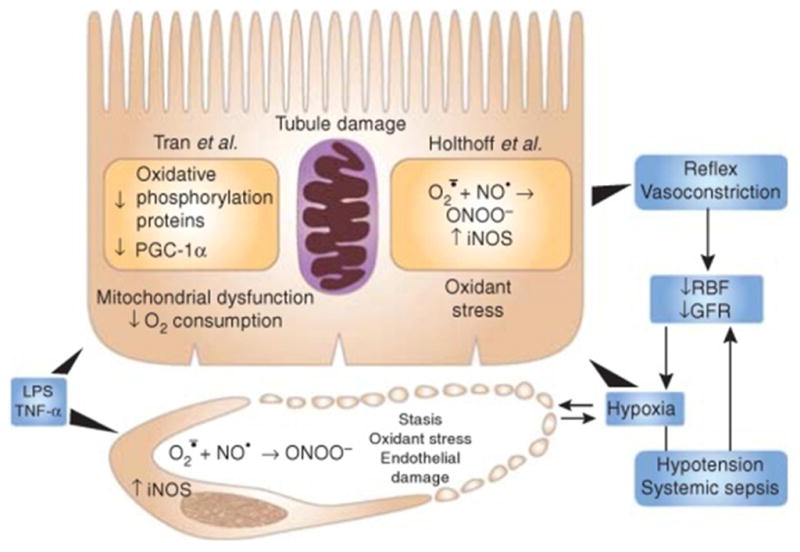
GFR, glomerular filtration rate; LPS, lipopolysaccharide; PGC-1α, PPAR-γ coactivator-1α RBF, renal blood flow; TNF-α, tumor necrosis factor-α
Contributor Information
Manjeri A. Venkatachalam, Department of Pathology, University of Texas Health Science Center, San Antonio, TX.
Joel M. Weinberg, Department of Medicine, University of Michigan Medical Center, Ann Arbor, MI.
References
- 1.Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351:159–169. doi: 10.1056/NEJMra032401. [DOI] [PubMed] [Google Scholar]
- 2.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 3.Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest. 2009;119:2868–2878. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El-Achkar TM, Hosein M, Dagher PC. Pathways of renal injury in systemic gram-negative sepsis. Eur J Clin Invest. 2008;38 (Suppl 2):39–44. doi: 10.1111/j.1365-2362.2008.02007.x. [DOI] [PubMed] [Google Scholar]
- 5.Wang W, Jittikanont S, Falk SA, Li P, et al. Interaction among nitric oxide, reactive oxygen species, and antioxidants during endotoxemia-related acute renal failure. Am J Physiol Renal Physiol. 2003;284:F532–537. doi: 10.1152/ajprenal.00323.2002. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez-Nino MD, Benito-Martin A, Goncalves S, Sanz AB, et al. TNF superfamily: a growing saga of kidney injury modulators. Mediators Inflamm. 2010 doi: 10.1155/2010/182958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noiri E, Kuwata S, Nosaka K, Tokunaga K, et al. Tumor necrosis factor-alpha mRNA expression in lipopolysaccharide-stimulated rat kidney. Chronological analysis of localization. Am J Pathol. 1994;144:1159–1166. [PMC free article] [PubMed] [Google Scholar]
- 8.Naito M, Bomsztyk K, Zager RA. Endotoxin mediates recruitment of RNA polymerase II to target genes in acute renal failure. J Am Soc Nephrol. 2008;19:1321–1330. doi: 10.1681/ASN.2007121368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunningham PN, Dyanov HM, Park P, Wang J, et al. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J Immunol. 2002;168:5817–5823. doi: 10.4049/jimmunol.168.11.5817. [DOI] [PubMed] [Google Scholar]
- 10.Holthoff JHWZ, Seely KA, Gokden N, Mayeux PR. Resveratrol improves renal microcirculation, protects the tubular epithelium, and prolongs survival in a mouse model of sepsis-induced acute kidney injury. Kidney International. 2011 doi: 10.1038/ki.2011.347. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu L, Gokden N, Mayeux PR. Evidence for the role of reactive nitrogen species in polymicrobial sepsis-induced renal peritubular capillary dysfunction and tubular injury. J Am Soc Nephrol. 2007;18:1807–1815. doi: 10.1681/ASN.2006121402. [DOI] [PubMed] [Google Scholar]
- 12.Seely KA, Holthoff JH, Burns ST, Wang Z, et al. Hemodynamic changes in the kidney in a pediatric rat model of sepsis-induced acute kidney injury. Am J Physiol Renal Physiol. 301:F209–217. doi: 10.1152/ajprenal.00687.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holthoff JH, Woodling KA, Doerge DR, Burns ST, et al. Resveratrol, a dietary polyphenolic phytoalexin, is a functional scavenger of peroxynitrite. Biochem Pharmacol. 80:1260–1265. doi: 10.1016/j.bcp.2010.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu L, Mayeux PR. Effects of the inducible nitric-oxide synthase inhibitor L-N(6)-(1-iminoethyl)-lysine on microcirculation and reactive nitrogen species generation in the kidney following lipopolysaccharide administration in mice. J Pharmacol Exp Ther. 2007;320:1061–1067. doi: 10.1124/jpet.106.117184. [DOI] [PubMed] [Google Scholar]
- 15.Ruggieri AJ, Levy RJ, Deutschman CS. Mitochondrial dysfunction and resuscitation in sepsis. Crit Care Clin. 2010;26:567–575. x–xi. doi: 10.1016/j.ccc.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arendshorst WJ, Finn WF, Gottschalk CW. Nephron stop-flow pressure response to obstruction for 24 hours in the rat kidney. J Clin Invest. 1974;53:1497–1500. doi: 10.1172/JCI107699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thurau K, Boylan JW. Acute renal success. The unexpected logic of oliguria in acute renal failure. Am J Med. 1976;61:308–315. doi: 10.1016/0002-9343(76)90365-x. [DOI] [PubMed] [Google Scholar]
- 18.Tran M, Tam D, Bardia A, Bhasin M, et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011 doi: 10.1172/JCI58662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rowlands DJ, Islam MN, Das SR, Huertas A, et al. Activation of TNFR1 ectodomain shedding by mitochondrial Ca2+ determines the severity of inflammation in mouse lung microvessels. J Clin Invest. 121:1986–1999. doi: 10.1172/JCI43839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weinberg JM. Mitochondrial biogenesis in kidney disease. J Am Soc Nephrol. 22:431–436. doi: 10.1681/ASN.2010060643. [DOI] [PubMed] [Google Scholar]