

Factor VIII (FVIII) is a key protein of the coagulation cascade where it acts as the cofactor of activated factor IX. Upon activation, FVIII dissociates from von Willebrand factor and binds to phospholipids at the surface of activated platelets to form the tenase complex, thus leading to the activation of factor X, to a burst of generation of thrombin and to the formation of fibrin that consolidates the platelet clot.1 Abnormalities in the gene encoding FVIII lead to hemophilia A (HA), a rare X-linked recessive hemorrhagic disorder that concerns one male in 5000. Depending on the residual activity of FVIII in plasma, HA is classified as severe (<1%), moderate (1–5%) or mild (5–25%).2 Although this classification correlates with the bleeding phenotype, about 10% of severe HA patients rarely bleed spontaneously, indicating that FVIII activity is not the only parameter defining the phenotypic severity of the disease.3
In their recent publication,4 van Bladel et al. investigated the basal state of activation and responsiveness of circulating platelets under conditions of abnormal hemostasis as a potential parameter influencing the bleeding phenotype of the patients. They found that the state of activation of platelets is up-regulated in patients with severe HA. The percentage of platelets expressing CD62P (P-selectin) at their surface was greater in patients with severe HA (15.9%) as compared to that in patients with mild/moderate HA (8.2%) and in healthy controls (6.4%). An earlier report had, however, failed to find signs of enhanced platelet pre-activation in patients with severe HA.5 In order to address the question in vivo in a system that is not perturbed by the different genetic or environmental factors that are typical of the human population, we studied the state of activation and responsiveness of platelets from wild-type mice and FVIII-deficient (FVIII°) mice, an experimental model of severe hemophilia A. To this end, wild-type and FVIII° mice were crossed on the C57Bl/6 background in our laboratory to ensure the lowest genetic variability with the exception of the HA-causing gene alteration.
Blood was collected on ACD-C buffer (124 mM sodium citrate, 130 mM citric acid, 110 mM dextrose, pH 6.5) from 12–14 week old mice by intra-cardiac puncture. Animals were handled according to local ethical authority recommendations (the Charles Darwin University Animal Ethics Committee, Ce5/2010/045). Platelets were purified in the presence of inhibitors (0.3 U/mL apyrase and 100 nM prostaglandin E1) as described.6 Purity of platelets was over 99% as assessed by the expression of CD41 and size estimate (side scatter) by flow cytometry (data not shown). Platelets were then stimulated by the thrombin receptor-activating peptide TRAP-4.7 The state of activation of platelets was estimated by the surface expression of CD62P, of the activated conformation of αIIbβ3 (GPIIb/IIIa) and by the surface binding of fibrinogen, measured by flow cytometry. Results are expressed as mean fluorescence intensities (MFI±SEM) on the total platelet population (Figure 1).
Figure 1.
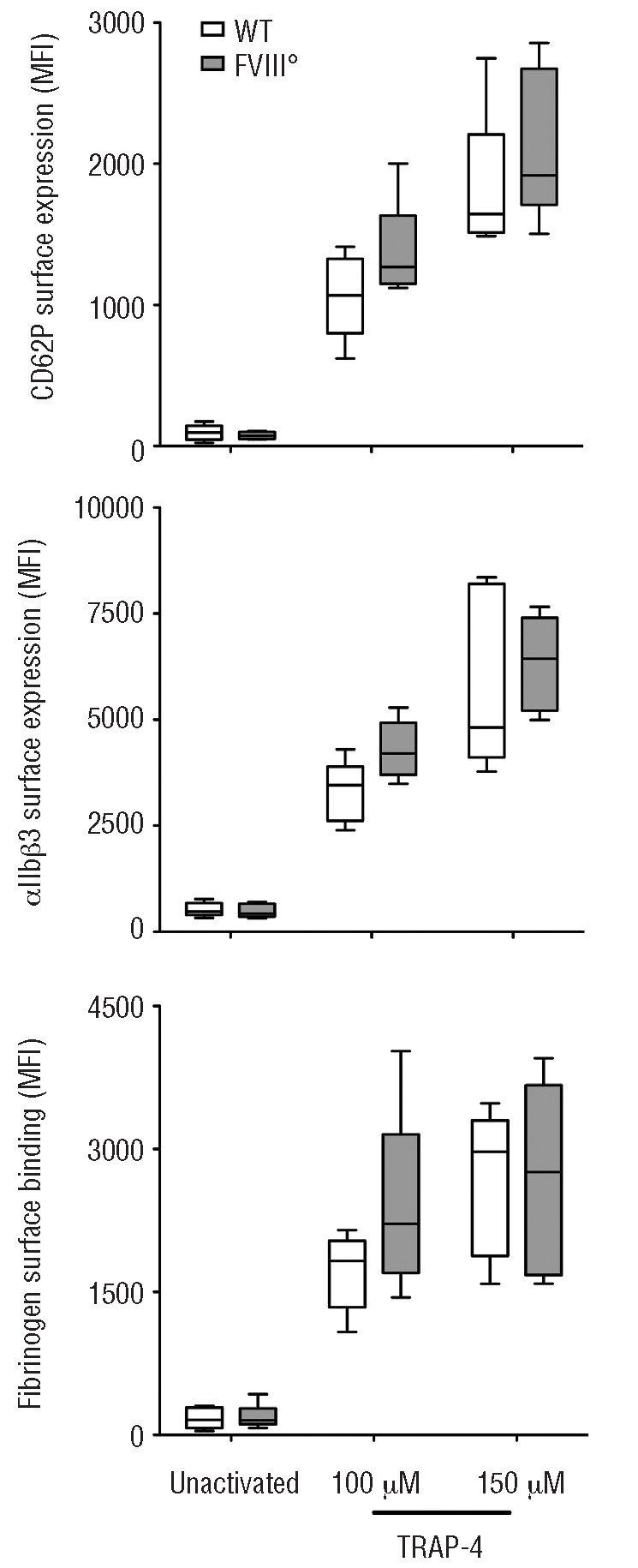
Basal activation status and responsiveness of platelets from FVIII-deficient mice (FVIII°) and their wild-type littermates (WT). Platelets were purified from the blood of WT (open bars) and FVIII° mice (full gray bars). Purified platelets were incubated alone or with 100 μM or 150 μM of TRAP-4 in tyrode buffer for 15 min at room temperature. The surface expression of CD62P (upper panel), αIIbβ3 (middle panel) and surface binding of fibrinogen (lower panel) were determined by flow cytometry using a PE-conjugated rat anti–mouse CD62P monoclonal antibody (Wug.E9), a PE-conjugated rat anti–mouse αIIbβ3 monoclonal antibody (JON/A, specific for the activated conformation of the mouse integrin), and oregon-green-labeled fibrinogen, respectively. Results (5–8 mice per group) are expressed as boxes and whiskers, showing the median, minimum and maximum MFI for each group. Differences were compared statistically using the non-parametric Mann-Whitney test.
Unstimulated platelets demonstrated low levels of expression of CD62P, αIIbβ3 and of surface-bound fibrinogen, that did not differ between wild-type mice and FVIII° mice (wild-type: 85±20, 479±60 and 173±49 MFI, respectively (mean±SEM); FVIII°: 71±9, 519±74, and 217±55 MFI respectively). Interestingly, similar levels of expression of CD62P and αIIbβ3 were measured on platelets in fresh blood (data not shown), suggesting that the purification procedure does not activate the platelets. In the case of wild-type mice, incubation of the platelets with TRAP-4 resulted in a drastic increase in the expression levels of CD62P, αIIbβ3 and in surface-bound fibrinogen (916±138, 3282±236, and 1714±182 MFI, respectively; P<0.05 in all cases as compared to unstimulated platelets) indicating that the purified platelets were functional and susceptible to activation. The incubation of platelets from FVIII° mice with TRAP-4 yielded expression levels of CD62P, αIIbβ3 and levels of surface-bound fibrinogen (1151±186, 4529±851 and 2103±255 MFI, respectively) that were statistically identical to those induced by TRAP-4 on platelets from their wild-type littermates. Similar results were obtained with different concentrations of TRAP-4 (Figure 1) and when thrombin was used instead of TRAP-4 (data not shown).
Taken together, our data indicate that the absence of pro-coagulant FVIII in unmanipulated adult animals is not associated either with alterations in the activation status of circulating platelets or with their ability to be activated. In patients with severe HA, the most significant morbidity is the development of arthropathy in joints that results from recurrent intra-articular hemorrhages. In our laboratory conditions, FVIII° mice do not bleed spontaneously and do not experience premature mortality. Indeed, FVIII° mice do not develop arthropathy unless bleeding is experimentally provoked in the articulations.8,9 Whether the state of activation and responsiveness of platelets differs under conditions of chronic hemorrhages in hemophilic mice is still to be investigated.
Footnotes
Funding: this work was supported by Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, Université Pierre et Marie Curie, Paris 6, and by a grant from CLS-Behring (Marburg, Germany). MT was the recipient of scholarships from Ministère de la Recherche (Paris) and from Fondation de la Recherche Médicale (FRM, Paris).
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Lenting PJ, van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998;92(11):3983–96. [PubMed] [Google Scholar]
- 2.Biggs R, Macfarlane RG. Haemophilia and related conditions: a survey of 187 cases. Br J Haematol. 1958;4(1):1–27. doi: 10.1111/j.1365-2141.1958.tb03830.x. [DOI] [PubMed] [Google Scholar]
- 3.Molho P, Rolland N, Lebrun T, Dirat G, Courpied JP, Croughs T, et al. Epidemiological survey of the orthopaedic status of severe haemophilia A and B patients in France. The French Study Group. secretariat.haemophiles@cch.ap-hop-paris.fr. Haemophilia. 2000;6(1):23–32. doi: 10.1046/j.1365-2516.2000.00358.x. [DOI] [PubMed] [Google Scholar]
- 4.van Bladel ER, Roest M, de Groot PG, Schutgens RE. Up-regulation of platelet activation in hemophilia A. Haematologica. 2011;96(6):888–95. doi: 10.3324/haematol.2011.042663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grunewald M, Siegemund A, Grunewald A, Konegen A, Koksch M, Griesshammer M. Absence of compensatory platelet activation in patients with severe haemophilia, but evidence for a platelet collagen-activation defect. Platelets. 2002;13(8):451–8. doi: 10.1080/0953710021000059422. [DOI] [PubMed] [Google Scholar]
- 6.Jandrot-Perrus M, Lagrue AH, Okuma M, Bon C. Adhesion and activation of human platelets induced by convulxin involve glyco-protein VI and integrin alpha2beta1. J Biol Chem. 1997;272(43):27035–41. doi: 10.1074/jbc.272.43.27035. [DOI] [PubMed] [Google Scholar]
- 7.Adam F, Kauskot A, Nurden P, Sulpice E, Hoylaerts MF, Davis RJ, et al. Platelet JNK1 is involved in secretion and thrombus formation. Blood. 2010;115(20):4083–92. doi: 10.1182/blood-2009-07-233932. [DOI] [PubMed] [Google Scholar]
- 8.Hakobyan N, Enockson C, Cole AA, Sumner DR, Valentino LA. Experimental haemophilic arthropathy in a mouse model of a massive haemarthrosis: gross, radiological and histological changes. Haemophilia. 2008;14(4):804–9. doi: 10.1111/j.1365-2516.2008.01689.x. [DOI] [PubMed] [Google Scholar]
- 9.Valentino LA, Hakobyan N, Kazarian T, Jabbar KJ, Jabbar AA. Experimental haemophilic synovitis: rationale and development of a murine model of human factor VIII deficiency. Haemophilia. 2004;10(3):280–7. doi: 10.1111/j.1365-2516.2004.00899.x. [DOI] [PubMed] [Google Scholar]