

Abstract
Background
Novel therapies capable of targeting drug resistant clonogenic MM cells are required for more effective treatment of multiple myeloma. This study investigates the cytotoxicity of natural killer cell lines against bulk and clonogenic multiple myeloma and evaluates the tumor burden after NK cell therapy in a bioluminescent xenograft mouse model.
Design and Methods
The cytotoxicity of natural killer cell lines was evaluated against bulk multiple myeloma cell lines using chromium release and flow cytometry cytotoxicity assays. Selected activating receptors on natural killer cells were blocked to determine their role in multiple myeloma recognition. Growth inhibition of clonogenic multiple myeloma cells was assessed in a methylcellulose clonogenic assay in combination with secondary replating to evaluate the self-renewal of residual progenitors after natural killer cell treatment. A bioluminescent mouse model was developed using the human U266 cell line transduced to express green fluorescent protein and luciferase (U266eGFPluc) to monitor disease progression in vivo and assess bone marrow engraftment after intravenous NK-92 cell therapy.
Results
Three multiple myeloma cell lines were sensitive to NK-92 and KHYG-1 cytotoxicity mediated by NKp30, NKp46, NKG2D and DNAM-1 activating receptors. NK-92 and KHYG-1 demonstrated 2- to 3-fold greater inhibition of clonogenic multiple myeloma growth, compared with killing of the bulk tumor population. In addition, the residual colonies after treatment formed significantly fewer colonies compared to the control in a secondary replating for a cumulative clonogenic inhibition of 89–99% at the 20:1 effector to target ratio. Multiple myeloma tumor burden was reduced by NK-92 in a xenograft mouse model as measured by bioluminescence imaging and reduction in bone marrow engraftment of U266eGFPluc cells by flow cytometry.
Conclusions
This study demonstrates that NK-92 and KHYG-1 are capable of killing clonogenic and bulk multiple myeloma cells. In addition, multiple myeloma tumor burden in a xenograft mouse model was reduced by intravenous NK-92 cell therapy. Since multiple myeloma colony frequency correlates with survival, our observations have important clinical implications and suggest that clinical studies of NK cell lines to treat MM are warranted.
Keywords: multiple myeloma, natural killer cell lines, cytotoxicity, immunotherapy, clonogenic, bioluminescent xenograft
Introduction
Multiple myeloma (MM) is an incurable plasma cell malignancy.1 Although clinical outcomes have improved considerably with combination therapy, including immunomodulatory agents, proteasome inhibitors, steroids and hematopoietic cell transplantation, 10-year survival remains around 20%.2–4 Clonogenic cells were first grown in soft agar from MM patient samples in 1977,5 demonstrating that colony formation is a property limited to a small population of cells. More recently, evidence for an MM stem cell has been reported, with subsets of MM cells identified that possess properties of normal stem cells, including Hoechst side population enrichment, G0 cell cycle and high aldehyde dehydrogenase activity.6–8 In addition, self-renewal of clonogenic MM cells was shown in vitro by serial replating of MM colonies and in vivo by primary and secondary engraftment in NOD/SCID mice.6,9,10
Furthermore, clonogenic MM cells have demonstrated drug resistance to conventional treatment, including dexamethasone, lenalidomide and bortezomib, suggesting that these therapies may target MM plasma cells to reduce tumor burden, but are ineffective in eradicating the disease.6 In addition, in vitro clonogenic growth from patient-derived bone marrow or peripheral blood samples correlated with significantly shorter survival of patients (n=14, mean survival 38 months from diagnosis) compared to those whose bone marrow samples could not form colonies (n=44, mean survival 66 months from diagnosis, P=0.009).10 Novel approaches are, therefore, needed to eliminate the clonogenic MM cells responsible for the maintenance and progression of MM.
Allogeneic and autologous primary NK cells demonstrate cytotoxicity against MM in vitro.11–15 Allogeneic hematopoietic cell transplantation is controversial in MM and often associated with high mortality, due in part to increased rates and severity of graft-versus-host disease (GvHD).16 However, increased survival was shown for MM patients undergoing HLA-matched sibling hematopoietic cell transplantation after initial autologous transplantation compared to patients receiving tandem autologous transplants.17 Taken together, these results suggest that NK cells may be potential mediators of a graft-versus-myeloma effect (GvM).
Adoptive immunotherapy with primary NK cells is limited by difficulties in cell expansion as well as variable activity from different patients.18 These limitations make established NK cell lines an appealing option for immunotherapy. NK-92 has demonstrated antitumor activity in a variety of malignancies, including MM in vitro and in human leukemia in SCID mice.19–21 NK-92 is the only NK cell line to have undergone clinical trials and has shown safety and expansion feasibility in a phase I trial of patients with advanced renal cell cancer and melanoma.22 Another NK cell line, KHYG-1, has broad cytotoxicity against leukemia cell lines and kills by a novel granzyme M dependent pathway.23 We, therefore, investigated the cytotoxicity of NK-92 and KHYG-1 against bulk and clonogenic MM cells to determine their therapeutic potential in MM.
Design and Methods
Cell growth conditions are described in the Online Supplementary Appendix.
Bulk tumor cytotoxicity assays
The chromium release assay was performed as previously described.24 In the flow cytometry cytotoxicity assay, effector and target cells were co-incubated under comparable conditions to the chromium release assay. After the 4-hour incubation at 37°C and 5% CO2, samples were labeled with CD7-biotin Streptavidin-PECy7 (eBioscience) to distinguish effector from target cells. Cell death was detected with Annexin V-FITC (Southern Biotech) and 7-AAD (eBioscience) and analyzed on a Beckman Coulter FC500 flow cytometer. A minimum of 10,000 target events were collected per sample and the results were analyzed using Flowjo v7.6 (Tree Star). Data presented are the mean ± SD of three separate experiments. The percentage of cytotoxicity was calculated as previously described.25
Cytotoxicity mechanism
Blocking experiments were performed using the following purified monoclonal blocking antibodies: DNAM-1 (clone DX11), NKp30 (clone P30-15), NKp44 (clone P44-8), NKp46 (clone 9E2), and NKG2D (clone 1D11) (BioLegend). After blocking specific receptors for 30 min at room temperature, the cytotoxicity detected by the flow cytometry cytotoxicity assay was compared to the cytotoxicty after blocking with a total mouse IgG isotype control at a 1:1 E:T ratio (Sigma-Aldrich). Blocking with a non-specific IgG antibody had minimal impact on the cytotoxicity compared to the cytotoxicity in the absence of blocking antibodies. All blocking antibodies were initially titrated to determine saturating concentrations between 10–30 μg/mL. Further information on the cytotoxicity mechanism is available in the Online Supplementary Appendix.
Methylcellulose clonogenic assays
In the methylcellulose clonogenic assay, effector and target cells were co-incubated for 4 h in comparable conditions to the chromium release assay then injected into MethoCult methylcellulose formation (Stemcell Technologies) as previously described and explained in more detail in the Online Supplementary Appendix.26
Secondary replating of methylcellulose colonies
Secondary replating was used to compare the self-renewal of residual MM colonies after treatment with NK-92 or KHYG-1 to the low density control. Colonies were enumerated after one week and the self-renewal of primary colonies was evaluated by replating pooled and individual colonies. Experimental controls and equations for calculating the percentage of primary clonogenic inhibition, percentage of secondary clonogenic inhibition and cumulative clonogenic inhibition are explained in more detail in the Online Supplementary Appendix.
Mice
This study was approved by the institutional Animal Care Committee (Protocol Number: 1991.4) and performed in accordance with the Canadian Council on Animal Care Guidelines. NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratories) were cared for in the Ontario Cancer Institute animal facility. Mice were monitored frequently for MM disease progression and sacrificed when they showed symptoms of hind limb paralysis, lethargy, weight loss or in the absence of symptoms at ten weeks.
NK-92 cell line therapy in vivo
Six to 10-week old male mice were injected intravenously with 2×106 U266eGFPluc MM cells in 200 μL of Plasma-Lyte (Baxter) on Day 0 to establish a MM xenograft model. Beginning on Day 7, treatment mice were injected intravenously with 10×106 NK-92 cells in 200 μL of Plasma-Lyte. A total of five doses were administered to each treatment mouse, each five days apart. NK-92 cell treatment mice and MM only control mice were imaged weekly starting at four weeks after MM injection and sacrificed at ten weeks after MM injection to compare bone marrow engraftment of MM cells by flow cytometry.
In vivo bioluminescence imaging
Information on in vivo bioluminescence imaging is described in more detail in the Online Supplementary Appendix.
Bone marrow engraftment by flow cytometry
At the time of sacrifice, bone marrow was harvested from the femur and tibia by flushing the bones with media. Red blood cells were lysed by adding ACK buffer (0.155 M NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA in distilled H2O) to the cell suspension for 15 min at room temperature and vortexing regularly. Cells were centrifuged at 600 g for 4 min then resuspended in PBS with 2% FBS and 1% EDTA. All cells were filtered and 7-AAD was added to samples to discriminate live and dead cells prior to analysis on an FC500 flow cytometer (Beckman Coulter). Bone marrow engraftment was quantified by the percentage of GFP positive cells. Analysis of MM bone marrow engraftment (GFP positive) was performed with Flowjo v7.6 software (Tree Star).
Statistics
In vitro data presented are the mean ± SD of three replicates representative of at least 2 separate experiments, unless stated otherwise. P values were calculated using a two-tailed Student’s t-test in Prism software to compare the mean of each group. In vivo bioluminescence data are presented as the mean ± SEM of one experiment and P values were calculated using the Mann-Whitney test in Prism software to compare the median of each group.
Results
Cytotoxicity of bulk multiple myeloma cells
In the chromium release assay, NK-92 effectively killed three MM cell lines at a 10:1 E:T ratio: U266 (80%), NCI-H929 (30%) and RPMI 8226 (25%) (Figure 1A). Interestingly, one of the MM cell lines, U266 was killed better by NK-92 than the positive control K562 at E:T ratios up to 20:1. KHYG-1 also showed cytotoxicity against the same panel of MM cell lines with lysis percentage at a 10:1 E:T ratio as follows: RPMI 8226 (50%), U266 (40%), NCI-H929 (30%) (Figure 1B). A dose response was observed for NK-92 and KHYG-1 cytotoxicity against MM cell lines in the chromium release assay. Similarly, in the flow cytometry cytotoxicity assay a dose response was observed with increasing E:T ratio (Figure 1C). The percentage of cytotoxicity of NK-92 against MM cell lines by flow cytometry at a 10:1 E:T ratio was: U266 (90%), RPMI 8226 (50%) and NCI-H929 (50%) (Figure 1D). The percentage of cytotoxicity of KHYG-1 against all three MM cell lines was 60–70% at the 10:1 E:T ratio (Figure 1E). These results reinforce our observations that NK-92 kills U266 better than H929 and RPMI 8226, whereas KHYG-1 had similar killing of all three MM cell lines. In addition, NK-92 killed U266 better than KHYG-1, whereas, KHYG-1 killed RPMI 8226 and NCI-H929 more effectively than NK-92.
Figure 1.

Bulk killing of RPMI 8226 (black), NCI-H929 (dark gray), and U266 (light gray) MM cell lines and K562 positive control (white) by NK-92 (A) and KHYG-1 (B). (C) Flow cytometry cytotoxicity gating strategy: CD7 negative target cells were selected and cell death was detected with 7-AAD and Annexin V. (D) NK-92 flow cytometry cytotoxicity. (E) KHYG-1 flow cytometry cytotoxicity.
Mechanism of cytotoxicity
To understand the mechanism of recognition of MM cells by NK-92 and KHYG-1, expression of activating receptors was evaluated on NK-92 and KHYG-1 (Online Supplementary Figure S1A). NK receptors were blocked with monoclonal antibodies prior to co-incubation of NK and MM cells in the flow cytometry cytotoxicity assay. Blocking NKp30 demonstrated a significant reduction in killing of RPMI 8226 (P=0.0001; P=0.0003), NCI-H929 (P=0.0012; 0.0009) and U266 (P=0.0004; P=0.0016) cells by NK-92 and KHYG-1, respectively (Figure 2A and Online Supplementary Figure S1A). This demonstrates NKp30 has an important role in MM recognition by NK cell lines. Blocking DNAM-1 caused a signficant reduction in the cytotoxicity of RPMI 8226 (P=0.0001) and NCI-H929 (P=0.0039) by NK-92 (Figure 2A), corresponding with the higher expression of CD112 by RPMI 8226 and NCI-H929 (Online Supplementary Figure S1B). In addition, blocking NKG2D caused a significant reduction in killing of U266 (P=0.0099) by NK-92 (Figure 2B), also corresponding to the highest expression of MICA/MICB by U266 compared to the other MM cell lines (Online Supplementary Figure S1B). Conversely, NKp46 was not important in NK-92 killing, but was involved with KHYG-1 killing of NCI-H929 (P=0.040) (Figure 2B).
Figure 2.
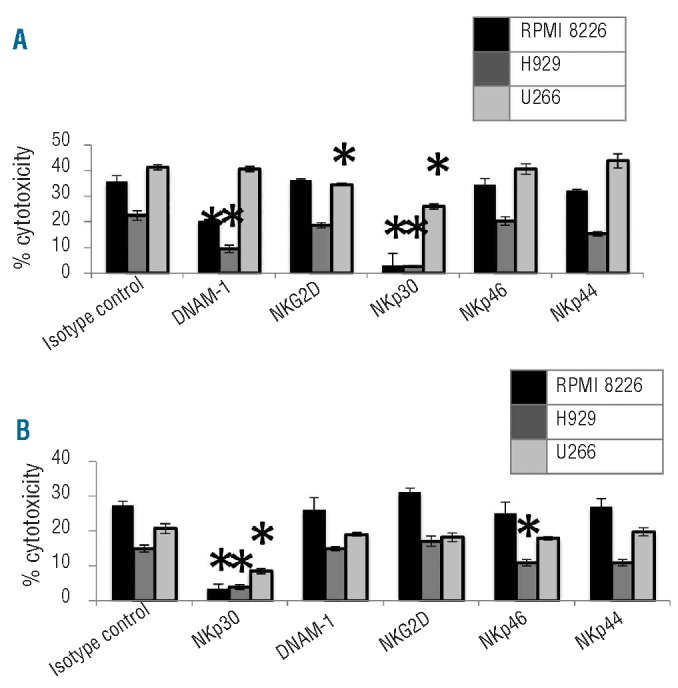
Functional relevance of blocking activating receptors on NK-92 and KHYG-1 involved with killing RPMI 8226 (black), NCI-H929 (dark gray) and U266 (light gray) MM cells compared to the killing when NK cells are blocked with an isotype control. (A) NKp30, DNAM-1 and NKG2D demonstrate a functional role in NK-92 killing of MM cell lines. (B) NKp30 and NKp46 demonstrate a functional role in KHYG-1 killing of MM cell lines. *Significant difference.
Cytotoxicity of NK cell lines against clonogenic multiple myeloma cells
After co-incubating NK and MM cells in comparable conditions to the chromium release assay (treatment), clonogenic capacity was compared with MM cells and NK cells incubated separately then co-injected into the same tube of methylcellulose (low density control). Representative pictures of RPMI 8226 colony formation showed fewer colonies after treatment with KHYG-1 (Figure 3A) compared to the number of colonies in the low density control (Figure 3B). The controls for this sample showed that NK cell lines did not form colonies in methylcellulose and RPMI 8226 cells alone formed a similar number of colonies to the low density control (data not shown). Similar results were obtained for RPMI 8226 treated with NK-92 and NCI-H929 treated with NK-92 and KHYG-1.
Figure 3.

Representative methyl-cellulose colony growth at two weeks. (A) RPMI 8226 MM cell lines treated with KHYG-1 at a 20:1 E:T ratio. (B) Low density control: MM cells and NK cells incubated separately then co-injected into methylcellulose. Preferential killing of the clonogenic fraction of MM cells detected in a methylcellulose cytotoxicity assay (black) compared to bulk killing detected in a chromium release assay (white) by NK cell lines. (C) Preferential killing of clonogenic RPMI 8226 cells by NK-92. (D) Preferential killing of clonogenic RPMI 8226 cells by KHYG-1.
Inhibition of clonogenic growth was compared with the cytotoxicity detected in the bulk population by the chromium release assay performed on the same day. At a 10:1 E:T ratio, there was a 70% and 90% inhibition of RPMI 8226 colony growth, in comparison to a chromium release assay cytotoxicity percentage of only 25% and 50% for NK-92 and KHYG-1, respectively (Figure 3C and 3D). Similarly, NK-92 and KHYG-1 mediated inhibition of 85% of NCI-H929 colony growth, whereas the percentage of cytotoxicity of bulk tumor cells was only 30% and 25%, respectively (Online Supplementary Figure S2A and B). These results indicate that NK-92 and KHYG-1 substantially inhibit clonogenic growth of RPMI 8226 and NCI-H929 MM cells.
Self-renewal of clonogenic multiple myeloma cells
When pooled colonies were replated at 1000 cells/mL, residual RPMI 8226 and NCI-H929 colonies previously treated with NK-92 formed significantly fewer colonies (RPMI 8226, P=0.0186; NCI-H929, P<0.0001) compared to the number of colonies formed by replating the low density control (Figure 4A and 4C). In addition, the residual RPMI 8226 colonies previously treated with KHYG-1 also formed significantly fewer colonies than the low density control (RPMI 8226, P=0.0007) (Figure 4E). However, the residual NCI-H929 colonies previously treated with KHYG-1 and the low density control colonies formed the same number of colonies (NCI-H929 P=1.00) in a secondary replating experiment (Figure 4G).
Figure 4.
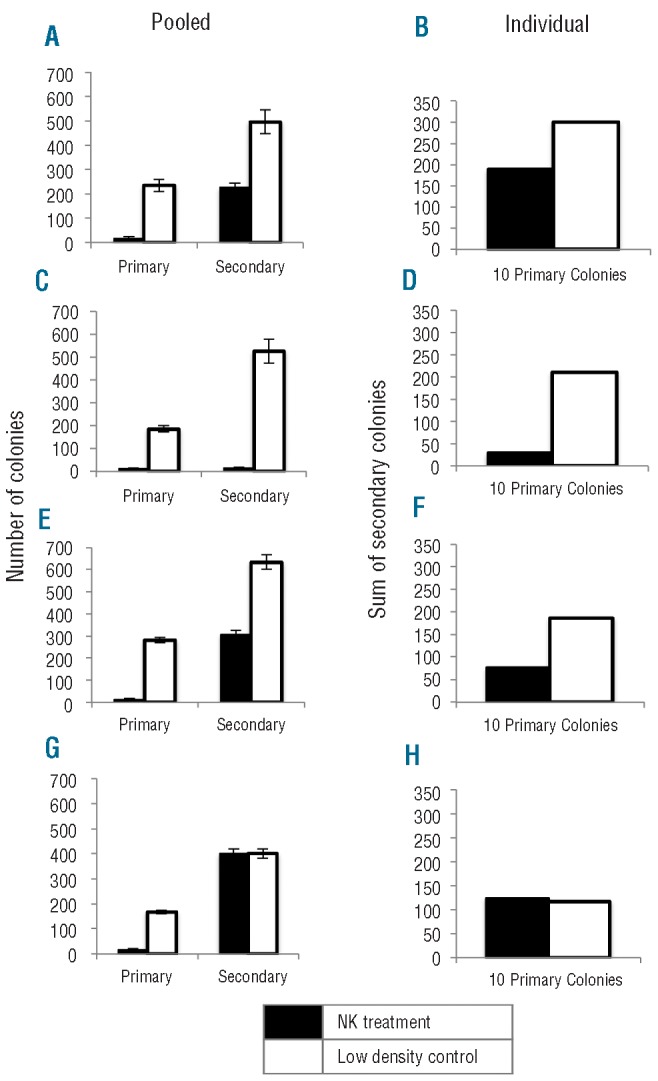
NK cell lines reduce the self renewal of MM colonies in a secondary replating experiment after treatment with NK cell lines at 20:1 E:T. Number of (A) pooled and (B) individual RPMI 8226 colonies formed from cells previously treated with NK-92 (black) or low density control (white). Number of (C) pooled and (D) individual NCI-H929 colonies formed from cells previously treated with NK-92 (black) or low density control (white). Number of (E) pooled and (F) individual RPMI 8226 colonies formed from cells previously treated with KHYG-1 (black) or low density control (white). Number of (G) pooled and (H) individual NCI-H929 colonies formed from cells previously treated with KHYG-1 (black) or low density control (white).
When individual colonies were replated, similar results to those obtained with pooled colonies were observed. The number of secondary colonies formed from a given colony was highly heterogenous and ranged between 0 and 84 secondary colonies. Ten residual RPMI 8226 and NCI-H929 colonies were replated and the sum of the secondary colonies after initial NK cell treatment was less than the sum of ten residual colonies from the low density control samples in which NK and MM cells were not initially co-incubated for 4 h in a 96-well plate (Figure 4B and D). This demonstrates that the self-renewal of residual MM colonies was reduced after treatment with NK-92. Similarly, the sum of secondary colonies formed from ten residual RPMI 8226 colonies after treatment with KHYG-1 also formed fewer colonies than low density control cells (Figure 4F). However, the sum of secondary colonies formed from NCI-H929 cells previously treated with KHYG-1 was the same as the low density control (Figure 4H). Therefore, KHYG-1 reduced the secondary clonogenic capacity of RPMI 8226 cells, but not NCI-H929 cells.
Cumulative inhibition of clonogenic growth
The cumulative clonogenic inhibition, which accounts for the self-renewal of secondary colonies, can be calculated from both methods of secondary replating at a 20:1 E:T ratio. Regardless of whether individual or pooled colonies were replated, similar results in the cumulative clonogenic inhibition calculation were observed. The cumulative clonogenic inhibition of RPMI 8226 was 96% and 98%, respectively (Online Supplementary Figure S3A) and the cumulative clonogenic inhibition of NCI-H929 was 99% and 89% by NK-92 and KHYG-1, respectively (Online Supplementary Figure S3B). The impact of including the self-renewal of residual primary colonies in the cumulative clonogenic inhibition calculation is shown in comparison to the clonogenic growth inhibition calculated from primary MM colony formation after NK cell treatment (Online Supplementary Figure S3C). Including self renewal capacity in this calculation increases the detected clonogenic inhibition by 0–6%.
NK-92 cell therapy in vivo
Given that NK-92 has been tested in phase I clinical trials with minimal toxicity, we further evaluated its efficacy in an in vivo model. We used a U266eGFPluc bioluminescent NSG mouse model to evaluate efficacy of NK-92 cell therapy in vivo. We injected 10×106 NK-92 cells every five days to a total dose of 5.0×107 cells seven days after MM injection. Tumor burden was monitored weekly by bioluminescence imaging four weeks after MM inoculation using the IVIS® Imaging System, and LivingImageTM Software was used to acquire images and quantify bioluminescence (Figure 5A and B). We showed that U266eGFPluc cells localized to BM and spine, reflecting MM pathophysiology (Figure 5C and D). Disease burden in the NK-92 treated group was consistently lower than controls over time and significantly lower at eight weeks (Dorsal and Ventral Mann-Whitney, P=0.0381). In a separate experiment, clinical disease progression in MM control mice correlated with IVIS signal intensity at week 11 (r2=0.4; F test P=0.0279; data not shown).
Figure 5.

In vivo cytotoxicity of NK-92 monitored over time by bioluminescence imaging from (A) dorsal and (B) ventral views with significantly lower tumor burden in NK-92 treated mice at eight weeks (Dorsal and Ventral P=0.0381). Tumor distribution at week 8 from (C) dorsal and (D) ventral views. BM engraftment of GFP+ MM cells at ten weeks for MM only control mice and NK-92 treated MM mice (E) (P=0.019) Line represents median of each sample; each dot represents BM engraftment of one mouse. Sample Flowjo images of GFP+ cells in MM only control mice and NK-92 treated mice (F). *Significant difference.
Engraftment was determined by sacrificing mice at ten weeks and analyzing BM for GFP+ cells by flow cytometry. Engraftment of MM cells in the BM for control mice was 5±1.9% and for NK-92 treated mice was 0.24±0.19% (mean±SEM). There was a trend toward a significant decrease in mean engraftment for the NK-92 group versus control (unpaired Student’s t-test P=0.055). One of 6 control mice had low engraftment with U266eGFPluc at ten weeks increasing the variance of the control mean and prompted an analysis of median engrafment. Using the Mann-Whitney test, there was a statistically significant decrease in median BM engraftment in the NK-92 treated group (Mann-Whitney P=0.019) (Figure 5E). Furthermore, all mice treated with NK-92 had less than 1% MM cells detected in the BM, which is below the typical definition of tumor cell engraftment.
In addition, GFP BM engraftment corresponded with bioluminescence detected in R and L BM by IVIS and representative samples of GFP BM engraftment detected by flow cytometry are shown for MM only control mice and NK-92 treated mice (Figure 5F).
Discussion
Our study demonstrates that both NK-92 and KHYG-1 are highly cytotoxic against MM cell lines with greater killing of the clonogenic fraction. Autologous, allogeneic, and KIR ligand-mismatched primary NK cell therapies and the NK cell line YT have been tested in MM, but the reported cytotoxicity is generally lower than our results for NK-92 and KHYG-1.12–15,28 NK-92 has previously demonstrated killing activity against a panel of MM cell lines using the chromium release assay.19,21 The in vitro cytotoxicity of KHYG-1 has been evaluated against leukemia, Burkitt’s lymphoma, and neuroblastoma cell lines, but KHYG-1 has not been tested against MM in vitro.23,29–31 Our present study is the first to demonstrate the cytotoxicity of KHYG-1 against MM cells and has shown that KHYG-1 kills the RPMI 8226 and NCI-H929 MM cell lines better than NK-92.
We evaluated the functional relevance of several activating receptors involved in NK-92 and KHYG-1 killing of MM cells. NK-92 killing of RPMI 8226 and NCI-H929 was DNAM-1 dependent, whereas killing of U266 was NKG2D dependent. A previous study also demonstrated a functional role for DNAM-1 or NKG2D in primary NK cell cytotoxicity against MM.32 Similarly, we have shown higher expression of Nectin-2, a ligand for DNAM-1, on RPMI 8226 and NCI-H929 compared to U266 and higher expression of MICA/MICB, a ligand of NKG2D, on U266 compared to RPMI 8226 and NCI-H929 cells. The greatest cytotoxicity was observed with the NK-92 killing of U266, suggesting an important role of NKG2D in this recognition. NKG2D and DNAM-1 do not have a functional role in KHYG-1 mediated killing.
The cytotoxicity of NK-92 and KHYG-1 against U266, RMPI 8226 and NCI-H929 was NKp30 dependent for all three MM cell line targets with a reduction in killing ranging from 40–85% upon blocking NKp30. Others have shown an important role of the NCRs in MM cytotoxicity by primary NK cells.33 However, this study blocked all three NCRs simultaneously, and this meant that the relative contributions of NKp30, NKp44 and NKp46 were unclear. In other studies, NKp30 and NKp46 were shown to be involved in recognition of MM cells by primary NK cells.32,34 Therefore, our work is the first to demonstrate an important functional role of NKp30 in MM recognition by NK cell lines.
Upregulation of DNAM-1 and NKG2D ligands on MM cell lines and an increase in NK-cell degranulation was shown after treatment of MM cells with sub-therapeutic doses of doxorubicin and melphalan.35 Another study demonstrated that HSP-90 inhibitors increased MICA/MICB expression on MM cells and that NK-cell degranulation increased in an NKG2D-dependent mechanism.36 Our study showed an important role of NKG2D and DNAM-1 in NK-92 killing of MM, suggesting that the cytotoxicity may be enhanced by pre-treating MM cells with doxorubicin, melphalan or HSP inhibitors.
To evaluate the ability of NK cells to target clonogenic cells, we had previously established an immune effector methylcellulose clonogenic assay with KG1 leukemia cells to compare with the standard chromium release and flow cytometric cytotoxicity assays.26 Using a clonogenic readout has the advantage of assessing the effect of a cytotoxic agent against the subset of cells capable of greater proliferation and can be used, therefore, to assess novel therapies. One limitation is that inhibition of clonogenic growth could be either from death of clonogenic cells or from pushing them into a cytostatic quiescent state. Rendering a cancer stem cell dead or cytostatic would clinically be the same, but a quiescent cancer stem cell could still recapitulate disease. To address this issue, we utilized a secondary replating assay to determine the residual colony forming capacity after NK cell exposure. Both NK-92 and KHYG-1 demonstrated clonogenic growth inhibition against the clonogenic population in the RPMI 8226 and NCI-H929 MM cell lines. Comparison of the chromium release assay and flow cytometric cytotoxicity assay with the methylcellulose clonogenic assays approximate the relative capacity of each NK cell line to mediate cytotoxicity against bulk and clonogenic cells. In some cases, the difference is 2–3 fold, providing some evidence of greater toxicity against a population with cancer stem cell properties, without requiring immunophenotypic markers. A previous study demonstrated in vitro cytotoxicity of autologous activated marrow-infiltrating T-lymphocytes (MILs) against CD138 positive plasma cells and growth inhibition of clonogenic MM cells37 similar to our findings with NK cell lines.
Secondary replating has been used to characterize clonogenic MM cells, but this method could also be useful for evaluating novel cancer stem cell targeting therapeutics in MM, as we have demonstrated in this study. Self-renewal of primary MM colonies has been evaluated by replating individual colonies and demonstrating that single primary MM colonies form between 0 to 65 secondary colonies,10 consistent with our findings. In addition, replating pooled colonies enriched the colony forming unit (CFU) frequency from 1% up to 20%.10 Most importantly, a therapy that can target clonogenic MM cells is clinically relevant as patient samples that form clonogenic cells in vitro are correlated with significantly shorter patient survival.10
To better quantify the total growth inhibition of clonogenic MM cells by NK cells, we considered the secondary MM clonogenic capacity in our cumulative clonogenic inhibition calculation. Residual RPMI 8226 and NCI-H929 colonies previously treated with NK-92 and RPMI 8226 cells previously treated with KHYG-1 formed significantly fewer secondary colonies compared with the number of secondary colonies in the low density control. These results indicate that after initial treatment, residual colonies have a reduced clonogenic capacity compared to the secondary replating of the low density control. Importantly, we observed similar results between secondary replating of pooled and individual colonies, indicating that the cells interspersed between colonies have no further clonogenic potential. This implies that NK cell cytotoxicity can affect long-term proliferative capacity of clonogenic cells detectable only in a secondary replating assay. This could be from low grade DNA damage that is not overtly cytotoxic but allows for only a limited number of cell cycles, or potentially from epigenetic changes that limit the proliferative capacity of clonogenic MM cells.
Secondary replating has been used to evaluate KIR ligand-mismatched NK cell cytotoxicity of leukemia stem cells in a clonogenic assay.38 That study was the first to evaluate the self-renewal of clonogenic cells after immune effector cell treatment, but cumulative clonogenic inhibition to account for self-renewal of residual colonies after NK cell treatment was not calculated. We determined the clonogenic inhibition of NCI-H929 by NK-92 to be 93% at 20:1 (E:T) ratio, and when the self renewal of residual NCI-H929 colonies was included, the cumulative clonogenic inhibition was 99%. When the self renewal of MM colonies was considered in the cumulative clonogenic inhibition calculation, the clonogenic inhibition increased by 0–6%.
Finally, a bioluminescent, xenograft MM mouse model was established to evaluate the cytotoxicity of NK-92 in vivo. The BM microenvironment is important for survival and proliferation of MM cells, reinforcing the importance of orthotopic mouse models for evaluating new therapies. Intravenous injection of the U266eGFPluc cell line in non-irradiated NSG mice facilitated engraftment in the bone marrow of long bones and the axial skeleton, similar to the engraftment of this cell line in previous studies with irradiated RAG2−/−γc−/− mice and irradiated NOG mice.39,40
The biodistribution of MM was visualized using real-time, non-invasive bioluminescence imaging (BLI) to quantify disease progression. In an intravenous injection model, BLI is valuable for quantifying the biodistribution of tumor cells, especially in MM when tumors may localize to the spine.39,41 In our study, NK-92 cell therapy reduced the tumor burden compared with controls as measured by BLI and by MM bone marrow engraftment at the study end point. This demonstrates that NK-92 can distribute to sites of MM involvement and affect myeloma progression.
NK-92 is the first NK cell line to be tested in clinical trials. A phase I clinical trial demonstrated large-scale expansion ability and safety of NK-92 in patients with advanced melanoma and renal cancer.22 Other ongoing phase I clinical trials are evaluating safety and the maximum tolerated dose of NK-92 in patients with refractory or relapsed AML and other advanced cancers, with preliminary observations suggesting that the infusion of up to 3×109 irradiated NK-92 cells is safe.18,42 We also are conducting a phase I trial of NK-92 in patients with relapsed/refractory hematologic malignancies at therapeutically relevant doses.43 However, given its ability to target drug resistant clonogenic cells, NK cell therapy is likely to be most useful in eradicating minimal residual disease after chemotherapy.
In conclusion, NK-92 and KHYG-1 demonstrate growth inhibition of clonogenic MM cells which are typically drug resistant. Given that the MM CFU frequency from bone marrow at diagnosis correlates with survival10 these findings have important clinical implications. We have also established a human MM cell line xenograft model in NSG mice that mimics clinical disease. MM progression was reduced by NK-92 as measured by BLI and by the reduction of engrafted U266eGFPluc cells. While standard of care therapies may prolong survival, relapse occurs in the majority of cases, indicating that MM stem cells are not eliminated.6 Our work suggests that clinical investigation of NK cell lines as therapeutic agents for the treatment of MM is warranted.
Acknowledgments
The authors would like to thank Dr. Suzanne Trudel, Princess Margaret Hospital, Toronto, ON, CAN for providing the RPMI 8226 and NCI-H929 MM cell lines and Dr. Hans Klingemann for NK-92.
Footnotes
Funding: this work was supported by an Ontario Regional Biotherapeutics (ORBiT) Program grant from the Ontario Institute for Cancer Research to AK and JM. BES was supported by the Natural Sciences Engineering and Research Council of Canada and an Ontario Graduate Scholarship, BAW was supported by a Terry Fox Clinical Research Fellowship from the National Cancer Institute of Canada and a Canadian Institute for Health Research Clinician Scientist Award, JM was supported by Spanish Health Office Grant BA09/90019 and CRIS Foundation, AK holds the Gloria and Seymour Epstein Chair in Cell Therapy and Transplantation at the University Health Network and the University of Toronto.
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Kyle RA, Rajkumar SV. Multiple myeloma. Blood. 2008;111(6):2962–72. doi: 10.1182/blood-2007-10-078022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046–60. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 3.Brenner H, Gondos A, Pulte D. Recent major improvement in long-term survival of younger patients with multiple myeloma. Blood. 2008;111(5):2521–6. doi: 10.1182/blood-2007-08-104984. [DOI] [PubMed] [Google Scholar]
- 4.Martinez-Lopez J, Blade J, Mateos MV, Grande C, Alegre A, García-Laraña J, et al. Long-term prognostic significance of response in multiple myeloma after stem cell transplantation. Blood. 2011;118(3):529–34. doi: 10.1182/blood-2011-01-332320. [DOI] [PubMed] [Google Scholar]
- 5.Hamburger A, Salmon SE. Primary bioassay of human myeloma stem cells. J Clin Invest. 1977;60(4):846–54. doi: 10.1172/JCI108839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008;68(1):190–7. doi: 10.1158/0008-5472.CAN-07-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng Y, Wen J, Mike P, Choi DS, Eshoa C, Shi ZZ, et al. Bone marrow stromal cells from myeloma patients support the growth of myeloma stem cells. Stem Cells Dev. 2010;19(9):1289–96. doi: 10.1089/scd.2010.0010. [DOI] [PubMed] [Google Scholar]
- 8.Jakubikova J, Adamia S, Kost-Alimova M, Klippel S, Cervi D, Daley JF, et al. Lenalidomide targets clonogenic side population in multiple myeloma: pathophysiologic and clinical implications. Blood. 2011;117(17):4409–19. doi: 10.1182/blood-2010-02-267344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, et al. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103(6):2332–6. doi: 10.1182/blood-2003-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi T, Lim B, Jamal N, Tritchler D, Lockwood G, McKinney S, et al. Colony growth and self renewal of plasma cell precursors in multiple myeloma. J Clin Oncol. 1985;3(12):1613–23. doi: 10.1200/JCO.1985.3.12.1613. [DOI] [PubMed] [Google Scholar]
- 11.Alici E, Konstantinidis KV, Sutlu T, Aints A, Gahrton G, Ljunggren HG, et al. Anti-myeloma activity of endogenous and adoptively transferred activated natural killer cells in experimental multiple myeloma model. Exp Hematol. 2007;35(12):1839–46. doi: 10.1016/j.exphem.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Alici E, Sutlu T, Bjorkstrand B, Gilljam M, Stellan B, Nahi H, et al. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood. 2008;111(6):3155–62. doi: 10.1182/blood-2007-09-110312. [DOI] [PubMed] [Google Scholar]
- 13.Shi J, Tricot G, Szmania S, Rosen N, Garg TK, Malaviarachchi PA, et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol. 2008;143(5):641–53. doi: 10.1111/j.1365-2141.2008.07340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frohn C, Hoppner M, Schlenke P, Kirchner H, Koritke P, Luhm J. Anti-myeloma activity of natural killer lymphocytes. Br J Haematol. 2002;119(3):660–4. doi: 10.1046/j.1365-2141.2002.03879.x. [DOI] [PubMed] [Google Scholar]
- 15.Feng X, Yan J, Wang Y, Zierath JR, Nordenskjold M, Henter JI, et al. The proteasome inhibitor bortezomib disrupts tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression and natural killer (NK) cell killing of TRAIL receptor-positive multiple myeloma cells. Mol Immunol. 2010;47(14):2388–96. doi: 10.1016/j.molimm.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Lokhorst H, Einsele H, Vesole D, Bruno B, San Miguel J, Perez-Simon JA, et al. International Myeloma Working Group consensus statement regarding the current status of allogeneic stem-cell transplantation for multiple myeloma. J Clin Oncol. 2010;28(29):4521–30. doi: 10.1200/JCO.2010.29.7929. [DOI] [PubMed] [Google Scholar]
- 17.Bruno B, Rotta M, Patriarca F, Mordini N, Allione B, Carnevale-Schianca F, et al. A Comparison of Allografting with Autografting for Newly Diagnosed Myeloma. NEJM. 2007;356(11):1110–20. doi: 10.1056/NEJMoa065464. [DOI] [PubMed] [Google Scholar]
- 18.Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res. 2001;10(4):535–44. doi: 10.1089/15258160152509145. [DOI] [PubMed] [Google Scholar]
- 19.Maki G, Hayes GM, Naji A, Tyler T, Carosella ED, Rouas-Freiss N, et al. NK resistance of tumor cells from multiple myeloma and chronic lymphocytic leukemia patients: implication of HLA-G. Leukemia. 2008;22(5):998–1006. doi: 10.1038/leu.2008.15. [DOI] [PubMed] [Google Scholar]
- 20.Yan Y, Steinherz P, Klingemann HG, Dennig D, Childs BH, McGuirk J, et al. Antileukemia activity of a natural killer cell line against human leukemias. Clin Cancer Res. 1998;4(11):2859–68. [PubMed] [Google Scholar]
- 21.Klingemann H, Wong E, Maki G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol Blood Marrow Transplant. 1996;2(2):68–75. [PubMed] [Google Scholar]
- 22.Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10(6):625–32. doi: 10.1080/14653240802301872. [DOI] [PubMed] [Google Scholar]
- 23.Suck G, Branch DR, Smyth MJ, Miller RG, Vergidis J, Fahim S, et al. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp Hematol. 2005;33(10):1160–71. doi: 10.1016/j.exphem.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 24.Johnson TR, Massey RJ, Deinhardt F. Lymphocyte and antibody cytotoxicity to tumor cells measured by a micro- 51 chromium release assay. Immunol Commun. 1972;1(3):247–61. doi: 10.3109/08820137209022939. [DOI] [PubMed] [Google Scholar]
- 25.Ozdemir O, Ravindranath Y, Savasan S. Cell-mediated cytotoxicity evaluation using monoclonal antibody staining for target or effector cells with annexinV/propidium iodide colabeling by fluorosphere-adjusted counts on three-color flow cytometry. Cytometry Part A: The Journal of the International Society for Analytical Cytology. 2003;56(1):53–60. doi: 10.1002/cyto.a.10081. [DOI] [PubMed] [Google Scholar]
- 26.Williams BA, Wang XH, Keating A. Clonogenic assays measure leukemia stem cell killing not detectable by chromium release and flow cytometric cytotoxicity assays. Cytotherapy. 2010;12(7):951–60. doi: 10.3109/14653241003628167. [DOI] [PubMed] [Google Scholar]
- 27.Harnack U, Johnen H, Pecher G. Natural killer cell line YT exerts cytotoxicity against CD86+ Myeloma Cells. Anticancer Res. 2011;31(2):475–80. [PubMed] [Google Scholar]
- 28.Yagita M, Huang CL, Umehara H, Matsuo Y, Tabata R, Miyake M, et al. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia. 2000;14(5):922–30. doi: 10.1038/sj.leu.2401769. [DOI] [PubMed] [Google Scholar]
- 29.Suck G, Branch DR, Aravena P, Mathieson M, Helke S, Keating A. Constitutively polarized granules prime KHYG-1 NK cells. Int Immunol. 2006;18(9):1347–54. doi: 10.1093/intimm/dxl071. [DOI] [PubMed] [Google Scholar]
- 30.Suck G, Branch DR, Keating A. Irradiated KHYG-1 retains cytotoxicity: potential for adoptive immunotherapy with a natural killer cell line. Int J Radiat Biol. 2006;82(5):355–61. doi: 10.1080/09553000600649653. [DOI] [PubMed] [Google Scholar]
- 31.Carlson LM, Pahlman S, De Geer A, Kogner P, Levitskaya J. Differentiation induced by physiological and pharmacological stimuli leads to increased antigenicity of human neuroblastoma cells. Cell Res. 2008;18(3):398–411. doi: 10.1038/cr.2008.27. [DOI] [PubMed] [Google Scholar]
- 32.El-Sherbiny YM, Meade JL, Holmes TD, McGonagle D, Mackie SL, Morgan AW, et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007;67(18):8444–9. doi: 10.1158/0008-5472.CAN-06-4230. [DOI] [PubMed] [Google Scholar]
- 33.Carbone E, Neri P, Mesuraca M, Fulciniti MT, Otsuki T, Pende D, et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood. 2005;105(1):251–8. doi: 10.1182/blood-2004-04-1422. [DOI] [PubMed] [Google Scholar]
- 34.Pogge von Strandmann E, Simhadri VR, von Tresckow B, Sasse S, Reiners KS, Hansen HP, et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity. 2007;27(6):965–74. doi: 10.1016/j.immuni.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113(15):3503–11. doi: 10.1182/blood-2008-08-173914. [DOI] [PubMed] [Google Scholar]
- 36.Fionda C, Soriani A, Malgarini G, Iannitto ML, Santoni A, Cippitelli M. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. J Immunol. 2009;183(7):4385–94. doi: 10.4049/jimmunol.0901797. [DOI] [PubMed] [Google Scholar]
- 37.Noonan K, Matsui W, Serafini P, Carbley R, Tan G, Khalili J, et al. Activated marrow-infiltrating lymphocytes effectively target plasma cells and their clonogenic precursors. Cancer Res. 2005;65(5):2026–34. doi: 10.1158/0008-5472.CAN-04-3337. [DOI] [PubMed] [Google Scholar]
- 38.Langenkamp U, Siegler U, Jorger S, Diermayr S, Gratwohl A, Kalberer CP, et al. Human acute myeloid leukemia CD34+CD38- stem cells are susceptible to allorecognition and lysis by single KIR-expressing natural killer cells. Haematologica. 2009;94(11):1590–4. doi: 10.3324/haematol.2009.005967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rozemuller H, van der Spek E, Bogers-Boer LH, Zwart MC, Verweij V, Emmelot M, et al. A bioluminescence imaging based in vivo model for preclinical testing of novel cellular immunotherapy strategies to improve the graft-versus-myeloma effect. Haematologica. 2008;93(7):1049–57. doi: 10.3324/haematol.12349. [DOI] [PubMed] [Google Scholar]
- 40.Miyakawa Y, Ohnishi Y, Tomisawa M, Monnai M, Kohmura K, Ueyama Y, et al. Establishment of a new model of human multiple myeloma using NOD/SCID/gammac(null) (NOG) mice. Biochem Biophys Res Commun. 2004;313(2):258–62. doi: 10.1016/j.bbrc.2003.11.120. [DOI] [PubMed] [Google Scholar]
- 41.Mitsiades CS, Mitsiades NS, Bronson RT, Chauhan D, Munshi N, Treon SP, et al. Fluorescence imaging of multiple myeloma cells in a clinically relevant SCID/NOD in vivo model: biologic and clinical implications. Cancer Res. 2003;63(20):6689–96. [PubMed] [Google Scholar]
- 42.ClinicalTrialsgov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Pittsburgh Uo ZRx-101 (NK-92) for the Treatment of Refractory or Relapsed Acute Myeloid Leukemia. Available from: http://www-clinicaltrialsgov/ct2/show/NCT00900809?term=NCT00900809&rank=1 NCT Identifier: NCT00900809. [cited 2011 Mar 16] [Google Scholar]
- 43.University Health, Network T. ClinicalTrialsgov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Safety Study Looking at the Use of a Natural Killer Cell Line Against Hematological Malignancies. Available from: http://wwwclinicaltrials-gov/ct2/show/NCT00990717?term=NCT00990717&rank=1 NCT Identifier: NCT00990717. [cited 2011 Mar 16] [Google Scholar]