

Abstract
Infantile spasms are an age-dependent epilepsy that are highly associated with cognitive impairment, autism, and movement disorders. Previous classification systems have focused on a distinction between symptomatic and cryptogenic etiologies, and have not kept pace with the recent discoveries of mutations in genes in key pathways of central nervous system development in patients with infantile spasms. Children with certain genetic syndromes are much more likely to have infantile spasms, and we review the literature to propose a genetic classification of these disorders. Children with these genetic associations with infantile spasms also have phenotypes beyond epilepsy that may be explained by recent advances in the understanding of underlying biological mechanisms. We therefore also propose a biologic classification of the genes highly associated with infantile spasms, and articulate models for infantile spasms pathogenesis based on that data. The two best described pathways of pathogenesis are abnormalities in the gene regulatory network of GABAergic forebrain development, and abnormalities in molecules expressed at the synapse. We intend for these genetic and biologic classifications to be flexible, and hope that they will encourage much needed progress in syndrome recognition, clinical genetic testing, and ultimately the development of new therapies that target specific pathways of pathogenesis.
Keywords: Infantile spasms, developmental epilepsy, autism, movement disorders, gene regulatory networks
Introduction
Infantile spasms (ISS) are characterized by clusters of epileptic spasms with ictal electrodecrement, usually occurring before the age of 1 year [1]. The incidence of ISS is 0.25–0.4 per 1,000 live births [2], and they are important because of their frequent association with severe developmental outcome, including autism [3].
There have been many variations in terminology describing ISS over the years, with the eponym “West syndrome” generally referring to the triad of spasms, hypsarrhythmia, and mental retardation or regression – despite the difficulty of detecting the latter in a young infant presenting with ISS. Hypsarrhythmia is regarded by some as a key feature in the diagnosis of ISS, but can be so variable that it remains surprisingly troublesome to define, and evolves over time [4]. As a consequence, the literature contains references to “atypical” and “modified” hypsarrhythmia [5, 6], and there is little consensus on the meaning of these terms, or on how much of the electroencephalographic record should be abnormal [4, 7, 8]. In fact, the international West Delphi Group consensus statement did not regard the presence of hypsarrhythmia as essential for the diagnosis of ISS [1], and correctly refocused the core of the electroclinical disorder on the ictal event associated with an electrodecrement on EEG in an appropriately aged child. ISS without hysparrhythmia are well documented [9]. We will continue to refer to the disorder as infantile spasms, while recognizing that in some children they may not occur during infancy, and may not manifest as classic flexor or extensor spasms, and hypsarrhythmia may only be an associated feature of the interictal EEG.
Infantile spasms and the genetics of brain development
In the past, the terms symptomatic, cryptogenic, and idiopathic were introduced to categorize ISS, and epilepsy in general [10]. The limitations of this approach to classification are well noted within the epilepsy community [11, 12], and these terms become even more untenable with growing evidence that ISS result from disturbances in key genetic pathways of brain development. Recent studies show that patients with ISS may have mutations in several genes including ARX, CDKL5, FOXG1, GRIN1, GRIN2A, MAGI2, MEF2C, SLC25A22, SPTAN1, and STXBP1 [13–24]. While not all patients with mutations in these genes have ISS, the phenotype is consistent enough to speak of them as ISS-associated genes, modified by incomplete penetrance and variable expressivity. Taken together, these single genes, as well as other candidate genes identified from pathogenic copy number variants, suggest that abnormalities in ventral forebrain development and synaptic functional pathways play critical roles in ISS pathogenesis [25]. We caution that the pathogenesis of ISS may mirror autism in its genetic complexity, where each genetic abnormality probably accounts for about 1% of the disorder [26, 27]. Therefore, many more ISS-associated genes are likely to be discovered, and the evolving genetic data demand a reassessment of this disorder.
New molecular insights from the recent genetic data imply that all forms of ISS may be “symptomatic”, and that a diagnosis of “cryptogenic” ISS should no longer be accepted. For this reason, the International League Against Epilepsy (ILAE) recently recommended replacing the symptomatic, cryptogenic, and idiopathic classification system of epilepsy syndromes [12]. We strongly agree with their recommendation, but argue that it is also possible to take the first steps toward genetic and biologic classifications of ISS.
The biologic link between infantile spasms and autism
Many clinical studies of ISS have focused on the subsequent intractable epilepsy that develops in 40–60% of affected children [28, 29]. Although important, this approach has underemphasized the broader phenotypes to which ISS may belong. In contrast, classification systems based on the emerging genetic and biologic data would incorporate the historical connections between ISS and phenotypes other than intractable epilepsy. These observations are not new, and the spectrum of neurologic associations with ISS has always been much broader than intractable epilepsy, as it was for William James West’s son.
West syndrome is an unusual eponym because it is named after the father of the proband. The English physician, William James West, is credited with writing the first description of ISS in English by reporting the history of his son, James [30]. Most authors since have focused only on West’s descriptions of the seizures. However, one report highlighted James’ later history, which included lack of speech and “frequent fits of idiotic laughter, and rollings of the head... delighted by music and gay colours”. This description leads “to the suspicion that James had features of autism” [31]. Langdon-Down, who cared for him in later life, reported that James and other children he observed with ISS had “a great tendency to automatism and rhythmical actions” including “salaams, horizontal swayings, and rotations of the head and body” [32]. These observations may be compatible with the stereotypies often seen with autism.
Later reports continued to show that autism could follow ISS [33–36]. This includes reports of tuberous sclerosis complex (TSC), ISS, and autism [37], with a similar correlation noted in patients with Down syndrome as well [38, 39]. Some argue that the subsequent autistic spectrum disorder is a consequence of severe epileptic encephalopathy [40, 41]. While this remains a compelling hypothesis, the most recent data suggest a primary biologic link between ISS and autism [25].
Infantile spasms and movement disorders
Several early reports of children with ISS described hyperkinesis, but the nature and significance of the finding remained unclear [36, 42]. The use of the term “cerebral palsy” as a putative “predisposing cause” may obscure recognition of a movement disorder as a specific feature associated with ISS [43, 44]. The discovery of ARX as the first gene clearly associated with (previously “cryptogenic”) ISS led to a wider appreciation that involuntary movements may be a key clinical feature of certain syndromes that include ISS [45]. The description of MEF2C deletions and mutations in patients with ISS and dyskinesia strengthens this association [20, 21, 46]. The identification of MEF2C as another ISS-associated gene is interesting as MEF2C may be a transcriptional target of ARX during ventral forebrain development when GABAergic interneurons proliferate and migrate [47]. It is perhaps not surprising that children with mutations in ARX and MEF2C have similar phenotypes. Additionally, dyskinetic movements have been reported in children with CDKL5 deletions [48] and STXBP1 mutations as well [49].
A genetic classification of ISS
We agree with the spirit of the recent ILAE recommendation that epilepsies should be divided into genetic, structural/metabolic, and unknown categories [12]. However, the distinction between genetic and structural/metabolic is artificial and should be modified, as most epilepsies associated with structural brain malformations or inborn errors of metabolism are also genetic. Those that are not primarily genetic, such as epilepsy due to brain injury and infection, are distinct and deserve a place of their own.
A genetic classification has practical benefits. When ISS are recognized as a symptom found in many developmental disorders, rather than seen in isolation, the many disorders associated with ISS can be divided into two main groups: those with the predisposing genotype known and those with the predisposing genotype unknown. Of course, we recognize several important subdivisions, as listed in Table 1. We think that this approach will facilitate future progress in several areas including genetics, pathogenesis, and treatment. Figure 1 illustrates the evaluation of ISS using the classification system presented in Table 1.
Table 1a.
Genetic classification of infantile spasms with known genotype and key phenotypes
| Clinical Syndrome | Gene or Locus | % with ISS | Brain imaging | Other neurodevelopmental phenotype | Reference(s) |
|---|---|---|---|---|---|
| 1. Infantile spasms as part of developmental disorder with predisposing genotype known | |||||
| 1a. Prototypic developmental epilepsies | |||||
| ISSX | ARX (less severe mutations) | High | Cortical atrophy, reduced WM, abnormal BG | EIEE, variable LGS, dystonia/hyperkinesis common | [45, 50, 51] |
| CDKL5 | High | Cortical atrophy, abnormal WM, calcifications | LGS, autistic features common | [15, 136] | |
| FOXG1 duplications | 50% | Variable dysgenesis of CC | Autistic features common | [18, 172] | |
| MEF2C | High | Atrophy, reduced WM | Variable subsequent epilepsy, severe cognitive impairment, hyperkinesis common | [20, 21] | |
| SLC25A22 | High | Atrophy, dysgenesis of CC, CBLH | EIEE, severe cognitive impairment, abnormal electroretinogram | [23, 174] | |
| SPTAN1 | High | Cortical atrophy and CBLA, hypomyelination, reduced WM, thin CC | EIEE | [22] | |
| STXBP1 | High | Atrophy, abnormal WM | EIEE, severe cognitive impairment | [17] | |
| 1b. Specific structural brain malformations | |||||
| XLAG | ARX (more severe mutations) | High | LIS | Neonatal seizures, LGS, severe cognitive impairment, genital hypoplasia | [58, 59] |
| ILS | DCX | Presumed high | LIS | LGS, severe cognitive impairment | [57] |
| ILS | PAFAH1B1/LIS1 | High | LIS | LGS, severe cognitive impairment, choreiform movements | [57] |
| Tuberous sclerosis complex | TSC1 TSC2 |
40% | Tubers | Variable LGS, cognitive impairment variable, autistic features common | [63] |
| TUBA1A | Presumed high | LIS, CBLH | LGS, severe cognitive impairment | [56] | |
| 1c. Inborn errors of metabolism | |||||
| Amino acidopathies (NKH, PKU) | GLDC, GCST, GCSH (NKH); PAH (PKU) | High | NKH: Variable agenesis of CC PKU: Abnormal WM | EME, subsequent epilepsy may be severe in NKH; cognitive impairment may be severe if metabolic disease uncontrolled | [72, 73, 77] |
| DEND | KCNJ11 | Several patient reports | Diabetes mellitus onset in infancy, severe cognitive impairment, athetosis | [78–80] | |
| Organic acidemias (MMA, MSUD, PA) | MUT (MMA); BCKDHA, BCKDHB, DBT, DLD (MSUD); PCCA, PCCB (PA) | Several patient reports | Subsequent epilepsy, cognitive impairment, and autistic features variable | [76, 175] | |
| Menkes disease | ATP7A | High | Cortical atrophy, abnormal WM, “ischemic changes” | Intractable epilepsy, severe cognitive impairment | [71, 74, 75] |
| 1d. Syndromes of genomic imbalance | |||||
| Deletion 1p36 syndrome | Deletion 1p36 | 20% | Cortical atrophy, abnormal WM, thin CC | EIEE; 35% develop LGS; moderate to severe cognitive impairment | [25, 176] |
| Williams syndrome plus | Deletion 7q11.23 (deletion of MAGI2, and/or other genes) | Several patient reports | Mild-moderate cognitive impairment, characteristic neurobehavioral profile | [16, 25, 98, 99, 177] | |
| Pallister-Killian syndrome | Tetrasomy 12p | Multiple patient reports | Cortical atrophy, delayed myelination | Severe cognitive impairment | [91, 94, 95] |
| Duplication 15q syndrome | Maternal duplication 15q11q13 | ~20% | Variable LGS; autistic features | [25, 93, 178] | |
| Miller-Dieker syndrome | Deletion 17p13 (deletion of LIS1) | High | LIS, mild CBVH | LGS common, severe cognitive impairment | [57, 89] |
| Down syndrome | Trisomy 21 | ~5% | CBLH reported | Variable subsequent epilepsy, LGS; cognitive impairment, autistic features common | [87, 88, 90, 92] |
| 1e. Syndromes with putative association with infantile spasms | |||||
| Autosomal recessive severe microcephaly with PNH | ARFGEF2 | Single patient report | PNH | Severe cognitive impairment, intractable epilepsy | [110] |
| Freeman-Sheldon syndrome | MYH3 | Patient report preceded genotyping | Distal arthrogryposis | [104] | |
| Mitochondrial encephalomyopathy with elevated methylmalonic acid | SUCLA2 | Single patient report | Variable cortical atrophy, delayed myelination | Severe hypotonia, choreathetosis | [82] |
| Neurogenic muscle weakness, ataxia, retinitis pigmentosa (NARP) | MT-ATP6 | Single pedigree | [81] | ||
| Neurofibromatosis type I | NF1 | Several patient reports | Variable cognitive impairment | [112, 113] | |
| Schinzel-Giedion syndrome | SETBP1 | Multiple patient reports | Cortical atrophy, abnormal WM | [105, 107, 108] | |
| Smith-Lemli-Opitz syndrome | DHCR7 | Patient reports preceded genotyping | Severe cognitive impairment | [103, 106] | |
| Smith-Magenis syndrome | Deletion 17p11.2 | Single patient report | Moderate-severe cognitive impairment, characteristic neurobehavioral profile | [115] | |
| Sotos syndrome | NSD1 | Patient reports preceded genotyping | [109] | ||
| X-linked PNH | FLNA | Patient reports | PNH | Intractable epilepsy common, variable cognitive impairment | [111] |
Figure 1.
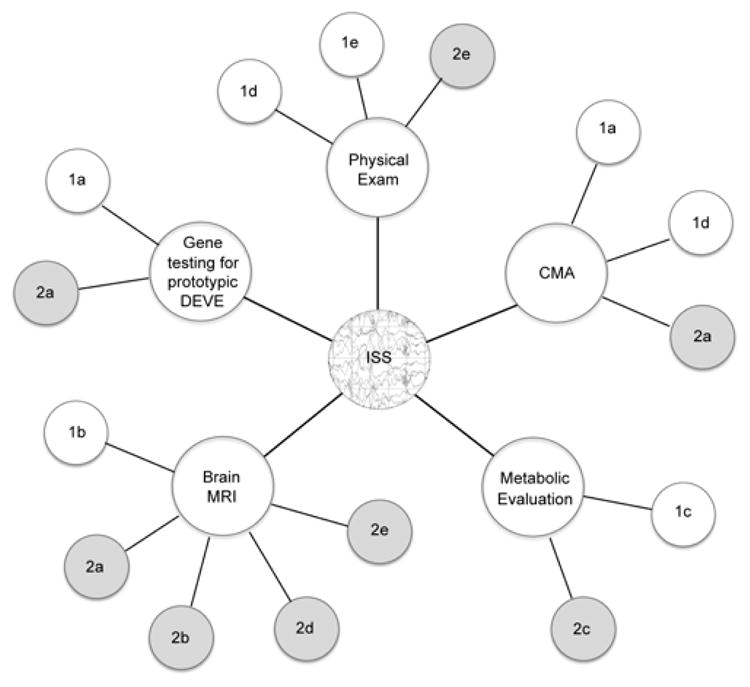
Evaluation of infantile spasms (ISS) using the classification system in Table 1. Diagnosis of ISS centers upon epileptic spasms associated with ictal electrodecrement. Evaluations may be carried out concurrently, depending upon the clinical scenario. Physical exam findings may lead to recognition of syndromes of genomic imbalance (Group 1d) such as Down syndrome, Williams syndrome, or Miller-Dieker syndrome; or may suggest other syndromes putatively associated with ISS (Group 1e); or recognizable genotype-unknown syndromes (Group 2e). Gene testing for the prototypic developmental epilepsies (DEVE) may find mutations in known associated genes (Group 1a); or if negative may suggest a genotype-unknown prototypic DEVE (Group 2a). Brain MRI may find specific structural brain malformations (Group 1b) and lead to specific follow-up genetic testing; or may reveal non-specific abnormalities associated with genotype-unknown prototypic DEVE (Group 2a), findings associated with genotype-unknown syndromes (Group 2b), extrinsic injury patterns (Group 2d), or recognizable abnormalities putatively associated with ISS (Group 2e). Metabolic evaluation may lead to identification of specific inborn errors of metabolism associated with ISS, leading to genetic testing for those disorders (Group 1c); or may reveal metabolic abnormalities associated with genotype-unknown conditions (Group 2c). Chromosomal microarray (CMA) may identify prototypic DEVE such as duplication of FOXG1 or deletion of CDKL5 or MEF2C (Group 1a); diagnose known syndromes of genomic imbalance associated with ISS (Group 1d); or may be negative suggesting a genotype-unknown prototypic DEVE (Group 2a).
1. ISS with predisposing genotype known
1a. Prototypic developmental epilepsies
This group provides the core data supporting our emerging understanding of ISS pathogenesis, and from this group the later biologic classification of ISS can also be built. These are the newly described genes in which mutations, intragenic expansions, deletions and/or duplications frequently are associated with spasms. Some children with ARX and STXBP1 mutations initially presented with early infantile epileptic encephalopathy (EIEE, Ohtahara syndrome) [17, 50–52] and illustrate a spectrum of infancy-onset epileptic encephalopathies. EIEE, early myoclonic encephalopathy (EME), and ISS remain enigmatic in their relationship to one another, and patient series of these infantile epilepsies overlap in genotype [49, 53–55], suggesting factors of incomplete penetrance and variable expressivity. The type of early-onset epilepsy may be related to severity of mutation, although more data are needed to show this relationship. Therefore, the term prototypic developmental epilepsies acknowledges the variability but likely biologic kinship of these phenotypes.
1b. Specific brain malformations
The two genes most commonly associated with classical lissencephaly, PAFAH1B1/LIS1 and DCX, as well as the less common TUBA1A are strongly associated with ISS [56] with a prevalence as high as 80% in children with PAFAH1B1/LIS1 deletions or mutations [57]. Severe mutations of ARX associated with X-linked lissencephaly with abnormal genitalia (XLAG) result in severe early-onset epilepsy that overlaps with EIEE. This pattern can evolve into ISS, but more commonly progresses to severe intractable epilepsy [58, 59]. ARX, DCX, and PAFAH1B1/LIS1 are all expressed in GABAergic interneurons [60–62], and we hypothesize that spasms are linked to deficits in this neuronal cell type, rather than the obvious cortical malformation.
This category also includes TSC, as mutations in TSC1 and TSC2 are associated with cortical tubers, subependymal nodules and other TSC-related brain malformations. Around 40% of patients with TSC may have ISS [63], although the prevalence varies and precise phenotype-genotype data are not available. Recent molecular and pathology evidence suggests tuber formation may be the result of a combination of germline and somatic TSC1/2 mutations [64]. The presence, location, and type of tubers correlate with ISS in some studies [65, 66]. A strong correlation has been established between TSC, ISS, and the subsequent development of autism [67–69], although the mechanism of this relationship remains unclear. Dysregulation of the mammalian target of rapamycin (mTOR) pathway in TSC is a key event leading to abnormal brain morphogenesis [70]. Given the phenotypic heterogeneity of TSC, ISS may occur when the mTOR pathway is perturbed in a specific neuronal population at a key neurodevelopmental stage.
1c. Inborn errors of metabolism
An association between ISS and certain inborn errors of metabolism has long been recognized. The best known examples include the aminoacidopathies phenylketonuria (PKU) and nonketotic hyperglycinemia (NKH); the organic acidemias methylmalonic acidemia (MMA), proprionic acidemia (PA) and maple-syrup urine disease (MSUD); and a disorder of copper metabolism, Menkes disease [71–77]. Mutations of KCNJ11 cause DEND syndrome (developmental delay, epilepsy, neonatal diabetes), which commonly features ISS and is a rare example of an ion channel gene associated with spasms [78–80]. The genetically-characterized mitochondrial disorders appear to be infrequently associated with ISS, with very few clinical reports available [81]. Mutations in SUCLA2 produce a specific mitochondrial encephalopathy, but only one reported patient had ISS [82].
Metabolic diseases presumably cause epilepsy by disturbing neuronal energetics or by toxicity from the offending metabolite [83]. However, PKU may have secondary effects on synapse function [84], while NKH and a few other metabolic disorders are associated with agenesis of the corpus callosum and cortical malformations [85]. It is possible that abnormalities in neuronal energetics, synapse function, and brain morphogenesis may all be interrelated in metabolic patients with ISS, and their symptoms reflect the susceptibility of specific neuronal populations to the offending metabolite at a key point in neurodevelopment. Not surprisingly, ISS are usually not the only epilepsy type associated with these metabolic disorders, and overlap exists with other early infant-onset epilepsies, such as EME.
1d. Syndromes of genomic imbalance
At least five well-known syndromes of chromosomal copy number variation are predisposing genotypes for ISS. These are deletion 1p36.3, Pallister-Killian syndrome (tetrasomy 12p), duplication of maternal 15q11q13, Miller-Dieker syndrome (deletion 17p13.3), and Down syndrome (trisomy 21) [25, 86–95]. The biological mechanism(s) of ISS in these syndromes are not known, except for Miller-Dieker syndrome in which the deletion of PAFAH1B1/LIS1 causes both abnormal primary glutamatergic and GABAergic interneuron migration [89, 96, 97]. Similarly, deletion of MAGI2 at 7q11.2 may be associated with ISS in Williams syndrome [16], although there are also reports of ISS in Williams syndrome patients with deletions that do not include MAGI2 [25, 98, 99]. Overall this suggests that copy number changes in key genes, in the right developmental context, influence the occurrence of ISS in these disorders. Further, “multiple hit” genomic events may account for some of the phenotypic variability seen in diseases mediated by copy number variants [100–102].
1e. Syndromes with putative association with ISS
ISS have occasionally been reported with several other well-characterized predisposing genotypes, often in reports predating molecular characterization of the disorder. For example, we found several reports of ISS in patients with Freeman-Sheldon, Schinzel-Giedieon, Smith-Lemli-Opitz, and Sotos syndrome [103–109]. There are also reports of ISS in patients with mutations in FLNA and ARFGEF2, genetic causes of periventricular nodular heterotopia [110, 111]. However, other seizure types are more common than ISS in most of these disorders, especially patients with heterotopia due to FLNA mutations, making it difficult to argue that these genes are reliably causative.
Several reports suggest an association between ISS and rasopathies (neurofibromatosis type I and cardio-facio-cutaneous syndrome), but not commonly enough to suggest a consistent link [112–114]. A single report each of ISS with Smith-Magenis and Pallister-Hall syndromes (the latter with hypothalamic hamartoma) suggest a rare association [115, 116].
2. ISS with predisposing genotype unknown
2a. Prototypic developmental epilepsies
This is a novel category introduced to refer to patients who present with ISS, but who lack features specific for any unifying diagnosis and accordingly have an unknown predisposing genotype. When reviewed at later ages, these children have features suggesting a global developmental disorder such as autistic features and dyskinesias that may not have been manifest at the time of presentation with ISS. Brain imaging studies may be normal, or may have findings that do not define a specific syndrome – such as cortical gray or white matter volume loss, asymmetric ventricles, simplified gyral pattern, hypogenesis of the corpus callosum, polymicrogyria, and/or cerebellar malformations. The subsequent epilepsy course after ISS is variable, with some children developing intractable epilepsy and others few seizures. Due to the spectrum of imaging and neurobehavioral findings in this group, this is not a replacement for the previous “cryptogenic” designation. Rather, we think that all or most of these patients will ultimately be found to have predisposing genotypes, particularly in early CNS developmental genes.
2b. Recognizable unifying phenotype and clear association with ISS
This group contains well-described developmental disorders with unknown genotype but unifying phenotypes that have ISS as a core finding. The two best examples are Aicardi and progressive encephalopathy with edema, hypsarrhythmia and optic atrophy (PEHO) syndromes. Aicardi syndrome consists of agenesis of the corpus callosum, polymicrogyria, chorioretinal lacunes, other more variable anomalies, with ISS being a prominent feature [117]. It affects only females, suggesting a gene located on the X chromosome. PEHO syndrome is less common, appears to overlap with a PEHO-like phenotype, and most resembles a neurodegenerative disorder with marked brain and optic nerve atrophy [118–120].
Several more heterogeneous conditions have also been associated with a high incidence of ISS. Around 7% of patients with focal cortical dysplasia (FCD) may develop ISS [121], and the cortical abnormalities may be below the resolution of MRI [122]. Interestingly, persistence of immature GABAergic networks appear to play a role in focal cortical dysplasia [123], and dysfunction in the mTOR signaling pathway is a shared feature between FCD and tuberous sclerosis complex [124]. Finally, isolated hemimegalencephaly (HMEG) is included in this category because without cutaneous or systemic findings it is a clinical entity distinct from the many syndromes that also feature this malformation [125, 126].
2c. Biochemical/metabolic abnormalities putatively associated with ISS
As a clear relationship between ISS and inborn errors of metabolism has been established, we also recognize a category of diseases where a biochemical defect is demonstrable, but the genetic etiology remains unclear. Patients with pyridoxine-responsive ISS have been reported [127], but none with mutations in the one known pyridoxine-dependent epilepsy gene ALDHA7 [128, 129]. Several reports link mitochondrial respiratory chain dysfunction and ISS, but without a clear genetic mutation identified, making it unclear if the mitochondrial dysfunction is primary or secondary [130–133]. Finally, neonatal hypoglycemia has long been offered as a cause of ISS [44, 134], sometimes in the setting of hyperinsulinism [135]. The evaluations reported in these patients are often unclear or incomplete, so it is difficult to make a clear causal connection between the two.
2d. Extrinsic injury patterns
Extrinsic brain injuries such as hypoxic-ischemic encephalopathy (HIE), near-miss sudden infant death syndrome, stroke, and infection are commonly accepted as causes of ISS. These infants rarely undergo further evaluation. A closer survey of the patients reported, however, leads to some interesting observations. One is that brain imaging patterns such as atrophy, calcifications, and white matter hyperintensities previously interpreted as due to ischemic injury or infection can overlap with predisposing genotypes such as CDKL5 and SPTAN1 mutations [15, 22, 136, 137]. Clinicians should therefore be aware of the conditions that may present “HIE or infection look-alikes” on imaging.
Reports remain in which ischemic events were well documented with no other predisposing condition identified, and it would appear that 5–10% of infants with ISS in these series have a presumed ischemic or infectious etiology [138–142]. Our concern is that this literature is retrospective, and may not accurately reflect the prevalence of ISS among children with acquired brain injuries. About 25% of children with perinatal hypoxia will have subsequent cerebral palsy, cognitive impairment, and epilepsy [143], but ISS do not appear to be a significant seizure type described in this population [144]. Despite the increased survival of very-low birth weight infants at risk for HIE, the rate of ISS has not increased [145], suggesting that other factors contribute to the development of spasms in this population. Perhaps hypoxia represents a “second hit” in a population made vulnerable to spasms by yet-undiscovered predisposing genotypes. While some may comment that there is no scientific data for such an assertion, we argue that the data are insufficient in either case. The study that prospectively follows a large cohort of patients with perinatal hypoxia and documents the prevalence of ISS has not yet been done.
Similar observations may be made about the many case series documenting infection as the cause of ISS [146–148]. While epilepsy can certainly be a long-term complication of CNS bacterial or viral infection during infancy, ISS as a specific consequence are not captured in outcome data [149–153]. If ISS are rare sequelae of CNS infection, predisposing genotype(s) may exist that lead to the expression of spasms in infants who have experienced CNS infection. It is also possible that ischemic or infectious events at a critical period in perinatal development may selectively damage specific neuronal populations – for example emerging GABAergic interneuron synaptic networks – and result in the later emergence of ISS. Therefore, children with extrinsic injury patterns are prime candidates for further study.
2e. Recognizable unifying phenotype and putative association with infantile spasms
This group of disorders contains syndromic phenotypes with unknown causative genotypes, in which ISS have been mentioned only in occasional clinical reports. In this category are frontal-perisylvian periventricular nodular heterotopia-polymicrogyria (PNH-PMG), hypomelanosis of Ito, macrocephaly polymicrogyria polydactyly hydrocephalus syndrome (MPPH) with the overlapping disorder macrocephaly capillary malformation syndrome (MCAP), and sebaceus nevus syndrome [154–157]. Also noted here are various nonsyndromic CNS neoplasms that have been reported with ISS, including choroid plexus papilloma, hypothalamic hamartoma (also described in Pallister-Killian syndrome), and ganglioglioma [158–160]. The association of these conditions and syndromes with ISS comes from a few patient reports, and requires further study to understand the exact relationship.
Interesting exceptions
Several developmental disorders with epilepsy are not associated with ISS, or a surprisingly small number of patients have been reported, suggesting some biological phenomena that may protect against the development of spasms. These disorders include Rett syndrome due to MECP2 mutations [161], and given that CDKL5 probably interacts with MECP2 [162], it is not clear why the epilepsy phenotypes in these two developmental disorders are so different.
The 15q11q13 locus is also intriguing in this regard, as duplications of maternal 15q11q13 are a common cause of both epilepsy – including ISS – and autism, while deletion of maternal 15q11q13 causes Angelman syndrome and paternal deletion causes Prader-Willi syndrome. Very few examples of ISS have been reported in Angelman syndrome patients [163]. ISS has not been reported in Prader-Willi syndrome, though epilepsy is 16 times more prevalent than in the general population [164].
Dravet syndrome due to SCN1A mutations poses another interesting situation. Infants with Dravet syndrome typically present with fever-induced status epilepticus followed by intractable multifocal epilepsy [165]. Dravet syndrome has a severe behavioral phenotype, as well as an ataxia/movement disorder [166]. Despite the fact that GABAergic interneurons express SCN1A at high levels, and the mouse model of severe myoclonic epilepsy of infancy demonstrates abnormal interneuron excitability, there is only a single patient report of ISS in this population [167–169]. Further inquiry into these exceptions may yield important insights into the biology of ISS.
Single genes associated with ISS allow biologic classification
A biologic classification system for ISS begins with the recognition that this phenotype in many, if not most, disorders results from specific disturbances of gene regulation of brain development and function. The known ISS associated genes can be classified in a biologic model drawing from Gene Ontology (GO) [170], as well as gene expression data combined with CNS developmental stages. This biologic model can help formulate hypotheses to explain the varied phenotypes seen with ISS, and hopefully identify new treatment targets.
Single genes highly associated with ISS can be classified biologically if data exist regarding the gene expression pattern in specific cell types across the spectrum of CNS development. As these data are incomplete in humans, they must sometimes be extrapolated from animal models, usually the mouse. When these data are combined with the GO molecular function of the gene product, a role for the gene product in neurodevelopment can be described. This allows for classification systems built on biological relationships between genes and gene products.
A biologic classification of ISS
One of the benefits of a biologic classification system is that hypotheses to explain phenotype differences among patients with mutations in ISS associated genes can be made. It is hoped that grouping by biologic mechanism will also aid identification of future disease-specific therapies. Table 2 summarizes the ISS phenotype subgroups after this biologic classification. The molecular function of each gene highly associated with ISS was determined from the GO database (www.geneontology.org), and the primary literature is referenced for evidence of role(s) in or effect(s) on specific stages of brain development. Information from the mouse gene expression databases at BGEM (www.stjudebgem.org), GENSAT (www.gensat.org), and MGI (www.informatics.jax.org) was accessed as well.
Table 2.
Biologic classification of infantile spasms. Genes are organized into groups by Gene Ontology molecular function and roles in or effect on central nervous system development.
| Group | Neurodevelopmental phenotype besides ISS | Gene(s) | GO Molecular Function | Role in/Effect on CNS development | References |
|---|---|---|---|---|---|
| A |
|
ARX FOXG1 MEF2C |
Transcription factors |
|
[181–184] |
| B1 |
|
DCX PAFAH1B1/LIS1 TUBA1A |
Binding |
|
[61, 185–189] |
| B2 |
|
TSC1 TSC2 |
Signal transduction |
|
[190, 191] |
| C1 |
|
GLDC PAH |
Binding, catalytic activity |
|
[192, 193] |
| C2 |
|
ATP7A KCNJ11 |
Transporter, catalytic activity |
|
[194–196] |
| D |
|
GRIN1 GRIN2A MAGI2 SPTAN1 STXBP1 |
Binding |
|
[19, 22, 24, 55, 197, 198] |
| E |
|
SLC25A22 | Transporter |
|
[23] |
Abbreviations: iN = interneurons; IPC = intermediate progenitor cells; N = neurons; NPC = neuronal progenitor cells
The groups are organized so that Group A comprises transcription factors, Group B comprises genes important in proliferation and cell migration, and later groups reflect the terminal differentiation stages of synapse development. The two best characterized groups are the gene regulatory network of ventral forebrain development and the pre- and post-synaptic protein networks, illustrated in Figure 2.
Figure 2.
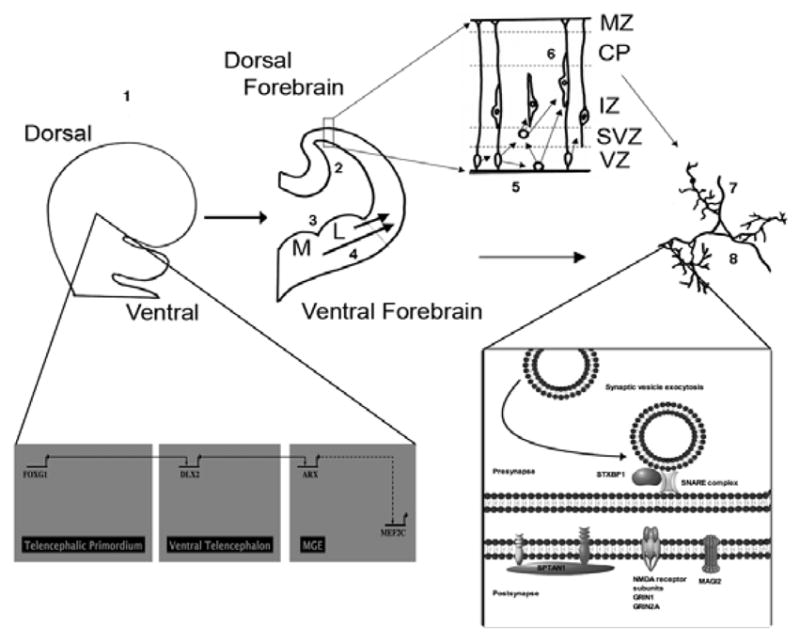
The two best characterized subgroups of ISS-associated genes illustrated with the stages of central nervous system (CNS) development. Group A includes transcription factors essential to CNS dorsal-ventral differentiation (FOXG1), affecting neuroprogenitor cells broadly, with downstream effects on synapse function, as well as ARX, which is involved in ventral GABAergic interneuron differentiation, migration, and synaptogenesis. MEF2C is probably involved in this process as well. Group D genes (GRIN1, GRIN2A, MAGI2, SPTAN1, STXBP1) encode binding proteins or receptors with direct roles in presynaptic and/or postsynaptic development and function. 1 = Dorsal-ventral differentiation; 2 = Dorsal neuroprogenitor cell proliferation; 3 = Ventral neuroprogenitor cell proliferation; 4 = GABAergic interneuron migration; 5 = Intermediate progenitor cell proliferation; 6 = Glutaminergic neuron migration; 7 = Axonogenesis; 8 = Synaptogenesis; Red = primary role in developmental stage; Pink = Secondary effect on developmental stage; L = lateral ganglionic eminence; M/MGE = medial ganglionic eminence; MZ = marginal zone; CP = cortical plate; IZ = intermediate zone; SVZ = subventricular zone; VZ = ventricular zone
Group A comprises patients who have abnormalities in transcription factors that are essential for ventral CNS development. These include duplications of FOXG1, mutations of ARX, and deletions of MEF2C. Abnormalities in FOXG1 affect development of major telencephalic structures, including the neocortex, hippocampus, and the lateral and medial ganglionic eminences [171]. Patients have mental retardation with an autistic phenotype and stereotypic movements [18, 172]. ARX is an important regulator of GABAergic interneuron differentiation and migration [173]. More severe mutations in ARX lead to lissencephaly, intractable epilepsy, and a phenotype overlap with Group B1. While MEF2C expression is altered in the absence of ARX [47], its exact position in the gene regulatory network of forebrain development remains unclear. Both ARX and MEF2C patients can have ISS associated with prominent movement disorders.
Group B1 comprises patients with mutations in DCX, PAFAH1B1/LIS1, and TUBA1A, which encode binding proteins that are expressed throughout much of forebrain development, in both glutamatergic and GABAergic cell types. Synaptogenesis appears to be disrupted by a secondary effect of abnormal neuronogenesis and migration. These genes also play direct roles in axonogenesis and synapse formation. Group B1 patients have severe brain malformations that include lissencephaly and variable cerebellar hypoplasia. Group B2 is related conceptually to Group B1 because genes in both groups are involved in neuronal proliferation and migration. Similar to Group B1 patients, TSC patients also have characteristic brain malformations, and can have severe cognitive impairment, and intractable epilepsy, but the neurobehavioral phenotype in TSC often distinctly involves autism.
Group C1 patients have mutations in genes such as GLDC and PAH that play roles in neuronal and non-neuronal cell metabolism but not directly in brain morphogenesis. Mutations in these genes appear to have secondary effects on synapse function, best documented in PKU. These patients have metabolic diseases that, if controlled, as a general rule (but not always) have less severe cognitive impairment and less epilepsy. Group C2 comprises patients with metabolic disorders such as DEND and Menkes disease where the causative gene also has a role in neurodevelopment and the phenotype is characterized by a severe encephalopathy. The genes that cause these disorders have ion transport and/or catalytic activity and play primary roles in synapse development.
Group D comprises patients with abnormalities of MAGI2, SPTAN1, and STXBP1, which encode proteins with direct roles in synaptic development and function. Group D patients tend to have intractable epilepsy, with the MAGI2 deletion patients so far reported also having Williams syndrome. The recent description of patients with mutations of GRIN1 [19] and GRIN2A [24] associated with ISS illustrates that further discoveries in synapse development and function likely lay ahead.
Future directions
ISS have long posed classification difficulties, and may be most accurately conceptualized as a phenotype “at the tip of the iceberg” of a broader group of developmental disorders that overlap with autism, intractable epilepsy, and movement disorders. This review does not claim to resolve all of these issues, but seeks to open a discussion in new directions. Knowledge is accumulating rapidly about the genetic associations of ISS, and ISS appear to be a genetically heterogeneous condition that can result from abnormalities in key brain development pathways. Like autism, each associated gene may account for only 1% of the condition. Taken together, however, these are not “rare causes”, as is currently commonly perceived. Instead, animal models of the genetic syndromes highly associated with ISS will likely prove helpful in improving our understanding of pathogenesis of ISS, and hopefully will lead to new therapies.
We propose the first genetic and biologic classifications of ISS. These classifications are designed to be flexible, and will allow the incorporation of new causes as they are discovered. They will help the clinician better direct diagnostic testing, and provide more accurate prognostic information and genetic counseling. Of equal importance, a biologic classification system for ISS is essential for the development of new disease-specific therapies, and helps to explain the phenotypes of various ISS syndromes.
Table 1b.
Genetic classification of infantile spasms with unknown genotype status with key phenotypes.
| Clinical Syndrome | Gene or Locus | % with ISS | Brain imaging | Other neurodevelopmental phenotype | Reference(s) |
|---|---|---|---|---|---|
| 2. Infantile spasms as part of developmental disorder with predisposing genotype unknown | |||||
| 2a. Prototypic developmental epilepsies | |||||
| Developmental disorder with ISS | Unknown | All, by definition | Normal, or non-specific (WM changes, asymmetric LV, atrophy, ACC, CBLH, CBVH) | Developmental disorder that can have subsequent epilepsy, cognitive impairment, autistic features, and/or movement disorder – presumably depending on underlying genotype not yet discovered | Novel category |
| 2b. Recognizable unifying phenotype and clear association with infantile spasms | |||||
| Aicardi syndrome | Unknown | High | ACC, HET, PMG-like, CBLH, CBVH | Variable subsequent epilepsy, moderate to severe cognitive impairment, can have autistic features | [117] |
| Focal cortical dysplasia | Unknown | ~7% | Cortical dysplasia | Focal epilepsy common | [121] |
| Isolated HMEG | Unknown | High | HMEG | Focal epilepsy common | [125, 126] |
| PEHO and PEHO-like | Unknown | High | Microcephaly, CBLA | Severe cognitive impairment with regression | [119, 120] |
| 2c. Metabolic abnormalities | |||||
| Mitochondrial dysfunction | Unknown | 10–20% | Cortical atrophy, infarct, CBLA, BGTH increased signal | [130–133] | |
| Neonatal hypoglycemia | Unknown | Multiple patient reports | [134] | ||
| Pyridoxine-dependent/responsive epilepsies | Unknown (not ALDH7A1) | ~10% | Cortical atrophy, CBLA | [128, 129] | |
| 2d. Extrinsic injury pattern | |||||
| Perinatal stroke or other CNS hypoxic event | Unknown | Unknown | Porencephaly, some PMG, abnormal WM, ischemic changes | Variable rate of intractable epilepsy, dystonia may be present | [139–142] |
| Post-infectious (TORCH, other viruses, bacterial meningitis) | Unknown | Unknown | Microcephaly, calcifications | [146–148] | |
| Schizencephaly | Unknown (not EMX2) | Unknown | SCH, PMG, ASP-SOD | Variable subsequent epilepsy, often focal | [179] |
| 2e. Recognizable unifying phenotype and putative association with infantile spasms | |||||
| Frontal-perisylvian PNH-PMG | Unknown | 60% | PNH, PMG | [180] | |
| Hypomelanosis of Ito | Unknown | <10% | [155] | ||
| MPPH/MCAP | Unknown | 30% | MEG, PMG, hydrocephalus, CBTH | [156] | |
| Sebaceus nevus syndrome | Unknown | High | HMEG, ACC, CBVH | [154, 157] | |
Abbreviations: ACC = agenesis of corpus callosum; ASP-SOD = absent septum pellucidum-septo-optic-dysplasia; BG = basal ganglia; BGTH = basal ganglia-thalami; CBLA = cerebellar atrophy; CBLH = diffuse (proportionate vermis and hemispheres) cerebellar hypoplasia; CBTH = cerebellar tonsillar herniation; CBVH = cerebellar vermis hypoplasia; CC = corpus callosum; DEND = developmental delay, epilepsy, and neonatal diabetes; EIEE = early infantile epileptic encephalopathy (aka Ohatahara syndrome); EME = early myoclonic encephalopathy; HET = heterotopia; HMEG = hemimegalencephaly; LGS = Lennox-Gastaut syndrome; LIS = lissencephaly; LV = lateral ventricles; MEG = megalencephaly; MMA = methylmalonic aciduria; MSUD = maple syrup urine disease; MPPH/MCAP = megalencephaly perisylvian polymicrogryia with postaxial polydactyly and hydrocephalus/macrocephaly capillary malformation syndrome; NKH = nonketotic hyperglycinemia; PA = propionic aciduria; PKU = phenylketonuria; PMG = polymicrogyria; PNH = periventricular nodular heterotopia; SCH = schizencephaly; WM = white matter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lux AL, Osborne JP. A proposal for case definitions and outcome measures in studies of infantile spasms and West syndrome: consensus statement of the West Delphi group. Epilepsia. 2004;45:1416–1428. doi: 10.1111/j.0013-9580.2004.02404.x. [DOI] [PubMed] [Google Scholar]
- 2.Trevathan E, Murphy CC, Yeargin-Allsopp M. The descriptive epidemiology of infantile spasms among Atlanta children. Epilepsia. 1999;40:748–751. doi: 10.1111/j.1528-1157.1999.tb00773.x. [DOI] [PubMed] [Google Scholar]
- 3.Saemundsen E, Ludvigsson P, Rafnsson V. Risk of autism spectrum disorders after infantile spasms: a population-based study nested in a cohort with seizures in the first year of life. Epilepsia. 2008;49:1865–1870. doi: 10.1111/j.1528-1167.2008.01688.x. [DOI] [PubMed] [Google Scholar]
- 4.Philippi H, Wohlrab G, Bettendorf U, Borusiak P, Kluger G, Strobl K, Bast T. Electroencephalographic evolution of hypsarrhythmia: toward an early treatment option. Epilepsia. 2008;49:1859–1864. doi: 10.1111/j.1528-1167.2008.01715.x. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe K, Negoro T, Aso K, Matsumoto A. Reappraisal of interictal electroencephalograms in infantile spasms. Epilepsia. 1993;34:679–685. doi: 10.1111/j.1528-1157.1993.tb00446.x. [DOI] [PubMed] [Google Scholar]
- 6.Endoh F, Yoshinaga H, Kobayashi K, Ohtsuka Y. Electroencephalographic changes before the onset of symptomatic West syndrome. Brain Dev. 2007;29:630–638. doi: 10.1016/j.braindev.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Kramer U, Sue WC, Mikati MA. Hypsarrhythmia: frequency of variant patterns and correlation with etiology and outcome. Neurology. 1997;48:197–203. doi: 10.1212/wnl.48.1.197. [DOI] [PubMed] [Google Scholar]
- 8.Muzykewicz DA, Costello DJ, Halpern EF, Thiele EA. Infantile spasms in tuberous sclerosis complex: prognostic utility of EEG. Epilepsia. 2009;50:290–296. doi: 10.1111/j.1528-1167.2008.01788.x. [DOI] [PubMed] [Google Scholar]
- 9.Caraballo RH, Ruggieri V, Gonzalez G, Cersósimo R, Gamboni B, Rey A, Poveda JCP, Dalla Bernardina B. Infantile spams without hypsarrhythmia: A study of 16 cases [Internet] Seizure. 2010 Dec; doi: 10.1016/j.seizure.2010.11.018. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21167750. [DOI] [PubMed]
- 10.Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia. 1989;30:389–399. doi: 10.1111/j.1528-1157.1989.tb05316.x. [DOI] [PubMed] [Google Scholar]
- 11.Lux AL, Osborne JP. The influence of etiology upon ictal semiology, treatment decisions and long-term outcomes in infantile spasms and West syndrome. Epilepsy Res. 2006;70 (Suppl 1):S77–86. doi: 10.1016/j.eplepsyres.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 12.Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, Engel J, French J, Glauser TA, Mathern GW, Moshé SL, Nordli D, Plouin P, Scheffer IE. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- 13.Strømme P, Mangelsdorf ME, Scheffer IE, Gécz J. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev. 2002;24:266–268. doi: 10.1016/s0387-7604(02)00079-7. [DOI] [PubMed] [Google Scholar]
- 14.Kato M, Das S, Petras K, Sawaishi Y, Dobyns WB. Polyalanine expansion of ARX associated with cryptogenic West syndrome. Neurology. 2003;61:267–276. doi: 10.1212/01.wnl.0000068012.69928.92. [DOI] [PubMed] [Google Scholar]
- 15.Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OLD, Archer H, Evans J, Clarke A, Pelka GJ, Tam PPL, Watson C, Lahooti H, Ellaway CJ, Bennetts B, Leonard H, Gécz J. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet. 2004;75:1079–1093. doi: 10.1086/426462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marshall CR, Young EJ, Pani AM, Freckmann M-L, Lacassie Y, Howald C, Fitzgerald KK, Peippo M, Morris CA, Shane K, Priolo M, Morimoto M, Kondo I, Manguoglu E, Berker-Karauzum S, Edery P, Hobart HH, Mervis CB, Zuffardi O, Reymond A, Kaplan P, Tassabehji M, Gregg RG, Scherer SW, Osborne LR. Infantile spasms is associated with deletion of the MAGI2 gene on chromosome 7q11.23–q21.11. Am J Hum Genet. 2008;83:106–111. doi: 10.1016/j.ajhg.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, Uruno K, Kumada S, Nishiyama K, Nishimura A, Okada I, Yoshimura Y, Hirai S-ichi, Kumada T, Hayasaka K, Fukuda A, Ogata K, Matsumoto N. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40:782–788. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- 18.Brunetti-Pierri N, Paciorkowski AR, Ciccone R, Mina ED, Bonaglia MC, Borgatti R, Schaaf CP, Sutton VR, Xia Z, Jelluma N, Ruivenkamp C, Bertrand M, de Ravel TJL, Jayakar P, Belli S, Rocchetti K, Pantaleoni C, D’Arrigo S, Hughes J, Cheung SW, Zuffardi O, Stankiewicz P. Duplications of FOXG1 in 14q12 are associated with developmental epilepsy, mental retardation, and severe speech impairment. Eur J Hum Genet. 2011;19:102–107. doi: 10.1038/ejhg.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding Y-X, Zhang Y, He B, Yue W-H, Zhang D, Zou L-P. A possible association of responsiveness to adrenocorticotropic hormone with specific GRIN1 haplotypes in infantile spasms. Dev Med Child Neurol. 2010;52:1028–1032. doi: 10.1111/j.1469-8749.2010.03746.x. [DOI] [PubMed] [Google Scholar]
- 20.Le Meur N, Holder-Espinasse M, Jaillard S, Goldenberg A, Joriot S, Amati-Bonneau P, Guichet A, Barth M, Charollais A, Journel H, Auvin S, Boucher C, Kerckaert J-P, David V, Manouvrier-Hanu S, Saugier-Veber P, Frébourg T, Dubourg C, Andrieux J, Bonneau D. MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J Med Genet. 2010;47:22–29. doi: 10.1136/jmg.2009.069732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nowakowska BA, Obersztyn E, Szymańska K, Bekiesińska-Figatowska M, Xia Z, Ricks CB, Bocian E, Stockton DW, Szczałuba K, Nawara M, Patel A, Scott DA, Cheung SW, Bohan TP, Stankiewicz P. Severe mental retardation, seizures, and hypotonia due to deletions of MEF2C. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:1042–1051. doi: 10.1002/ajmg.b.31071. [DOI] [PubMed] [Google Scholar]
- 22.Saitsu H, Tohyama J, Kumada T, Egawa K, Hamada K, Okada I, Mizuguchi T, Osaka H, Miyata R, Furukawa T, Haginoya K, Hoshino H, Goto T, Hachiya Y, Yamagata T, Saitoh S, Nagai T, Nishiyama K, Nishimura A, Miyake N, Komada M, Hayashi K, Hirai S-I, Ogata K, Kato M, Fukuda A, Matsumoto N. Dominant-negative mutations in alpha-II spectrin cause West syndrome with severe cerebral hypomyelination, spastic quadriplegia, and developmental delay. Am J Hum Genet. 2010;86:881–891. doi: 10.1016/j.ajhg.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molinari F, Kaminska A, Fiermonte G, Boddaert N, Raas-Rothschild A, Plouin P, Palmieri L, Brunelle F, Palmieri F, Dulac O, Munnich A, Colleaux L. Mutations in the mitochondrial glutamate carrier SLC25A22 in neonatal epileptic encephalopathy with suppression bursts. Clin Genet. 2009;76:188–194. doi: 10.1111/j.1399-0004.2009.01236.x. [DOI] [PubMed] [Google Scholar]
- 24.Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, Milh M, Kortüm F, Fritsch A, Pientka FK, Hellenbroich Y, Kalscheuer VM, Kohlhase J, Moog U, Rappold G, Rauch A, Ropers H-H, von Spiczak S, Tönnies H, Villeneuve N, Villard L, Zabel B, Zenker M, Laube B, Reis A, Wieczorek D, Van Maldergem L, Kutsche K. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010;42:1021–1026. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- 25.Paciorkowski AR, Thio LL, Rosenfeld JA, Gajecka M, Gurnett CA, Kulkarni S, Chung WK, Marsh ED, Gentile M, Reggin JD, Wheless JW, Balasubramanian S, Kumar R, Christian SL, Marini C, Guerrini R, Maltsev N, Shaffer LG, Dobyns WB. Copy number variants and infantile spasms: evidence for abnormalities in ventral forebrain development and pathways of synaptic function [Internet] Eur J Hum Genet. 2011 doi: 10.1038/ejhg.2011.121. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21694734. [DOI] [PMC free article] [PubMed]
- 26.Mefford HC, Cooper GM, Zerr T, Smith JD, Baker C, Shafer N, Thorland EC, Skinner C, Schwartz CE, Nickerson DA, Eichler EE. A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Genome Res. 2009;19:1579–1585. doi: 10.1101/gr.094987.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BHY, Cochrane L, Corsello C, Crawford EL, Crossett A, Cytrynbaum C, Dawson G, de Jonge M, Delorme R, Drmic I, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA, Folstein SE, Fombonne E, Freitag CM, Gilbert J, Gillberg C, Glessner JT, Goldberg J, Green A, Green J, Guter SJ, Hakonarson H, Heron EA, Hill M, Holt R, Howe JL, Hughes G, Hus V, Igliozzi R, Kim C, Klauck SM, Kolevzon A, Korvatska O, Kustanovich V, Lajonchere CM, Lamb JA, Laskawiec M, Leboyer M, Le Couteur A, Leventhal BL, Lionel AC, Liu X-Q, Lord C, Lotspeich L, Lund SC, Maestrini E, Mahoney W, Mantoulan C, Marshall CR, McConachie H, McDougle CJ, McGrath J, McMahon WM, Merikangas A, Migita O, Minshew NJ, Mirza GK, Munson J, Nelson SF, Noakes C, Noor A, Nygren G, Oliveira G, Papanikolaou K, Parr JR, Parrini B, Paton T, Pickles A, Pilorge M, Piven J, Ponting CP, Posey DJ, Poustka A, Poustka F, Prasad A, Ragoussis J, Renshaw K, Rickaby J, Roberts W, Roeder K, Roge B, Rutter ML, Bierut LJ, Rice JP, Salt J, Sansom K, Sato D, Segurado R, Sequeira AF, Senman L, Shah N, Sheffield VC, Soorya L, Sousa I, Stein O, Sykes N, Stoppioni V, Strawbridge C, Tancredi R, Tansey K, Thiruvahindrapduram B, Thompson AP, Thomson S, Tryfon A, Tsiantis J, Van Engeland H, Vincent JB, Volkmar F, Wallace S, Wang K, Wang Z, Wassink TH, Webber C, Weksberg R, Wing K, Wittemeyer K, Wood S, Wu J, Yaspan BL, Zurawiecki D, Zwaigenbaum L, Buxbaum JD, Cantor RM, Cook EH, Coon H, Cuccaro ML, Devlin B, Ennis S, Gallagher L, Geschwind DH, Gill M, Haines JL, Hallmayer J, Miller J, Monaco AP, Nurnberger JI, Paterson AD, Pericak-Vance MA, Schellenberg GD, Szatmari P, Vicente AM, Vieland VJ, Wijsman EM, Scherer SW, Sutcliffe JS, Betancur C. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Partikian A, Mitchell WG. Neurodevelopmental and epilepsy outcomes in a North American cohort of patients with infantile spasms. J Child Neurol. 2010;25:423–428. doi: 10.1177/0883073809341664. [DOI] [PubMed] [Google Scholar]
- 29.Lagae L, Verhelst H, Ceulemans B, De Meirleir L, Nassogne M-C, De Borchgrave V, D’Hooghe M, Foulon M, Van Bogaert P. Treatment and long term outcome in West syndrome: the clinical reality. A multicentre follow up study. Seizure. 2010;19:159–164. doi: 10.1016/j.seizure.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 30.West WJ. On a peculiar form of infantile convulsions. Lancet. 1841:724–725. [Google Scholar]
- 31.Lux AL. West & son: the origins of West syndrome. Brain Dev. 2001;23:443–446. doi: 10.1016/s0387-7604(01)00266-2. [DOI] [PubMed] [Google Scholar]
- 32.Pies NJ, Beardsmore CW. West & West syndrome--a historical sketch about the eponymous doctor, his work and his family. Brain Dev. 2003;25:84–101. doi: 10.1016/s0387-7604(02)00161-4. [DOI] [PubMed] [Google Scholar]
- 33.Gibbs EL, Fleming MM, Gibbs FA. Diagnosis and prognosis of hypsarhythmia and infantile spasms. Pediatrics. 1954;13:66–73. [PubMed] [Google Scholar]
- 34.Jeavons PM, Bower BD. The natural history of infantile spasms. Arch Dis Child. 1961;36:17–22. doi: 10.1136/adc.36.185.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thornton EM, Pampiglione G. Psychiatric disorders following infantile spasms. Lancet. 1979;1:1297. doi: 10.1016/s0140-6736(79)92258-x. [DOI] [PubMed] [Google Scholar]
- 36.Riikonen R, Amnell G. Psychiatric disorders in children with earlier infantile spasms. Dev Med Child Neurol. 1981;23:747–760. doi: 10.1111/j.1469-8749.1981.tb02063.x. [DOI] [PubMed] [Google Scholar]
- 37.Taft LT, Cohen HJ. Hypsarrhythmia and infantile autism: a clinical report. J Autism Child Schizophr. 1971;1:327–336. doi: 10.1007/BF01557352. [DOI] [PubMed] [Google Scholar]
- 38.Rasmussen P, Börjesson O, Wentz E, Gillberg C. Autistic disorders in Down syndrome: background factors and clinical correlates. Dev Med Child Neurol. 2001;43:750–754. doi: 10.1017/s0012162201001372. [DOI] [PubMed] [Google Scholar]
- 39.Molloy CA, Murray DS, Kinsman A, Castillo H, Mitchell T, Hickey FJ, Patterson B. Differences in the clinical presentation of Trisomy 21 with and without autism. J Intellect Disabil Res. 2009;53:143–151. doi: 10.1111/j.1365-2788.2008.01138.x. [DOI] [PubMed] [Google Scholar]
- 40.Primec ZR, Stare J, Neubauer D. The risk of lower mental outcome in infantile spasms increases after three weeks of hypsarrhythmia duration. Epilepsia. 2006;47:2202–2205. doi: 10.1111/j.1528-1167.2006.00888.x. [DOI] [PubMed] [Google Scholar]
- 41.Pellock JM, Hrachovy R, Shinnar S, Baram TZ, Bettis D, Dlugos DJ, Gaillard WD, Gibson PA, Holmes GL, Nordl DR, O’Dell C, Shields WD, Trevathan E, Wheless JW. Infantile spasms: a U.S. consensus report. Epilepsia. 2010;51:2175–2189. doi: 10.1111/j.1528-1167.2010.02657.x. [DOI] [PubMed] [Google Scholar]
- 42.Jeavons PM, Harper JR, Bower BD. Long-term prognosis in infantile spasms: a follow-up report on 112 cases. Dev Med Child Neurol. 1970;12:413–421. doi: 10.1111/j.1469-8749.1970.tb01934.x. [DOI] [PubMed] [Google Scholar]
- 43.Pollack MA, Zion TE, Kellaway P. Long-term prognosis of patients with infantile spasms following ACTH therapy. Epilepsia. 1979;20:255–260. doi: 10.1111/j.1528-1157.1979.tb04802.x. [DOI] [PubMed] [Google Scholar]
- 44.Rantala H, Shields WD, Christenson PD, Nielsen C, Buch D, Jacobsen V, Zachau-Christiansen B, Uhari M, Cherry JD. Risk factors of infantile spasms compared with other seizures in children under 2 years of age. Epilepsia. 1996;37:362–366. doi: 10.1111/j.1528-1157.1996.tb00572.x. [DOI] [PubMed] [Google Scholar]
- 45.Guerrini R, Moro F, Kato M, Barkovich AJ, Shiihara T, McShane MA, Hurst J, Loi M, Tohyama J, Norci V, Hayasaka K, Kang UJ, Das S, Dobyns WB. Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology. 2007;69:427–433. doi: 10.1212/01.wnl.0000266594.16202.c1. [DOI] [PubMed] [Google Scholar]
- 46.Zweier M, Gregor A, Zweier C, Engels H, Sticht H, Wohlleber E, Bijlsma EK, Holder SE, Zenker M, Rossier E, Grasshoff U, Johnson DS, Robertson L, Firth HV, Ekici AB, Reis A, Rauch A. Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum Mutat. 2010;31:722–733. doi: 10.1002/humu.21253. [DOI] [PubMed] [Google Scholar]
- 47.Fulp CT, Cho G, Marsh ED, Nasrallah IM, Labosky PA, Golden JA. Identification of Arx transcriptional targets in the developing basal forebrain. Hum Mol Genet. 2008;17:3740–3760. doi: 10.1093/hmg/ddn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mei D, Marini C, Novara F, Bernardina BD, Granata T, Fontana E, Parrini E, Ferrari AR, Murgia A, Zuffardi O, Guerrini R. Xp22. 3 genomic deletions involving the CDKL5 gene in girls with early onset epileptic encephalopathy. Epilepsia. 2010;51:647–654. doi: 10.1111/j.1528-1167.2009.02308.x. [DOI] [PubMed] [Google Scholar]
- 49.Deprez L, Weckhuysen S, Holmgren P, Suls A, Van Dyck T, Goossens D, Del-Favero J, Jansen A, Verhaert K, Lagae L, Jordanova A, Van Coster R, Yendle S, Berkovic SF, Scheffer I, Ceulemans B, De Jonghe P. Clinical spectrum of early-onset epileptic encephalopathies associated with STXBP1 mutations. Neurology. 2010;75:1159–1165. doi: 10.1212/WNL.0b013e3181f4d7bf. [DOI] [PubMed] [Google Scholar]
- 50.Kato M, Saitoh S, Kamei A, Shiraishi H, Ueda Y, Akasaka M, Tohyama J, Akasaka N, Hayasaka K. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome) Am J Hum Genet. 2007;81:361–366. doi: 10.1086/518903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Absoud M, Parr JR, Halliday D, Pretorius P, Zaiwalla Z, Jayawant S. A novel ARX phenotype: rapid neurodegeneration with Ohtahara syndrome and a dyskinetic movement disorder. Dev Med Child Neurol. 2010;52:305–307. doi: 10.1111/j.1469-8749.2009.03470.x. [DOI] [PubMed] [Google Scholar]
- 52.Fullston T, Brueton L, Willis T, Philip S, MacPherson L, Finnis M, Gecz J, Morton J. Ohtahara syndrome in a family with an ARX protein truncation mutation (c.81C>G/p.Y27X) Eur J Hum Genet. 2010;18:157–162. doi: 10.1038/ejhg.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Djukic A, Lado FA, Shinnar S, Moshé SL. Are early myoclonic encephalopathy (EME) and the Ohtahara syndrome (EIEE) independent of each other? Epilepsy Res. 2006;70 (Suppl 1):S68–76. doi: 10.1016/j.eplepsyres.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 54.Otsuka M, Oguni H, Liang J-S, Ikeda H, Imai K, Hirasawa K, Imai K, Tachikawa E, Shimojima K, Osawa M, Yamamoto T. STXBP1 mutations cause not only Ohtahara syndrome but also West syndrome--result of Japanese cohort study. Epilepsia. 2010;51:2449–2452. doi: 10.1111/j.1528-1167.2010.02767.x. [DOI] [PubMed] [Google Scholar]
- 55.Saitsu H, Kato M, Okada I, Orii KE, Higuchi T, Hoshino H, Kubota M, Arai H, Tagawa T, Kimura S, Sudo A, Miyama S, Takami Y, Watanabe T, Nishimura A, Nishiyama K, Miyake N, Wada T, Osaka H, Kondo N, Hayasaka K, Matsumoto N. STXBP1 mutations in early infantile epileptic encephalopathy with suppression-burst pattern. Epilepsia. 2010;51:2397–2405. doi: 10.1111/j.1528-1167.2010.02728.x. [DOI] [PubMed] [Google Scholar]
- 56.Morris-Rosendahl DJ, Najm J, Lachmeijer AMA, Sztriha L, Martins M, Kuechler A, Haug V, Zeschnigk C, Martin P, Santos M, Vasconcelos C, Omran H, Kraus U, Van der Knaap MS, Schuierer G, Kutsche K, Uyanik G. Refining the phenotype of alpha-1a Tubulin (TUBA1A) mutation in patients with classical lissencephaly. Clin Genet. 2008;74:425–433. doi: 10.1111/j.1399-0004.2008.01093.x. [DOI] [PubMed] [Google Scholar]
- 57.Guerrini R, Filippi T. Neuronal migration disorders, genetics, and epileptogenesis. J Child Neurol. 2005;20:287–299. doi: 10.1177/08830738050200040401. [DOI] [PubMed] [Google Scholar]
- 58.Dobyns WB, Berry-Kravis E, Havernick NJ, Holden KR, Viskochil D. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am J Med Genet. 1999;86:331–337. [PubMed] [Google Scholar]
- 59.Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, Omichi K, Suzuki R, Kato-Fukui Y, Kamiirisa K, Matsuo M, Kamijo S-ichi, Kasahara M, Yoshioka H, Ogata T, Fukuda T, Kondo I, Kato M, Dobyns WB, Yokoyama M, Morohashi K-ichirou. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359–369. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- 60.Fleck MW, Hirotsune S, Gambello MJ, Phillips-Tansey E, Suares G, Mervis RF, Wynshaw-Boris A, McBain CJ. Hippocampal abnormalities and enhanced excitability in a murine model of human lissencephaly. J Neurosci. 2000;20:2439–2450. doi: 10.1523/JNEUROSCI.20-07-02439.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McManus MF, Nasrallah IM, Pancoast MM, Wynshaw-Boris A, Golden JA. Lis1 is necessary for normal non-radial migration of inhibitory interneurons. Am J Pathol. 2004;165:775–784. doi: 10.1016/S0002-9440(10)63340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marsh E, Fulp C, Gomez E, Nasrallah I, Minarcik J, Sudi J, Christian SL, Mancini G, Labosky P, Dobyns W, Brooks-Kayal A, Golden JA. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain. 2009;132:1563–1576. doi: 10.1093/brain/awp107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51:1236–1241. doi: 10.1111/j.1528-1167.2009.02474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crino PB, Aronica E, Baltuch G, Nathanson KL. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology. 2010;74:1716–1723. doi: 10.1212/WNL.0b013e3181e04325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Doherty C, Goh S, Young Poussaint T, Erdag N, Thiele EA. Prognostic significance of tuber count and location in tuberous sclerosis complex. J Child Neurol. 2005;20:837–841. doi: 10.1177/08830738050200101301. [DOI] [PubMed] [Google Scholar]
- 66.Chu-Shore CJ, Major P, Montenegro M, Thiele E. Cyst-like tubers are associated with TSC2 and epilepsy in tuberous sclerosis complex. Neurology. 2009;72:1165–1169. doi: 10.1212/01.wnl.0000345365.92821.86. [DOI] [PubMed] [Google Scholar]
- 67.Baker P, Piven J, Sato Y. Autism and tuberous sclerosis complex: prevalence and clinical features. J Autism Dev Disord. 1998;28:279–285. doi: 10.1023/a:1026004501631. [DOI] [PubMed] [Google Scholar]
- 68.Staley BA, Montenegro MA, Major P, Muzykewicz DA, Halpern EF, Kopp CMC, Newberry P, Thiele EA. Self-injurious behavior and tuberous sclerosis complex: frequency and possible associations in a population of 257 patients. Epilepsy Behav. 2008;13:650–653. doi: 10.1016/j.yebeh.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 69.Hunt A, Dennis J. Psychiatric disorder among children with tuberous sclerosis. Dev Med Child Neurol. 1987;29:190–198. doi: 10.1111/j.1469-8749.1987.tb02135.x. [DOI] [PubMed] [Google Scholar]
- 70.Schick V, Majores M, Engels G, Hartmann W, Elger CE, Schramm J, Schoch S, Becker AJ. Differential Pi3K-pathway activation in cortical tubers and focal cortical dysplasias with balloon cells. Brain Pathol. 2007;17:165–173. doi: 10.1111/j.1750-3639.2007.00059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sfaello I, Castelnau P, Blanc N, Ogier H, Evrard P, Arzimanoglou A. Infantile spasms and Menkes disease. Epileptic Disord. 2000;2:227–230. [PubMed] [Google Scholar]
- 72.Chen PT, Young C, Lee WT, Wang PJ, Peng SS, Shen YZ. Early epileptic encephalopathy with suppression burst electroencephalographic pattern--an analysis of eight Taiwanese patients. Brain Dev. 2001;23:715–720. doi: 10.1016/s0387-7604(01)00285-6. [DOI] [PubMed] [Google Scholar]
- 73.Zhongshu Z, Weiming Y, Yukio F, Cheng-LNing Z, Zhixing W. Clinical analysis of West syndrome associated with phenylketonuria. Brain Dev. 2001;23:552–557. doi: 10.1016/s0387-7604(01)00260-1. [DOI] [PubMed] [Google Scholar]
- 74.Venta-Sobero JA, Porras-Kattz E, Gutiérrez-Moctezuma J. West syndrome as an epileptic presentation in Menkes’ disease. Two cases report. Rev Neurol. 2004;39:133–136. [PubMed] [Google Scholar]
- 75.Bahi-Buisson N, Kaminska A, Nabbout R, Barnerias C, Desguerre I, De Lonlay P, Mayer M, Plouin P, Dulac O, Chiron C. Epilepsy in Menkes disease: analysis of clinical stages. Epilepsia. 2006;47:380–386. doi: 10.1111/j.1528-1167.2006.00432.x. [DOI] [PubMed] [Google Scholar]
- 76.Campeau PM, Valayannopoulos V, Touati G, Bahi-Buisson N, Boddaert N, Plouin P, Rabier D, Benoist J-F, Dulac O, de Lonlay P, Desguerre I. Management of West syndrome in a patient with methylmalonic aciduria. J Child Neurol. 2010;25:94–97. doi: 10.1177/0883073809336119. [DOI] [PubMed] [Google Scholar]
- 77.Rossi S, Daniele I, Bastrenta P, Mastrangelo M, Lista G. Early myoclonic encephalopathy and nonketotic hyperglycinemia. Pediatr Neurol. 2009;41:371–374. doi: 10.1016/j.pediatrneurol.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 78.Bahi-Buisson N, Eisermann M, Nivot S, Bellanné-Chantelot C, Dulac O, Bach N, Plouin P, Chiron C, de Lonlay P. Infantile spasms as an epileptic feature of DEND syndrome associated with an activating mutation in the potassium adenosine triphosphate (ATP) channel, Kir6.2. J Child Neurol. 2007;22:1147–1150. doi: 10.1177/0883073807306272. [DOI] [PubMed] [Google Scholar]
- 79.Shimomura K, Hörster F, de Wet H, Flanagan SE, Ellard S, Hattersley AT, Wolf NI, Ashcroft F, Ebinger F. A novel mutation causing DEND syndrome: a treatable channelopathy of pancreas and brain. Neurology. 2007;69:1342–1349. doi: 10.1212/01.wnl.0000268488.51776.53. [DOI] [PubMed] [Google Scholar]
- 80.Della Manna T, Battistim C, Radonsky V, Savoldelli RD, Damiani D, Kok F, Pearson ER, Ellard S, Hattersley AT, Reis AF. Glibenclamide unresponsiveness in a Brazilian child with permanent neonatal diabetes mellitus and DEND syndrome due to a C166Y mutation in KCNJ11 (Kir6. 2) gene. Arq Bras Endocrinol Metabol. 2008;52:1350–1355. doi: 10.1590/s0004-27302008000800024. [DOI] [PubMed] [Google Scholar]
- 81.Mäkelä-Bengs P, Suomalainen A, Majander A, Rapola J, Kalimo H, Nuutila A, Pihko H. Correlation between the clinical symptoms and the proportion of mitochondrial DNA carrying the 8993 point mutation in the NARP syndrome. Pediatr Res. 1995;37:634–639. doi: 10.1203/00006450-199505000-00014. [DOI] [PubMed] [Google Scholar]
- 82.Ostergaard E, Hansen FJ, Sorensen N, Duno M, Vissing J, Larsen PL, Faeroe O, Thorgrimsson S, Wibrand F, Christensen E, Schwartz M. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130:853–861. doi: 10.1093/brain/awl383. [DOI] [PubMed] [Google Scholar]
- 83.Pascual JM, Campistol J, Gil-Nagel A. Epilepsy in inherited metabolic disorders. Neurologist. 2008;14:S2–S14. doi: 10.1097/01.nrl.0000340787.30542.41. [DOI] [PubMed] [Google Scholar]
- 84.Glushakov AV, Glushakova O, Varshney M, Bajpai LK, Sumners C, Laipis PJ, Embury JE, Baker SP, Otero DH, Dennis DM, Seubert CN, Martynyuk AE. Long-term changes in glutamatergic synaptic transmission in phenylketonuria. Brain. 2005;128:300–307. doi: 10.1093/brain/awh354. [DOI] [PubMed] [Google Scholar]
- 85.Dobyns WB. Agenesis of the corpus callosum and gyral malformations are frequent manifestations of nonketotic hyperglycinemia. Neurology. 1989;39:817–820. doi: 10.1212/wnl.39.6.817. [DOI] [PubMed] [Google Scholar]
- 86.Battaglia A, Hoyme HE, Dallapiccola B, Zackai E, Hudgins L, McDonald-McGinn D, Bahi-Buisson N, Romano C, Williams CA, Brailey LL, Braley LL, Zuberi SM, Carey JC. Further delineation of deletion 1p36 syndrome in 60 patients: a recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. 2008;121:404–410. doi: 10.1542/peds.2007-0929. [DOI] [PubMed] [Google Scholar]
- 87.Tatsuno M, Hayashi M, Iwamoto H, Suzuki Y, Kuroki Y. Epilepsy in childhood Down syndrome. Brain Dev. 1984;6:37–44. doi: 10.1016/s0387-7604(84)80008-x. [DOI] [PubMed] [Google Scholar]
- 88.Romano C, Tiné A, Fazio G, Rizzo R, Colognola RM, Sorge G, Bergonzi P, Pavone L. Seizures in patients with trisomy 21. Am J Med Genet Suppl. 1990;7:298–300. doi: 10.1002/ajmg.1320370758. [DOI] [PubMed] [Google Scholar]
- 89.Dobyns WB, Curry CJ, Hoyme HE, Turlington L, Ledbetter DH. Clinical and molecular diagnosis of Miller-Dieker syndrome. Am J Hum Genet. 1991;48:584–594. [PMC free article] [PubMed] [Google Scholar]
- 90.Pueschel SM, Louis S, McKnight P. Seizure disorders in Down syndrome. Arch Neurol. 1991;48:318–320. doi: 10.1001/archneur.1991.00530150088024. [DOI] [PubMed] [Google Scholar]
- 91.Schinzel A. Tetrasomy 12p (Pallister-Killian syndrome) J Med Genet. 1991;28:122–125. doi: 10.1136/jmg.28.2.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stafstrom CE, Konkol RJ. Infantile spasms in children with Down syndrome. Dev Med Child Neurol. 1994;36:576–585. doi: 10.1111/j.1469-8749.1994.tb11894.x. [DOI] [PubMed] [Google Scholar]
- 93.Repetto GM, White LM, Bader PJ, Johnson D, Knoll JH. Interstitial duplications of chromosome region 15q11q13: clinical and molecular characterization. Am J Med Genet. 1998;79:82–89. doi: 10.1002/(sici)1096-8628(19980901)79:2<82::aid-ajmg2>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 94.Sánchez-Carpintero R, McLellan A, Parmeggiani L, Cockwell AE, Ellis RJ, Cross JH, Eckhardt S, Guerrini R. Pallister-Killian syndrome: an unusual cause of epileptic spasms. Dev Med Child Neurol. 2005;47:776–779. doi: 10.1017/S0012162205001623. [DOI] [PubMed] [Google Scholar]
- 95.Yamamoto H, Fukuda M, Murakami H, Kamiyama N, Miyamoto Y. A case of Pallister-Killian syndrome associated with West syndrome. Pediatr Neurol. 2007;37:226–228. doi: 10.1016/j.pediatrneurol.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 96.Cardoso C, Leventer RJ, Ward HL, Toyo-Oka K, Chung J, Gross A, Martin CL, Allanson J, Pilz DT, Olney AH, Mutchinick OM, Hirotsune S, Wynshaw-Boris A, Dobyns WB, Ledbetter DH. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet. 2003;72:918–930. doi: 10.1086/374320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, Ayala R, Tsai L-H, Dobyns W, Ledbetter D, Hirotsune S, Wynshaw-Boris A. 14-3-3epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet. 2003;34:274–285. doi: 10.1038/ng1169. [DOI] [PubMed] [Google Scholar]
- 98.Komoike Y, Fujii K, Nishimura A, Hiraki Y, Hayashidani M, Shimojima K, Nishizawa T, Higashi K, Yasukawa K, Saitsu H, Miyake N, Mizuguchi T, Matsumoto N, Osawa M, Kohno Y, Higashinakagawa T, Yamamoto T. Zebrafish gene knockdowns imply roles for human YWHAG in infantile spasms and cardiomegaly. Genesis. 2010;48:233–243. doi: 10.1002/dvg.20607. [DOI] [PubMed] [Google Scholar]
- 99.Röthlisberger B, Hoigné I, Huber AR, Brunschwiler W, Capone Mori A. Deletion of 7q11.21–q11.23 and infantile spasms without deletion of MAGI2. Am J Med Genet A. 2010;152A:434–437. doi: 10.1002/ajmg.a.33220. [DOI] [PubMed] [Google Scholar]
- 100.Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, Mefford HC, Kidd JM, Browning SR, Browning BL, Dickel DE, Levy DL, Ballif BC, Platky K, Farber DM, Gowans GC, Wetherbee JJ, Asamoah A, Weaver DD, Mark PR, Dickerson J, Garg BP, Ellingwood SA, Smith R, Banks VC, Smith W, McDonald MT, Hoo JJ, French BN, Hudson C, Johnson JP, Ozmore JR, Moeschler JB, Surti U, Escobar LF, El-Khechen D, Gorski JL, Kussmann J, Salbert B, Lacassie Y, Biser A, McDonald-McGinn DM, Zackai EH, Deardorff MA, Shaikh TH, Haan E, Friend KL, Fichera M, Romano C, Gécz J, DeLisi LE, Sebat J, King M-C, Shaffer LG, Eichler EE. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vorstman JAS, van Daalen E, Jalali GR, Schmidt ERE, Pasterkamp RJ, de Jonge M, Hennekam EAM, Janson E, Staal WG, van der Zwaag B, Burbach JPH, Kahn RS, Emanuel BS, van Engeland H, Ophoff RA. A double hit implicates DIAPH3 as an autism risk gene [Internet] Mol Psychiatry. 2010 Mar; doi: 10.1038/mp.2010.26. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20308993. [DOI] [PMC free article] [PubMed]
- 102.O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, Eichler EE. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marion RW, Alvarez LA, Marans ZS, Lantos G, Chitayat D. Computed tomography of the brain in the Smith-Lemli-Opitz syndrome. J Child Neurol. 1987;2:198–200. doi: 10.1177/088307388700200305. [DOI] [PubMed] [Google Scholar]
- 104.Sackey A, Coulter B, Fryer A, Van Velzen D. Epilepsy in the Freeman Sheldon syndrome. J Child Neurol. 1995;10:335–337. doi: 10.1177/088307389501000421. [DOI] [PubMed] [Google Scholar]
- 105.Kondoh T, Kamimura N, Tsuru A, Matsumoto T, Matsuzaka T, Moriuchi H. A case of Schinzel-Giedion syndrome complicated with progressive severe gingival hyperplasia and progressive brain atrophy. Pediatr Int. 2001;43:181–184. doi: 10.1046/j.1442-200x.2001.01348.x. [DOI] [PubMed] [Google Scholar]
- 106.Sugai K, Fukuyama Y, Yasuda K, Fujimoto S, Ohtsu M, Ohta H, Ogawa A, Hamano S, Hirano S, Yoshioka H, Ishikawa A, Seki T, Itokazu N, Tawa R. Clinical and pedigree study on familial cases of West syndrome in Japan. Brain Dev. 2001;23:558–564. doi: 10.1016/s0387-7604(01)00262-5. [DOI] [PubMed] [Google Scholar]
- 107.Grosso S, Pagano C, Cioni M, Di Bartolo RM, Morgese G, Balestri P. Schinzel-Giedion syndrome: a further cause of West syndrome. Brain Dev. 2003;25:294–298. doi: 10.1016/s0387-7604(02)00232-2. [DOI] [PubMed] [Google Scholar]
- 108.Minn D, Christmann D, De Saint-Martin A, Alembik Y, Eliot M, Mack G, Fischbach M, Flament J, Veillon F, Dollfus H. Further clinical and sensorial delineation of Schinzel-Giedion syndrome: report of two cases. Am J Med Genet. 2002;109:211–217. doi: 10.1002/ajmg.10348. [DOI] [PubMed] [Google Scholar]
- 109.Ray M, Malhi P, Bhalla AK, Singhi PD. Cerebral gigantism with West syndrome. Indian Pediatr. 2003;40:673–675. [PubMed] [Google Scholar]
- 110.Sheen VL, Ganesh VS, Topcu M, Sebire G, Bodell A, Hill RS, Grant PE, Shugart YY, Imitola J, Khoury SJ, Guerrini R, Walsh CA. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 2004;36:69–76. doi: 10.1038/ng1276. [DOI] [PubMed] [Google Scholar]
- 111.Masruha MR, Caboclo LOSF, Carrete H, Cendes IL, Rodrigues MG, Garzon E, Yacubian EMT, Sakamoto AC, Sheen V, Harney M, Neal J, Hill RS, Bodell A, Walsh C, Vilanova LCP. Mutation in filamin A causes periventricular heterotopia, developmental regression, and West syndrome in males. Epilepsia. 2006;47:211–214. doi: 10.1111/j.1528-1167.2006.00390.x. [DOI] [PubMed] [Google Scholar]
- 112.Korf BR, Carrazana E, Holmes GL. Patterns of seizures observed in association with neurofibromatosis 1. Epilepsia. 1993;34:616–620. doi: 10.1111/j.1528-1157.1993.tb00437.x. [DOI] [PubMed] [Google Scholar]
- 113.Ruggieri M, Iannetti P, Clementi M, Polizzi A, Incorpora G, Spalice A, Pavone P, Praticò AD, Elia M, Gabriele AL, Tenconi R, Pavone L. Neurofibromatosis type 1 and infantile spasms. Childs Nerv Syst. 2009;25:211–216. doi: 10.1007/s00381-008-0706-5. [DOI] [PubMed] [Google Scholar]
- 114.Aizaki K, Sugai K, Saito Y, Nakagawa E, Sasaki M, Aoki Y, Matsubara Y. Cardio-facio-cutaneous syndrome with infantile spasms and delayed myelination. Brain Dev. 2011;33:166–169. doi: 10.1016/j.braindev.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 115.Hino-Fukuyo N, Haginoya K, Uematsu M, Nakayama T, Kikuchi A, Kure S, Kamada F, Abe Y, Arai N, Togashi N, Onuma A, Tsuchiya S. Smith-Magenis syndrome with West syndrome in a 5-year-old girl: a long-term follow-up study. J Child Neurol. 2009;24:868–873. doi: 10.1177/0883073808330186. [DOI] [PubMed] [Google Scholar]
- 116.Wakamoto H, Sumi A, Motoki T, Ohmori H. Positron emission tomography with glucose hypermetabolism of a hypothalamic hamartoma in infantile spasms associated with Pallister-Hall syndrome. Brain Dev. 2010;32:677–680. doi: 10.1016/j.braindev.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 117.Aicardi J. Aicardi syndrome. Brain Dev. 2005;27:164–171. doi: 10.1016/j.braindev.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 118.Shevell MI, Colangelo P, Treacy E, Polomeno RC, Rosenblatt B. Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO syndrome) Pediatr Neurol. 1996;15:337–339. doi: 10.1016/s0887-8994(96)00161-0. [DOI] [PubMed] [Google Scholar]
- 119.Klein A, Schmitt B, Boltshauser E. Progressive encephalopathy with edema, hypsarrhythmia and optic atrophy (PEHO) syndrome in a Swiss child. Eur J Paediatr Neurol. 2004;8:317–321. doi: 10.1016/j.ejpn.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 120.Field MJ, Grattan-Smith P, Piper SM, Thompson EM, Haan EA, Edwards M, James S, Wilkinson I, Adès LC. PEHO and PEHO-like syndromes: report of five Australian cases. Am J Med Genet A. 2003;122A:6–12. doi: 10.1002/ajmg.a.20216. [DOI] [PubMed] [Google Scholar]
- 121.Fauser S, Huppertz H-J, Bast T, Strobl K, Pantazis G, Altenmueller D-M, Feil B, Rona S, Kurth C, Rating D, Korinthenberg R, Steinhoff BJ, Volk B, Schulze-Bonhage A. Clinical characteristics in focal cortical dysplasia: a retrospective evaluation in a series of 120 patients. Brain. 2006;129:1907–1916. doi: 10.1093/brain/awl133. [DOI] [PubMed] [Google Scholar]
- 122.Chugani HT, Shields WD, Shewmon DA, Olson DM, Phelps ME, Peacock WJ. Infantile spasms: I. PET identifies focal cortical dysgenesis in cryptogenic cases for surgical treatment. Ann Neurol. 1990;27:406–413. doi: 10.1002/ana.410270408. [DOI] [PubMed] [Google Scholar]
- 123.Cepeda C, André VM, Wu N, Yamazaki I, Uzgil B, Vinters HV, Levine MS, Mathern GW. Immature neurons and GABA networks may contribute to epileptogenesis in pediatric cortical dysplasia. Epilepsia. 2007;48:79–85. doi: 10.1111/j.1528-1167.2007.01293.x. [DOI] [PubMed] [Google Scholar]
- 124.Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51:27–36. doi: 10.1111/j.1528-1167.2009.02341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tjiam AT, Stefanko S, Schenk VW, de Vlieger M. Infantile spasms associated with hemihypsarrhythmia and hemimegalencephaly. Dev Med Child Neurol. 1978;20:779–798. doi: 10.1111/j.1469-8749.1978.tb15310.x. [DOI] [PubMed] [Google Scholar]
- 126.Kobayashi K, Ohtsuka Y, Ohno S, Ohmori I, Ogino T, Yoshinaga H, Tanaka A, Hiraki Y, Oka E. Clinical spectrum of epileptic spasms associated with cortical malformation. Neuropediatrics. 2001;32:236–244. doi: 10.1055/s-2001-19117. [DOI] [PubMed] [Google Scholar]
- 127.Blennow G, Starck L. High dose B6 treatment in infantile spasms. Neuropediatrics. 1986;17:7–10. doi: 10.1055/s-2008-1052491. [DOI] [PubMed] [Google Scholar]
- 128.Basura GJ, Hagland SP, Wiltse AM, Gospe SM. Clinical features and the management of pyridoxine-dependent and pyridoxine-responsive seizures: review of 63 North American cases submitted to a patient registry. Eur J Pediatr. 2009;168:697–704. doi: 10.1007/s00431-008-0823-x. [DOI] [PubMed] [Google Scholar]
- 129.Bennett CL, Chen Y, Hahn S, Glass IA, Gospe SM. Prevalence of ALDH7A1 mutations in 18 North American pyridoxine-dependent seizure (PDS) patients. Epilepsia. 2009;50:1167–1175. doi: 10.1111/j.1528-1167.2008.01816.x. [DOI] [PubMed] [Google Scholar]
- 130.Shah NS, Mitchell WG, Boles RG. Mitochondrial disorders: a potentially under-recognized etiology of infantile spasms. J Child Neurol. 2002;17:369–372. doi: 10.1177/088307380201700511. [DOI] [PubMed] [Google Scholar]
- 131.Sadleir LG, Connolly MB, Applegarth D, Hendson G, Clarke L, Rakshi C, Farrell K. Spasms in children with definite and probable mitochondrial disease. Eur J Neurol. 2004;11:103–110. doi: 10.1046/j.1351-5101.2003.00724.x. [DOI] [PubMed] [Google Scholar]
- 132.Lee YM, Kang HC, Lee JS, Kim SH, Kim EY, Lee SK, Slama A, Kim HD. Mitochondrial respiratory chain defects: underlying etiology in various epileptic conditions. Epilepsia. 2008;49:685–690. doi: 10.1111/j.1528-1167.2007.01522.x. [DOI] [PubMed] [Google Scholar]
- 133.Khurana DS, Salganicoff L, Melvin JJ, Hobdell EF, Valencia I, Hardison HH, Marks HG, Grover WD, Legido A. Epilepsy and respiratory chain defects in children with mitochondrial encephalopathies. Neuropediatrics. 2008;39:8–13. doi: 10.1055/s-2008-1076737. [DOI] [PubMed] [Google Scholar]
- 134.Riikonen R, Donner M. Incidence and aetiology of infantile spasms from 1960 to 1976: a population study in Finland. Dev Med Child Neurol. 1979;21:333–343. doi: 10.1111/j.1469-8749.1979.tb01625.x. [DOI] [PubMed] [Google Scholar]
- 135.Kumaran A, Kar S, Kapoor RR, Hussain K. The clinical problem of hyperinsulinemic hypoglycemia and resultant infantile spasms. Pediatrics. 2010;126:e1231–1236. doi: 10.1542/peds.2009-2775. [DOI] [PubMed] [Google Scholar]
- 136.Bahi-Buisson N, Nectoux J, Rosas-Vargas H, Milh M, Boddaert N, Girard B, Cances C, Ville D, Afenjar A, Rio M, Héron D, N’guyen Morel MA, Arzimanoglou A, Philippe C, Jonveaux P, Chelly J, Bienvenu T. Key clinical features to identify girls with CDKL5 mutations. Brain. 2008;131:2647–2661. doi: 10.1093/brain/awn197. [DOI] [PubMed] [Google Scholar]
- 137.Rosas-Vargas H, Bahi-Buisson N, Philippe C, Nectoux J, Girard B, N’Guyen Morel MA, Gitiaux C, Lazaro L, Odent S, Jonveaux P, Chelly J, Bienvenu T. Impairment of CDKL5 nuclear localisation as a cause for severe infantile encephalopathy. J Med Genet. 2008;45:172–178. doi: 10.1136/jmg.2007.053504. [DOI] [PubMed] [Google Scholar]
- 138.Alvarez LA, Shinnar S, Moshé SL. Infantile spasms due to unilateral cerebral infarcts. Pediatrics. 1987;79:1024–1026. [PubMed] [Google Scholar]
- 139.Constantinou JE, Gillis J, Ouvrier RA, Rahilly PM. Hypoxic-ischaemic encephalopathy after near miss sudden infant death syndrome. Arch Dis Child. 1989;64:703–708. doi: 10.1136/adc.64.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cusmai R, Ricci S, Pinard JM, Plouin P, Fariello G, Dulac O. West syndrome due to perinatal insults. Epilepsia. 1993;34:738–742. doi: 10.1111/j.1528-1157.1993.tb00455.x. [DOI] [PubMed] [Google Scholar]
- 141.Soman TB, Moharir M, DeVeber G, Weiss S. Infantile spasms as an adverse outcome of neonatal cortical sinovenous thrombosis. J Child Neurol. 2006;21:126–131. doi: 10.1177/08830738060210021001. [DOI] [PubMed] [Google Scholar]
- 142.Golomb MR, Garg BP, Williams LS. Outcomes of children with infantile spasms after perinatal stroke. Pediatr Neurol. 2006;34:291–295. doi: 10.1016/j.pediatrneurol.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 143.Volpe J. Neurology of the newborn. 5. Philadelphia: Saunders/Elsevier; 2008. [Google Scholar]
- 144.Pisani F, Orsini M, Braibanti S, Copioli C, Sisti L, Turco EC. Development of epilepsy in newborns with moderate hypoxic-ischemic encephalopathy and neonatal seizures. Brain Dev. 2009;31:64–68. doi: 10.1016/j.braindev.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 145.Riikonen R. Decreasing perinatal mortality: unchanged infantile spasm morbidity. Dev Med Child Neurol. 1995;37:232–238. doi: 10.1111/j.1469-8749.1995.tb11997.x. [DOI] [PubMed] [Google Scholar]
- 146.Riikonen R. Cytomegalovirus infection and infantile spasms. Dev Med Child Neurol. 1978;20:570–579. doi: 10.1111/j.1469-8749.1978.tb15275.x. [DOI] [PubMed] [Google Scholar]
- 147.Ushijima H, Bosu K, Abe T, Shinozaki T. Suspected rotavirus encephalitis. Arch Dis Child. 1986;61:692–694. doi: 10.1136/adc.61.7.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Riikonen R. Infantile spasms: infectious disorders. Neuropediatrics. 1993;24:274–280. doi: 10.1055/s-2008-1071556. [DOI] [PubMed] [Google Scholar]
- 149.Edwards MS, Rench MA, Haffar AA, Murphy MA, Desmond MM, Baker CJ. Long-term sequelae of group B streptococcal meningitis in infants. J Pediatr. 1985;106:717–722. doi: 10.1016/s0022-3476(85)80342-5. [DOI] [PubMed] [Google Scholar]
- 150.Corey L, Whitley RJ, Stone EF, Mohan K. Difference between herpes simplex virus type 1 and type 2 neonatal encephalitis in neurological outcome. Lancet. 1988;1:1–4. doi: 10.1016/s0140-6736(88)90997-x. [DOI] [PubMed] [Google Scholar]
- 151.Franco SM, Cornelius VE, Andrews BF. Long-term outcome of neonatal meningitis. Am J Dis Child. 1992;146:567–571. doi: 10.1001/archpedi.1992.02160170047014. [DOI] [PubMed] [Google Scholar]
- 152.Lahat E, Barr J, Barkai G, Paret G, Brand N, Barzilai A. Long term neurological outcome of herpes encephalitis. Arch Dis Child. 1999;80:69–71. doi: 10.1136/adc.80.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.de Louvois J, Halket S, Harvey D. Neonatal meningitis in England and Wales: sequelae at 5 years of age. Eur J Pediatr. 2005;164:730–734. doi: 10.1007/s00431-005-1747-3. [DOI] [PubMed] [Google Scholar]
- 154.Dodge NN, Dobyns WB. Agenesis of the corpus callosum and Dandy-Walker malformation associated with hemimegalencephaly in the sebaceous nevus syndrome. Am J Med Genet. 1995;56:147–150. doi: 10.1002/ajmg.1320560206. [DOI] [PubMed] [Google Scholar]
- 155.Pascual-Castroviejo I, Roche C, Martinez-Bermejo A, Arcas J, Lopez-Martin V, Tendero A, Esquiroz JL, Pascual-Pascual SI. Hypomelanosis of ITO. A study of 76 infantile cases. Brain Dev. 1998;20:36–43. doi: 10.1016/s0387-7604(97)00097-1. [DOI] [PubMed] [Google Scholar]
- 156.Mirzaa G, Dodge NN, Glass I, Day C, Gripp K, Nicholson L, Straub V, Voit T, Dobyns WB. Megalencephaly and perisylvian polymicrogyria with postaxial polydactyly and hydrocephalus: a rare brain malformation syndrome associated with mental retardation and seizures. Neuropediatrics. 2004;35:353–359. doi: 10.1055/s-2004-830497. [DOI] [PubMed] [Google Scholar]
- 157.Menascu S, Donner EJ. Linear nevus sebaceous syndrome: case reports and review of the literature. Pediatr Neurol. 2008;38:207–210. doi: 10.1016/j.pediatrneurol.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 158.Branch CE, Dyken PR. Choroid plexus papilloma and infantile spasms. Ann Neurol. 1979;5:302–304. doi: 10.1002/ana.410050315. [DOI] [PubMed] [Google Scholar]
- 159.Gabriel YH. Unilateral hemispheric ganglioglioma with infantile spasms. Ann Neurol. 1980;7:287–288. doi: 10.1002/ana.410070316. [DOI] [PubMed] [Google Scholar]
- 160.Kerrigan JF, Ng Y-tze, Prenger E, Krishnamoorthy KS, Wang NC, Rekate HL. Hypothalamic hamartoma and infantile spasms. Epilepsia. 2007;48:89–95. doi: 10.1111/j.1528-1167.2006.00835.x. [DOI] [PubMed] [Google Scholar]
- 161.Glaze DG, Percy AK, Skinner S, Motil KJ, Neul JL, Barrish JO, Lane JB, Geerts SP, Annese F, Graham J, McNair L, Lee H-S. Epilepsy and the natural history of Rett syndrome. Neurology. 2010;74:909–912. doi: 10.1212/WNL.0b013e3181d6b852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ricciardi S, Kilstrup-Nielsen C, Bienvenu T, Jacquette A, Landsberger N, Broccoli V. CDKL5 influences RNA splicing activity by its association to the nuclear speckle molecular machinery. Hum Mol Genet. 2009;18:4590–4602. doi: 10.1093/hmg/ddp426. [DOI] [PubMed] [Google Scholar]
- 163.Uemura N, Matsumoto A, Nakamura M, Watanabe K, Negoro T, Kumagai T, Miura K, Ohki T, Mizuno S, Okumura A, Aso K, Hayakawa F, Kondo Y. Evolution of seizures and electroencephalographical findings in 23 cases of deletion type Angelman syndrome. Brain Dev. 2005;27:383–388. doi: 10.1016/j.braindev.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 164.Fan Z, Greenwood R, Fisher A, Pendyal S, Powell CM. Characteristics and frequency of seizure disorder in 56 patients with Prader-Willi syndrome. Am J Med Genet A. 2009;149A:1581–1584. doi: 10.1002/ajmg.a.32934. [DOI] [PubMed] [Google Scholar]
- 165.Wallace RH, Hodgson BL, Grinton BE, Gardiner RM, Robinson R, Rodriguez-Casero V, Sadleir L, Morgan J, Harkin LA, Dibbens LM, Yamamoto T, Andermann E, Mulley JC, Berkovic SF, Scheffer IE. Sodium channel alpha1-subunit mutations in severe myoclonic epilepsy of infancy and infantile spasms. Neurology. 2003;61:765–769. doi: 10.1212/01.wnl.0000086379.71183.78. [DOI] [PubMed] [Google Scholar]
- 166.Harkin LA, McMahon JM, Iona X, Dibbens L, Pelekanos JT, Zuberi SM, Sadleir LG, Andermann E, Gill D, Farrell K, Connolly M, Stanley T, Harbord M, Andermann F, Wang J, Batish SD, Jones JG, Seltzer WK, Gardner A, Sutherland G, Berkovic SF, Mulley JC, Scheffer IE. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130:843–852. doi: 10.1093/brain/awm002. [DOI] [PubMed] [Google Scholar]
- 167.Ragona F, Brazzo D, De Giorgi I, Morbi M, Freri E, Teutonico F, Gennaro E, Zara F, Binelli S, Veggiotti P, Granata T. Dravet syndrome: early clinical manifestations and cognitive outcome in 37 Italian patients. Brain Dev. 2010;32:71–77. doi: 10.1016/j.braindev.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 168.Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 169.Yamakawa K. Molecular basis of severe myoclonic epilepsy in infancy. Brain Dev. 2009;31:401–404. doi: 10.1016/j.braindev.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 170.Blake JA, Harris MA. The Gene Ontology (GO) project: structured vocabularies for molecular biology and their application to genome and expression analysis. Curr Protoc Bioinformatics. 2008;7(Unit 7.2) doi: 10.1002/0471250953.bi0702s00. [DOI] [PubMed] [Google Scholar]
- 171.Hébert JM, Fishell G. The genetics of early telencephalon patterning: some assembly required. Nat Rev Neurosci. 2008;9:678–685. doi: 10.1038/nrn2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yeung A, Bruno D, Scheffer IE, Carranza D, Burgess T, Slater HR, Amor DJ. 4. 45 Mb microduplication in chromosome band 14q12 including FOXG1 in a girl with refractory epilepsy and intellectual impairment. Eur J Med Genet. 2009;52:440–442. doi: 10.1016/j.ejmg.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 173.Colasante G, Sessa A, Crispi S, Calogero R, Mansouri A, Collombat P, Broccoli V. Arx acts as a regional key selector gene in the ventral telencephalon mainly through its transcriptional repression activity. Dev Biol. 2009;334:59–71. doi: 10.1016/j.ydbio.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 174.Molinari F. Mitochondria and neonatal epileptic encephalopathies with suppression burst. J Bioenerg Biomembr. 2010;42:467–471. doi: 10.1007/s10863-010-9323-6. [DOI] [PubMed] [Google Scholar]
- 175.Aldamiz-Echevarría Azuar L, Prats Viñas JM, Sanjurjo Crespo P, Prieto Perera JA, Labayru Echeverría MT. Infantile spasms as the first manifestation of propionic acidemia. An Pediatr (Barc) 2005;63:548–550. doi: 10.1016/s1695-4033(05)70255-1. [DOI] [PubMed] [Google Scholar]
- 176.Battaglia A, Hoyme HE, Dallapiccola B, Zackai E, Hudgins L, McDonald-McGinn D, Bahi-Buisson N, Romano C, Williams CA, Brailey LL, Braley LL, Zuberi SM, Carey JC. Further delineation of deletion 1p36 syndrome in 60 patients: a recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. 2008;121:404–410. doi: 10.1542/peds.2007-0929. [DOI] [PubMed] [Google Scholar]
- 177.Tsao CY, Westman JA. Infantile spasms in two children with Williams syndrome. Am J Med Genet. 1997;71:54–56. [PubMed] [Google Scholar]
- 178.Bolton PF, Dennis NR, Browne CE, Thomas NS, Veltman MW, Thompson RJ, Jacobs P. The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am J Med Genet. 2001;105:675–685. doi: 10.1002/ajmg.1551. [DOI] [PubMed] [Google Scholar]
- 179.Denis D, Chateil JF, Brun M, Brissaud O, Lacombe D, Fontan D, Flurin V, Pedespan J. Schizencephaly: clinical and imaging features in 30 infantile cases. Brain Dev. 2000;22:475–483. doi: 10.1016/s0387-7604(00)00173-x. [DOI] [PubMed] [Google Scholar]
- 180.Wieck G, Leventer RJ, Squier WM, Jansen A, Andermann E, Dubeau F, Ramazzotti A, Guerrini R, Dobyns WB. Periventricular nodular heterotopia with overlying polymicrogyria. Brain. 2005;128:2811–2821. doi: 10.1093/brain/awh658. [DOI] [PubMed] [Google Scholar]
- 181.Colombo E, Collombat P, Colasante G, Bianchi M, Long J, Mansouri A, Rubenstein JLR, Broccoli V. Inactivation of Arx, the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene, leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. J Neurosci. 2007;27:4786–4798. doi: 10.1523/JNEUROSCI.0417-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Colasante G, Collombat P, Raimondi V, Bonanomi D, Ferrai C, Maira M, Yoshikawa K, Mansouri A, Valtorta F, Rubenstein JLR, Broccoli V. Arx is a direct target of Dlx2 and thereby contributes to the tangential migration of GABAergic interneurons. J Neurosci. 2008;28:10674–10686. doi: 10.1523/JNEUROSCI.1283-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Li H, Radford JC, Ragusa MJ, Shea KL, McKercher SR, Zaremba JD, Soussou W, Nie Z, Kang Y-J, Nakanishi N, Okamoto S-ichi, Roberts AJ, Schwarz JJ, Lipton SA. Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proc Natl Acad Sci USA. 2008;105:9397–9402. doi: 10.1073/pnas.0802876105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ahlgren S, Vogt P, Bronner-Fraser M. Excess FoxG1 causes overgrowth of the neural tube. J Neurobiol. 2003;57:337–349. doi: 10.1002/neu.10287. [DOI] [PubMed] [Google Scholar]
- 185.Nacher J, Crespo C, McEwen BS. Doublecortin expression in the adult rat telencephalon. Eur J Neurosci. 2001;14:629–644. doi: 10.1046/j.0953-816x.2001.01683.x. [DOI] [PubMed] [Google Scholar]
- 186.Friocourt G, Liu JS, Antypa M, Rakic S, Walsh CA, Parnavelas JG. Both doublecortin and doublecortin-like kinase play a role in cortical interneuron migration. J Neurosci. 2007;27:3875–3883. doi: 10.1523/JNEUROSCI.4530-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Grabham PW, Seale GE, Bennecib M, Goldberg DJ, Vallee RB. Cytoplasmic dynein and LIS1 are required for microtubule advance during growth cone remodeling and fast axonal outgrowth. J Neurosci. 2007;27:5823–5834. doi: 10.1523/JNEUROSCI.1135-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fallet-Bianco C, Loeuillet L, Poirier K, Loget P, Chapon F, Pasquier L, Saillour Y, Beldjord C, Chelly J, Francis F. Neuropathological phenotype of a distinct form of lissencephaly associated with mutations in TUBA1A. Brain. 2008;131:2304–2320. doi: 10.1093/brain/awn155. [DOI] [PubMed] [Google Scholar]
- 189.Greenwood JSF, Wang Y, Estrada RC, Ackerman L, Ohara PT, Baraban SC. Seizures, enhanced excitation, and increased vesicle number in Lis1 mutant mice. Ann Neurol. 2009;66:644–653. doi: 10.1002/ana.21775. [DOI] [PubMed] [Google Scholar]
- 190.Onda H, Crino PB, Zhang H, Murphey RD, Rastelli L, Gould Rothberg BE, Kwiatkowski DJ. Tsc2 null murine neuroepithelial cells are a model for human tuber giant cells, and show activation of an mTOR pathway. Mol Cell Neurosci. 2002;21:561–574. doi: 10.1006/mcne.2002.1184. [DOI] [PubMed] [Google Scholar]
- 191.Knox S, Ge H, Dimitroff BD, Ren Y, Howe KA, Arsham AM, Easterday MC, Neufeld TP, O’Connor MB, Selleck SB. Mechanisms of TSC-mediated control of synapse assembly and axon guidance. PLoS ONE. 2007;2:e375. doi: 10.1371/journal.pone.0000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Takayanagi M, Kure S, Sakata Y, Kurihara Y, Ohya Y, Kajita M, Tada K, Matsubara Y, Narisawa K. Human glycine decarboxylase gene (GLDC) and its highly conserved processed pseudogene (psiGLDC): their structure and expression, and the identification of a large deletion in a family with nonketotic hyperglycinemia. Hum Genet. 2000;106:298–305. doi: 10.1007/s004390051041. [DOI] [PubMed] [Google Scholar]
- 193.Park J-W, Park E-S, Choi EN, Park H-Y, Jung S-C. Altered brain gene expression profiles associated with the pathogenesis of phenylketonuria in a mouse model. Clin Chim Acta. 2009;401:90–99. doi: 10.1016/j.cca.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 194.Niciu MJ, Ma X-M, El Meskini R, Pachter JS, Mains RE, Eipper BA. Altered ATP7A expression and other compensatory responses in a murine model of Menkes disease. Neurobiol Dis. 2007;27:278–291. doi: 10.1016/j.nbd.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhou M, Tanaka O, Suzuki M, Sekiguchi M, Takata K, Kawahara K, Abe H. Localization of pore-forming subunit of the ATP-sensitive K(+)-channel, Kir6.2, in rat brain neurons and glial cells. Brain Res Mol Brain Res. 2002;101:23–32. doi: 10.1016/s0169-328x(02)00137-7. [DOI] [PubMed] [Google Scholar]
- 196.El Meskini R, Crabtree KL, Cline LB, Mains RE, Eipper BA, Ronnett GV. ATP7A (Menkes protein) functions in axonal targeting and synaptogenesis. Mol Cell Neurosci. 2007;34:409–421. doi: 10.1016/j.mcn.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gulyás-Kovács A, de Wit H, Milosevic I, Kochubey O, Toonen R, Klingauf J, Verhage M, Sørensen JB. Munc18-1: sequential interactions with the fusion machinery stimulate vesicle docking and priming. J Neurosci. 2007;27:8676–8686. doi: 10.1523/JNEUROSCI.0658-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sumita K, Sato Y, Iida J, Kawata A, Hamano M, Hirabayashi S, Ohno K, Peles E, Hata Y. Synaptic scaffolding molecule (S-SCAM) membrane-associated guanylate kinase with inverted organization (MAGI)-2 is associated with cell adhesion molecules at inhibitory synapses in rat hippocampal neurons. J Neurochem. 2007;100:154–166. doi: 10.1111/j.1471-4159.2006.04170.x. [DOI] [PubMed] [Google Scholar]