

Abstract
Within the hierarchy of epithelial stem cells, normal progenitor cells may express regulated telomerase during renewal cycles of proliferation and differentiation. Dis-continuous telomerase activity may promote increased renewal capacity of progenitor cells, while deregulated/ continuous telomerase activity may promote immortalization when differentiation and/or senescent pathways are compromised. In the present work, we show that resveratrol activates, while progesterone inactivates, continuous telomerase activity within 24 h in subpopulations of human Li–Fraumeni syndrome-derived breast epithelial cells. Resveratrol results in immortalization of mixed progenitor cells with mutant p53, but not human epithelial cells with wild type p53. Our results demonstrate the potential for renewing progenitor cells with mutant p53 to immortalize after continuous telomerase expression when exposed to certain environmental compounds. Understanding the effects of telomerase modulators on endogenous telomerase activity in progenitor cells is relevant to the role of immortalization in the initiation and progression of cancer subtypes.
Keywords: telomerase, p53, progesterone, breast epithelial, aging
Introduction
Cell immortalization occurs when cells re-activate or upregulate the ribonucleoprotein enzyme, telomerase, through the catalytic subunit hTERT (Bayne and Liu, 2005; Dong et al., 2005). Telomerase facilitates cellular immortalization by maintaining telomere length and allows for proliferation (Masutomi et al., 2003; Dong et al., 2005; Shay and Wright, 2005). Immortalization events are rare and usually blocked by replicative senescence (continued cell viability without cell division), but immortalization can occur in vivo as part of carcinogenesis (Holt and Shay, 1999; Sun et al., 1999; Masutomi et al., 2003; Bayne and Liu, 2005; Dimri et al., 2005; Dong et al., 2005; Shay and Wright, 2005). Normal stem and/or stem-like cells with self-renewing capacity are usually telomerase competent and can express regulated telomerase in a cell cycle-dependent fashion during proliferation and differentiation, without immortalizing (Tanaka et al., 1998; Holt and Shay, 1999; Williams et al., 2001; Yaswen and Stampfer, 2002). As these cells self-renew, they maintain their mortality by progressive telomere shortening with increased age (Bayne and Liu, 2005; Shay and Wright, 2005).
Studies of breast tissue homeostasis and cell-type diversification indicate that the breast remains very dynamic, undergoing morphological and functional changes in response to circulating hormones (Sun et al., 1999; Fridriksdottir et al., 2005). Mature breast epithelium is divided into two main cell lineages, luminal epithelial and myoepithelial cells, that are distinguished by surface markers and cytoskeletal proteins (Sun et al., 1999). Rodent and cultured cell studies document an elaborate hierarchy of cell populations (progenitors) within the breast (Sun et al., 1999). Further, cultured cell studies indicate bi-potential (mixed colonies of luminal and myoepithelial) and/or heterogeneous progenitors based on expression of specific markers (Sun et al., 1999: Masutomi et al., 2003). Some of these progenitors may be steroid receptor-positive (Sun et al., 1999; Conneely et al., 2003). During renewal, breast stem and/or progenitors may express telomerase subject to hormonal-dependent cell cycle regulation (Tanaka et al., 1998; Williams et al., 2001; Stingl et al., 2005). The hTERT promoter contains various consensus sequences available for hormone receptor-mediated activation, and is both a direct and indirect target of estrogen and progesterone receptor-mediated activity (Bayne and Liu, 2005). As a direct estrogen receptor (ER) target gene, hTERT may be modulated by estrogens/hormone therapy, estrogen mimics through environmental sources, and/or estrogen modulators/cancer therapy (Bayne and Liu, 2005). However, normal cellular differentiation signaling pathways should facilitate transcriptional downregulation of telomerase activity, thus preventing immortalization/proliferation of self-renewing cells upon inappropriate exposure to hTERT modulators (Holt and Shay, 1999; Bayne and Liu, 2005; Shay and Wright, 2005).
In tumorigenesis, luminal breast epithelial cells proliferate possibly due to early loss of p53 activity and subsequent upregulation of telomerase (Kim et al., 2002; Yasmen and Stampfer, 2002; Masutomi and Hahn, 2003). If differentiation and/or senescence signaling pathways are compromised due to loss of p53 and/or p16ink4a, then hTERT modulators could mediate un-regulated telomerase activity and promote immortalization in progenitors (Holt and Shay, 1999; Hahn, 2004). We examined whether certain progenitors found in breast tissue that are hTERT competent and exhibit regulated telomerase activity are at risk upon exposure to hTERT/telomerase modulators for immortalization, and tumorigenesis (Petersen et al., 2003; Bayne and Liu, 2005).
Results
Resveratrol activates telomerase in a fraction of HME50 cells, but not through the ER
We used Li–Fraumeni syndrome-derived breast epithelial cell strains that contain a p53 germline mutation at codon 133 (HME50) to analyse the ability of epithelial lineage cells that are telomerase competent to become telomerase positive through possible hTERT activators (Shay et al., 1995). HME50 cells contain epithelial progenitor cells that can gain unlimited proliferative capacity (spontaneously immortalize) in vitro at a frequency of about 1 × 10−6, only after crisis (state of widespread apoptosis due to short unprotected telomeres) (Shay et al., 1995). We screened HME50 cells and/or derived clones for hormone regulation of telomerase activity using the telomeric repeat amplification protocol (TRAP) assay after treatment with various molar concentrations of 17-β estradiol (E2) and various E2 modulators, including resveratrol, to determine if an ER modulator induced hTERT expression (Supplementary Figure 1; Shay et al., 1995). We discovered that 10−8 m resveratrol (trans-3, 4′, 5-trihydroxystilbene; polyphenol in grapes and red wine), but not E2 activated telomerase within 24 h in a fraction of cells (~10% relative to control) (Figure 1a; Supplementary Figure 2) (Signorelli and Ghidoni, 2005). Further, we show that telomerase activation was not dependent on ongoing resveratrol treatments since these cells continued to express the same level of telomerase activity after a month in culture without further resveratrol treatments (Figure 1b). We confirmed by limiting dilution and ring cloning that only a fraction of HME50 cells expressed detectable telomerase activity both during and post-10−8 m resveratrol treatment (Figure 1c). None of the clones isolated from untreated HME50 cells or untreated controls expressed detectable telomerase activity (Figures 1a–c).
Figure 1.

Resveratrol mediates telomerase activity, but not through the ER (estrogen receptor). (a) 17β-estradiol does not activate telomerase in HME50 cells (population doubling (PD) 39.72) after 16 days in culture with either no treatment (control) or treatment with 17β-estradiol (E2) every 3 days; and/or 4-hydroxytamoxifen (4HT) every 3 days for 16 days in culture did not effect 10−8 m resveratrol activation of telomerase. (b) TRAP (telomerase) assay shows telomerase activity after 48, 72 and 96 h treatment with 10−8 m resveratrol (Rv) in HME50 cells, and after cells were split into two T25 flask per time point, then cultured for 30 days with or without continued treatment. HME50 cells were plated at 100 000 cells per well, treated with or without Rv every 3 days, then analysed. (c) 10% of clones exhibit telomerase activity after HME50 cells (PD 12.5) were plated at 3000 cells per 15 cm dish, then treated with 10−8 m concentrations of reveratrol and selected by ring cloning.
Since studies show resveratrol-mediated transcriptional activation of both ER-α and -β in vitro, we assayed for ligand-activated ER-mediated transcriptional activity of hTERT in HME50 cells (Signorelli and Ghidoni, 2005). HME50 cells were screened with varying molar concentrations of the natural ligand E2 and E2 antagonist 4-hydroxytamoxifen (4HT). We found no E2-induced telomerase activity in the HME50 cells, other than the resveratrol-induced telomerase activity (Figure 1a). The ER antagonist 4HT blocks E2 and resveratrol direct activation of ER in the breast, and more specifically E2-induced hTERT activation in breast cancer cells (Bayne and Liu, 2005; Dong et al., 2005). However, 4HT did not block resveratrol-mediated telomerase activity (Figure 1a). Immunohistochemistry, immunofluorescence staining, western analysis and reverse transcriptase–PCR indicate that HME50 cells have lost ER expression (data not shown). Together, these data show that resveratrol mediates telomerase activity, but not directly through ER-mediated transcriptional activity.
Resveratrol activation of telomerase is blocked by progesterone through the progesterone receptor
Progesterone (Pg) acting through the progesterone receptor (PR) is a hormonal regulator involved in stem cell growth and differentiation, and can downregulate telomerase activity (Wang et al., 2000; Dong et al., 2005). Similarly, we observed that 10−8 m Pg acting through the PR-expressing HME50 cells inhibited the ability of resveratrol to induce telomerase activity during and post-resveratrol treatment (Figures 2a and b). The inhibitory effects of Pg were blocked by 10−6 m antiprogestin, CP-8754 indicating that Pg was acting directly through PR to inhibit resveratrol-mediated telomerase activity in HME50 cells (Figure 2b) (Tabata et al., 2002). HME50 cells remained sensitive to progesterone inhibition during and post-resveratrol treatments for over 11 days, also indicating that the cells expressing telomerase activity were still sensitive to differentiation signaling (Figure 2c).
Figure 2.

Progesterone inhibits resveratrol-mediated telomerase activity in HME50 cell lines through a PR (progesterone receptor). (a) Western blot shows HME50 cells are PR positive. (b) After 48 h treatment with 10−8 m progesterone (Pg) in combination with 10−8 m resveratrol (Rv), HME50-8 cells show no telomerase activity as compared with resveratrol-treated cells; and progesterone antagonist CP-8754 blocks progesterone inhibition of 10−8 m resveratrol and telomerase activity (10−6antagonist with 10−8 m progesterone with 10−8 m resveratrol (A/Pg/Rv), and 10−8 m progesterone with 10−6 m antagonist (Pg/A)). (c) Progesterone continues to block telomerase activity in HME50 cells (PD10.6) even after 11 days of Rv treatment. 1 × 106 cells were each plated in two T75 s, then one T75 received 10−8 m Rv while another did not (control). After 11 days, each flask was split into two T25 flasks; one control T25 continued to receive no treatment while the other 10−8 m Pg. In the resveratrol-treated group, one T25 continued to receive 10−8 m Rv, while the other received 10−8 m Pg/10−8 m Rv).
Resveratrol activates telomerase in cells with spontaneous immortalization potential
Using previously characterized HME50-derived clone lines (HME50-3, -5, -6, -8 and -9), we determined that cells with spontaneous immortalization potential were targets of resveratrol-mediated telomerase activity (Shay et al., 1995). Clonally derived strains HME50-5, -8 and -9, similar to parental HME50 cells, can spontaneously immortalize after crisis at a very low frequency (reflective of the very rare event), while HME50-3 and -6 clonal strains do not spontaneously immortalize (Shay et al., 1995). These clonally-derived lines were initially isolated from the parental line to maintain the p53 germline mutation, and both wild-type and mutant p53 conformations were present in parental HME50 cells up to 22 population doublings (PD) (Shay et al., 1995). We observed that 10−8 m resveratrol-activated telomerase in HME50-5, -8 and -9 clonal cells at PD greater than 22 (Figure 3). We also found that Pg-inhibited resveratrol-mediated telomerase activity in PR-positive clones, similar to parental HME50 cells (Figures 2a–c). HME50-3 and -6 clonal cell strains that did not spontaneously immortalize were not affected by resveratrol (Figure 3).
Figure 3.
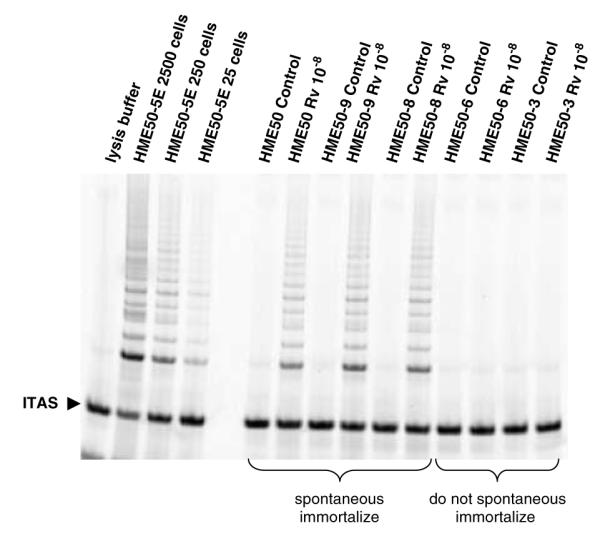
Continuous resveratrol (Rv)-mediated telomerase activity in cell clones either permissive or non-permissive for spontaneous immortalization. HME50-derived clonal populations HME50-9 and -8 show telomerase activity by TRAP analysis after 48 h when treated with resveratrol (Rv 10−8 M), while HME50-3 and -6 clone that never spontaneously immortalize do not have Rv-mediated telomerase activity.
HME50 cells with resveratrol-mediated telomerase activity immortalized
We examined whether the cells that expressed resveratrol-mediated telomerase activity would immortalize by serially passaging each cell line, during- and post-resveratrol treatments along with the untreated or treated cells that did not respond to resveratrol. We found that 250/2500 cells sampled in HME50, -5, -8 and -9, relative to control, continued to express telomerase activity for at least 30 days in culture after resveratrol treatment, and independent of the age of cells as determined by PD (results not shown; based on greater than five experiments at differing PDs). After 30 days, resveratrol-treated HME50 and HME50-5E cells (Figures 1a and b) appeared to undergo a growth arrest or crisis phase, then proliferated in multiple colonies per flask and were telomerase-positive. In all experiments, untreated cells entered crisis and cell death occurred as represented by untreated HME50-8 and HME50-9 cells, which gradually ceased proliferating after ~39PDs (Figure 4a). However, resveratrol-treated cells (HME50-8R and HME50-9R) continued to proliferate beyond their normal senescent checkpoint and are now at over 100PDs (Figure 4a). HME50-9R cells appeared to go through a proliferative arrest before multiple telomerase-positive colonies emerged, however HME50-8R cells did not (Figure 4a). Interestingly, until the subpopulation of telomerase-positive cells exceeded ~15% (at least 30 days), they were responsive to progesterone blockage of telomerase activity (unpublished data).
Figure 4.

(a) Growth curves show proliferation of 10−8 m resveratrol (Rv)-treated cells (HME50-8R; solid triangles and HME50-9R; solid squares) versus untreated cells (HME50-8C; empty triangles and HME50-9C; empty squares). (b) HME50-8R and HME50-9R cells show a 200-fold excess in hTERT mRNA levels over a spontaneous immortalized epithelial cell line (HME50-5E). GAPDH was used as the loading control and fold difference is relative to control. Water is the negative control. (c) Telomere restriction fragments for treated HME50-8R cells (left panel) and treated HME50-9R cells (right panel) show that telomeres are elongated and maintained in the HME50-8R cells expressing hTERT as analysed by Southern blot analysis at increasing PDs. In HME50-9R the telomeres gradually shorten but cell proliferation continued. C = control cells R = resveratrol-treated cells. Size markers are in kilobases (kb), and all cells were proliferating at each PD.
Since loss or mutation in p53 contributes to immortalization potential, we measured TRAP activity after resveratrol treatment in other characterized telomerase-competent HME cells without a p53 mutation (HME30 cells) (Campisi, 2005; Dong et al., 2005). HME30 cells with wild-type p53 did not respond to resveratrol or show any telomerase activity (Supplementary Figure 3).
Next, we determined that resveratrol-mediated telomerase activity corresponded with increased hTERT transcription by comparing mRNA in mortal versus immortal cell lines. While no hTERT mRNA was detected in mortal HME50-5 cells, both immortal HME50-8R and HME50-9R cells expressed hTERT mRNA levels that was higher than normal as represented by HME50-5E (Figure 4b).
Based on TRF-length analysis during progressive cell-passaging, telomeres were being maintained in the resveratrol-treated HME50-8R cells (Masutomi et al., 2003; Shay and Wright, 2005). In HME50-8R cells there appears to be some very short telomeres and some that are elongated (Figure 4c, left panel). This suggests that some cells may be dropping out of the population with critically short telomeres while others were being maintained. In HME50-9R cells, while telomeres appeared to elongate between PD38 and 47 by PD 62 the telomeres were shortening (Figure 4c). However, with continued culture, HME50-9R populations with shorter overall telomere length (~4.3 kb) continued to proliferate (Figure 4c, right panel).
Immortalized HME50-8R and HME50-9R cells are mixed progenitors
To determine the stem and/or progenitor cell type in the resveratrol-immortalized cells, we compared lineage-specific surface antigens in all immortal and mortal HME50 cell lines (Trask et al., 1990; Petersen et al., 2003; Bankfalvi et al., 2004; Fridriksdottir et al., 2005). Using cytokeratin (CK)-specific monoclonal antibodies, we analysed by immunofluorescence staining HME50-8 and -8R cells with CK-19 (basal stem cell marker, also found in breast cancer), CK-18 (luminal cell marker, also found in breast cancer), CK-14 (myoepithelial cell marker, not found in breast cancer) and CK5/CK8 (progenitor cell markers associated with CK14 and CK18, respectively) (Figure 5a) (Bankfalvi et al., 2004; Stingl et al., 2005). We observed that immortal HME50-8R cells were a mixed progenitor hierarchy with 60% luminal (CK8/18+), 20% myoepithelial (CK5/14+) and some suprabasal cells (CK5/CK14+ and/or CK19/CK5/CK14+) (Figure 5b) (Fridriksdottir et al., 2005; Stingl et al., 2005). Within these mixed progenitors, we found that HME50-8R luminal epithelial lineage progenitors doubled their population size, while myoepithelial progenitors maintained the same population size, when compared to mortal HME50-8 cells (Figure 5b). Correspondingly, we show increased CK18 protein expression in these cells and HME50-9R cells relative to mortal cells through Western analysis (Figure 5c). Immortalized cells with basal marker CK19+ decreased ~30%, indicating possible loss of suprabasal cells (Figures 5a and b) (Bankfalvi et al., 2004; Stingl et al., 2005). Since stem cell expansion is based on symmetrical division, in that one cells gives rise to two, the immortal luminal progenitors appear to have divided symmetrically (Bankfalvi et al., 2004; Stingl et al., 2005). The immortal myoepithelial progenitors appear to have divided asymmetrically, based on a 1:1 cell division ratio (Figure 5a) (Bankfalvi et al., 2004; Stingl et al., 2005). Similar to other epithelial proliferation studies, our data shows that cell division predominately occurred among luminal cells in the resveratrol-immortalized cells (Stingl et al., 2005).
Figure 5.

Mixed progenitors are the target of resveratrol-mediated telomerase activity in HME50 cells. (a) Representative immunofluorescence images show CK18 in the untreated mortal (HME50-8C) versus resveratrol-treated immortal (HME50-8R) cells using mouse monoclonal antibodies against cytokeratins (CK) CK18, CK14, or CK19 and CK5/8 (red), as indicated merged with the DNA (nuclear) stain DAPI. (b) Using the immunofluorescence images, the number of cells expressing cytokeratin over total nuclei were counted and the percentage of cells expressing CK19 (CK19+), 18 (CK18+), or 14 (CK14+) and 5/8 (CK5/8+) show that progenitor subtypes immortalized, with luminal progenitors cell numbers increasing (c) western analysis also shows increased expression of CK18 in HME50-8R, and HME50-9R that coincides with the increase in percentages of CK18 staining cells. HME50, HME50-6C and HME50-6R, that did express telomerase and did not immortalize, did not show the increase in CK18 expression.
Discussion
Oncogenic events, such as the loss of p53, or the inappropriate activation of hTERT, may target epithelial stem/progenitor cells and give rise to immortalized cells with unique characteristics including dysregulated proliferation rates (Sun et al., 1999; Masutomi et al., 2003; Bankfalvi et al., 2004; Dimri et al., 2005; Dong et al., 2005; Signorelli and Ghidoni, 2005). In the present study, we found that resveratrol, at physiologically relevant levels, activated telomerase in p53 heterozygous mammary epithelial-mixed progenitors, resulting in up-regulation of hTERT and immortalization, a critical if not rate-limiting steps in cancer progression (Holt and Shay, 1999; Sun et al., 1999; Masutomi et al., 2003; Bankfalvi et al., 2004; Dimri et al., 2005; Dong et al., 2005; Signorelli and Ghidoni, 2005).
One possibility to explain these findings is that telomerase might be hormone-activated through direct ER-mediated activation of hTERT, but we found no evidence for resveratrol acting as a phytoestrogen/ER to activate telomerase at the effective concentrations (Bayne and Liu, 2005; Signorelli and Ghidoni, 2005). Further, we found that HME50 cells were ER-negative (data not shown). However, we did find hormone deactivation of telomerase activity through Pg/PR in PR-positive HME50 cell lines (Figure 2) (Wang et al., 2000; Bayne and Liu, 2005). Pg can induce differentiation, and inhibit resveratrol-mediated telomerase activity (Wang et al., 2000; Lebeau et al., 2002; Leonhardt and Edwards, 2002; Li and O’Malley, 2003). Pg can also increase apoptosis, and thereby induce cell death in resveratrol-treated cells, but our results did not show any cell loss with progesterone treatments (Conneely et al., 2003; Bayne and Liu, 2005). Quite the contrary, cells appeared to thrive during progesterone treatments. Pg/PR blockage of resveratrol may be mediated by PR isoforms (Lim et al., 1999; Mote et al., 2002; Leslie et al., 2005; Signorelli and Ghidoni, 2005). We found that resveratrol-immortalized cells express a predominance of PRA over B (characteristic of proliferative growth, and also found in breast cancers), while mortal HME50 parental cells express equal amounts of both isoforms (Figure 2a; unpublished data) (Lim et al., 1999; Mote et al., 2002; Leslie et al., 2005). These PR+/ER− (inactive ER) resveratrol-immortalized progenitors proliferate, despite a proposed linear differentiation pathway for mammary epithelial cells wherein PR+/ER− stem cells may become PR+/ER− and differentiate or PR+/ER− and remain quiescent (Counter et al., 1998).
Resveratrol is considered an important nutrient implicated in prosurvival pathway induction (Le Corre et al., 2005; Signorelli and Ghidoni, 2005). While most resveratrol-mediated effects such as increasing SIRT1 activity, are reported at high micromolar concentrations in transformed cells (Lanzilli et al., 2006) or animal models, a few studies report resveratrol effects at nanomolar and lower concentrations including inhibition of platelet aggregation, neuroprotection through ERK1 and 2 phosphorylation and quinone reductase 2 binding (Miloso et al., 1999; Bhat et al., 2001; Gusman et al., 2001; Bhat and Pezzuto, 2002; Dong, 2003; Signorelli and Ghidoni, 2005). Our data support the pleiotropic effects of resveratrol by showing nanomolar concentrations of resveratrol initiate prosurvival effects by upregulating or reactivating telomerase in progenitor cells (Miloso et al., 1999; Bhat et al., 2001; Gusman et al., 2001; Bhat and Pezzuto, 2002; Dong, 2003; Signorelli and Ghidoni, 2005).
Micromolar concentrations of resveratrol in certain cellular models may upregulate p53 by increasing cellular content and inducing post-translational modifications resulting in senescence and apoptosis (Campisi, 2005; Signorelli and Ghidoni, 2005). Inactivation of p53 is sufficient to disable both senescence and apoptosis (Zhivotovsky and Kroemer, 2004). Germ line or acquired p53 mutations such as found in Li– Fraumini syndrome and/or altered cellular localization of p53 protein are mechanisms of inhibiting p53 function (Moll et al., 1996; Ostermeyer et al., 1996; Saeki et al., 1997; Nikolaev et al., 2003). We found that mutant p53, but not wild-type p53, is upregulated and sequestered to the nucleus in resveratrol-immortalized HME50-8R and HME50-9R cells which could facilitate an inactive p53 (Supplementary Figure 4). Acetylation and/or phosphorylation of mutant p53 could result in nuclear accumulation of mutant p53 in the HME50 progenitor cells, and thus inactivate wild type p53 (Moll et al., 1996; Ostermeyer et al., 1996; Saeki et al., 1997; Nikolaev et al., 2003) (Supplementary Figure 3). Others found that loss of p53 function results in rapidly expressed telomerase activity and full immortality in HME cells (Stampfer et al., 2003). Similarly, resveratrol facilitated inactivation of p53 in the subpopulation of resveratrol-treated HME50 cells resulting in telomerase activity and cellular immortality (Rambhatla et al., 2001).
Studies suggest that resveratrol has different effects based on cell type, conditions and concentration (Signorelli and Ghidoni, 2005; Lanzilli et al., 2006). Higher concentrations are usually more effective on highly proliferative cancer cells (Le Corre et al., 2005; Signorelli and Ghidoni, 2005). We observed that low concentrations of resveratrol activated telomerase in slow growing mammary epithelial progenitors, as characterized by cytokeratin markers, but only p53+/− HME50 cells immortalized. We did observe that both luminal and myoepithelial (with possible basal cells) lineage progenitors are telomerase competent and capable of immortalization under conditions of mutant/inactive p53 (Stingl et al., 2005). The observations that only low concentrations of resveratrol produced these effects in these cells, though surprising, were highly reproducible. Dose–response effects of resveratrol were repeated many times, with several experiments maintained long-term. Further, control cells from one experiment were replated for a second series of experiments into two differing flasks, one flask was maintained as the control cells, while the other flask was treated with resveratrol and/or estradiol. Only the resveratrol-treated cells exhibited telomerase activity (within 24 h) and continued expressing telomerase activity. This was repeated many times and each time we used the control cells from the previous experiment. In all experiments, only the resveratrol-treated cells exhibited telomerase activity. None of the control cells without treatment ever exhibited telomerase activity. In all experiments, when the control cells stopped proliferating, the cells treated with resveratrol continued to grow under similar culture conditions.
In summary, our results indicate that certain compounds such as resveratrol and progesterone can mediate telomerase activity in self-renewing human cells. Further, continued telomerase activity in luminal and/or myoepithelial breast progenitor cells with inactive p53 may facilitate immortalization (Dong, 2003; Masutomi and Hahn, 2003). Mixed progenitors that provide normal epithelial proliferation are prime targets for immortalization and may be precursors of different subtypes of cancer (Dong, 2003; Bayne and Liu, 2005; Dimri et al., 2005). These resveratrol-immortalized mixed progenitors may provide models for understanding how different epithelial subtypes immortalize, and cancers originate.
Materials and methods
Cell and cell culture
Human mammary epithelial cells were derived from the noncancerous breast tissue of a 31-year-old female diagnosed with Li–Fraumini syndrome, and contain a germline mutation (Met133Thr) in the p53 gene that influences confirmation of the wild-type p53 protein (HME50) (Shay et al., 1995). HME50-3, -5, -6, -8 and -9 were derived from clones of HME50 parental cells (Shay et al., 1995) (cell strains) and grown in MCDB 170 media (Life Technologies Inc., (GIBCO BBRLL), Rockville, MD, USA) supplemented with 0.4% bovine pituitary extract (Hammond Cell Technology, Windsor, CA, USA), 10 ng ml−1 epidermal growth factor, 5 μg ml−1 insulin, 0.5 μg ml−1 hydrocortisone and 5 μg ml−1 transferin (Sigma Chemical Co., St Louis, MO, USA) (SF-170 media). Medium was changed every 2–3 days.
TRAP (telomerase) and TRF (telomere length) assays
Cells were treated with compounds, then 105 cells were harvested for telomerase activity (visualized by 6 base pair (bp) ladder) using the cell-based TRAP (telomere repeat amplification protocol) assay as described previously (Kim et al., 1994). Spontaneous immortalized HME50-5E or lung cancer H1299 cells were used as TRAP-positive controls, while lysis buffer/ ITAS served as a negative control.
Mean telomere length was measured by telomere restriction fragment (TRF) analysis (Southern Blot variation). Isolated DNAs from different cell preparations were digested with restriction enzymes mixtures and resolved on a 0.7% agarose gel. The dried gel was hybridized with a 32P-labeled oligonucleotide, (TTAGGG)3 and exposed to a Phosphorimager screen.
RT–PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA, USA). cDNAs were synthesized with Super-Script III (Invitrogen, Carlsbad, CA, USA), and hTERT levels were measured on the LightCycler (Roche, Basel, Switzerland) using a LightCycler FastStart DNA Master STBR Green I kit (Roche). LT5 and LT6 primers were used to detect hTERT transcript levels. Levels were standardized against GAPDH cDNA to calculate the relative expression ratios (Yu et al., 2001).
Western analysis and immunofluorescence
Cell lysates were collected, spun down and lysed in TRAP buffer (Kim et al., 1994). For use in western analysis, equal parts loading buffer were added to TRAP cell lysates. Proteins were separated on 10 and 8% sodium dodecyl sulfate– polyacrylamide gel electrophoresis gels, transferred to PVDF membranes (0.45 μm; Millipore, Billerica, MA, USA), and blocked in phosphate-buffered saline+ 0.05% Tween 20 (v/v) (PBS-T) and 5% (w/v) powdered non-fat milk for 1 h at room temperature (RT). Membranes were incubated with specific antibodies in PBS-T for 1–2 h or overnight at 4 1C, washed with PBS-T, then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Pierce; Amersham Biosciences Corp., Piscataway, NJ, USA) for 1.5 or 2 h at RT. Detection of peroxidase activity was by enhanced chemiluminescence (ECL Plus; Amersham Pharmacia, Piscataway, NJ, USA) followed by autoradiography. Epithelial cells were fixed for immunofluorescence in glass chamber slides (Lab-Tek, VWR International, West Chester, PA, USA) in methanol for 10–15 min; washed 3 × for 5 min each and stained with antibodies for 1 h or overnight. Secondary antibodies used for detection were Alexa 488-conjugated goat anti-mouse IgG and goat anti-rabbit IgG (Molecular Probes, Carlsbad, CA, USA), and FITC-conjugated donkey anti-goat IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Antibodies included: mouse anti-cytokeratins 19 (sc-6278), 18 (sc-6259), 14 (sc-17104), 5/8 (sc-8021) and rabbit polyclonal anti-PR (sc-538 and sc-539); mouse anti-SIRT1, (Upstate, Lake Placid, NY, USA); and mouse monoclonal anti-p53 (AB-5) and -p53 (Ab-6) (Oncogene, Calbiochem International, San Diego, CA, USA).
Supplementary Material
Acknowledgements
We acknowledge Jackie Swanik for editorial comments and William Walker for technical assistance. This work was supported by NCI-NO1-CN43301, NCI-CN-05106, Lung Cancer SPORE P50 CA75907, NSCOR NNJ05HD36G and the Ted Nash Foundation.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- Bankfalvi A, Ludwig A, de-Hesselle B, Buerger H, Buchwalow IB, Boecker W. Different proliferative activity of the glandular and myoepithelial lineages in benign and proliferative and early malignant breast diseases. Modern Path. 2004;17:1051–1061. doi: 10.1038/modpathol.3800082. [DOI] [PubMed] [Google Scholar]
- Bayne S, Liu JP. Hormones and growth factors regulate telomerase activity in ageing and cancer. Mol Cell Endocrinol. 2005;240:11–22. doi: 10.1016/j.mce.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Bhat KP, Kosmeder JW, II, Pezzuto JM. Biological effects of resveratrol. Antioxid Redox Signa. 2001;3:1041–1064. doi: 10.1089/152308601317203567. [DOI] [PubMed] [Google Scholar]
- Bhat KPL, Pezzuto JM. Cancer chemopreventive activity of resveratrol. Ann NY Acad Sci. 2002;957:210–229. doi: 10.1111/j.1749-6632.2002.tb02918.x. [DOI] [PubMed] [Google Scholar]
- Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Conneely OM, Jericevic BM, Lydon JP. Progesterone receptors in mammary gland development and tumorigenesis. J Mamm Gland Biol Neopl. 2003;8:205–214. doi: 10.1023/a:1025952924864. [DOI] [PubMed] [Google Scholar]
- Counter CM, Hahn WC, Wei W, Caddle SD, Beijersbergen RL, Lansdorp PM, et al. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci USA. 1998;95:14723–14728. doi: 10.1073/pnas.95.25.14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri G, Band H, Band V. Mammary epithelial cell transformation: insights from cell culture and mouse models. Breast Cancer Res. 2005;7:171–179. doi: 10.1186/bcr1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong CK, Masutomi K, Hahn WC. Telomerase: regulation, function and transformation. Crit Rev Oncol Hematol. 2005;54:85–93. doi: 10.1016/j.critrevonc.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Dong Z. Molecular mechanism of the chemopreventive effect of resveratrol. Mutat Res. 2003;523–524:145–150. doi: 10.1016/s0027-5107(02)00330-5. [DOI] [PubMed] [Google Scholar]
- Fridriksdottir AJ, Villadsen R, Gudjonsson T, Petersen OW. Maintenance of cell type diversification in the human breast. J Mammary Gland Biol Neoplasia. 2005;10:61–74. doi: 10.1007/s10911-005-2541-6. [DOI] [PubMed] [Google Scholar]
- Gusman J, Malonne H, Atassi G. A reappraisal of the potential chemopreventive and chemotherapeutic properties of resveratrol. Carcinogenesis. 2001;22:1111–1117. doi: 10.1093/carcin/22.8.1111. [DOI] [PubMed] [Google Scholar]
- Hahn WC. Cancer: surviving on the edge. Cancer Cell. 2004;6:215–222. doi: 10.1016/j.ccr.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Holt SE, Shay JW. Role of telomerase in cellular proliferation and cancer. J Cell Physiol. 1999;180:10–18. doi: 10.1002/(SICI)1097-4652(199907)180:1<10::AID-JCP2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Kim H, Farris J, Christman SA, Kong BW, Foster LK, O’Grady SM, et al. Events in the immortalizing process of primary human mammary epithelial cells by the catalytic subunit of human telomerase. Biochem J. 2002;365(Part 3):765–772. doi: 10.1042/BJ20011848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- Lanzilli G, Fuggetta MP, Tricarico M, Cottarelli A, Serafino A, Falchetti R, et al. Resveratrol down-regulates the growth and telomerase activity of breast cancer cells in vitro. G Int J Oncol. 2006;28:641–648. [PubMed] [Google Scholar]
- Le Corre L, Chalabi N, Delort L, Bignon YJ, Bernard-Gallon DJ. Resveratrol and breast cancer chemoprevention: molecular mechanisms. Mol Nutr Food Res. 2005;49:462–471. doi: 10.1002/mnfr.200400094. [DOI] [PubMed] [Google Scholar]
- Lebeau J, Fouchet P, Ory K, Chevillard S. Down-regulation of telomerase activity after progesterone treatment of human breast cancer cells: essential role of cell cycle status. Anticancer Res. 2002;22:2161–2166. [PubMed] [Google Scholar]
- Leonhardt SA, Edwards DP. Mechanism of action of progesterone antagonists. Exp Biol Med (Maywood) 2002;227:969–980. doi: 10.1177/153537020222701104. [DOI] [PubMed] [Google Scholar]
- Leslie KK, Stein MP, Kumar NS, Dai D, Stephens J, Wandinger-Ness A, et al. Progesterone receptor isoform identification and subcellular localization in endometrial cancer. Gyenol Oncol. 2005;96:32–41. doi: 10.1016/j.ygyno.2004.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, O’Malley BW. Unfolding the action of progesterone receptors. J Biol Chem. 2003;278:39261–39264. doi: 10.1074/jbc.R300024200. [DOI] [PubMed] [Google Scholar]
- Lim CS, Baumann CT, Htun H, Xian W, Irie M, Smith CL, et al. Differential localization and activity of the A- and B-forms of the human progesterone receptor using green fluorescent protein chimeras. Mol Endocrinol. 1999;13:366–375. doi: 10.1210/mend.13.3.0247. [DOI] [PubMed] [Google Scholar]
- Masutomi K, Hahn WC. Telomerase and tumorigenesis. Cancer Lett. 2003;194:163–172. doi: 10.1016/s0304-3835(02)00703-6. [DOI] [PubMed] [Google Scholar]
- Masutomi K, Yu EY, Khurts S, Ben-Porath I, Currier JL, Metz GB, et al. Telomerase maintains telomere structure in normal cells. Cell. 2003;114:241–253. doi: 10.1016/s0092-8674(03)00550-6. [DOI] [PubMed] [Google Scholar]
- Miloso M, Bertelli AA, Nicolini G, Tredici G. Resveratrol-induced activation of the mitogen-activated protein kinases, ERK1 and ERK2, in human neuroblastoma SH-SY5Y cells. Neurosci Lett. 1999;264:141–144. doi: 10.1016/s0304-3940(99)00194-9. [DOI] [PubMed] [Google Scholar]
- Moll UM, Ostermeyer AG, Haladay R, Winkfield B, Frazier M, Zambetti G. Cytoplasmic sequestration of wild-type p53 protein impairs the G1 checkpoint after DNA damage. Mol Cell Biol. 1996;16:1126–1137. doi: 10.1128/mcb.16.3.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mote PA, Bartow S, Tran N, Clarke CL. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat. 2002;72:163–172. doi: 10.1023/a:1014820500738. [DOI] [PubMed] [Google Scholar]
- Nikolaev AY, Li M, Puskas N, Qin J, Gu W. Parc: a cytoplasmic anchor for p53. Cell. 2003;112:1–2. doi: 10.1016/s0092-8674(02)01255-2. [DOI] [PubMed] [Google Scholar]
- Ostermeyer AG, Runko E, Winkfield B, Ahn B, Moll UM. Cytoplasmically sequestered wild-type p53 protein in neuroblastoma is relocated to the nucleus by a C-terminal peptide. Proc Natl Acad Sci USA. 1996;93:15190–15194. doi: 10.1073/pnas.93.26.15190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OW, Gudjonsson T, Villadsen R, Bissell MJ, Ronnov-Jessen L. Epithelial progenitor cell lines as models of normal breast morphogenesis and neoplasia. Cell Prolif. 2003;36:33–44. doi: 10.1046/j.1365-2184.36.s.1.4.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambhatla L, Bohn SA, Stadler PB, Boyd JT, Coss RA, Sherley JL. Cellular senescence: ex vivo p53-dependent asymmetric cell kinetics. J Biomed Biotechnol. 2001;1:28–37. doi: 10.1155/S1110724301000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki Y, Tamura K, Yamamoto Y, Hatada T, Furuyama J, Utsunomiya J. Germline p53 mutation at codon 133 in a cancer-prone family. J Mol Med. 1997;75:50–56. doi: 10.1007/s001090050086. [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE. Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis. 2005;26:867–874. doi: 10.1093/carcin/bgh296. [DOI] [PubMed] [Google Scholar]
- Shay JW, Tomlinson G, Piatyszek MA, Gollahon LS. Spontaneous in vitro immortalization of breast epithelial cells from a patient with Li-Fraumeni syndrome. Mol Cell Biol. 1995;15:425–432. doi: 10.1128/mcb.15.1.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signorelli P, Ghidoni R. Resveratrol as an anticancer nutrient: molecular basis, open questions and promises. J Nutr Biochem. 2005;16:449–466. doi: 10.1016/j.jnutbio.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Stampfer MR, Garbe J, Nijjar T, Wigington D, Swisshelm K, Yaswen P. Loss of p53 function accelerates acquisition of telomerase activity in indefinite lifespan human mammary epithelial cell lines. Oncogene. 2003;22:5238–5251. doi: 10.1038/sj.onc.1206667. [DOI] [PubMed] [Google Scholar]
- Stingl J, Raouf A, Emerman JT, Eaves CJ. Epithelial progenitors in the normal human mammary gland. J Mammary Gland Biol Neoplasia. 2005;10:49–59. doi: 10.1007/s10911-005-2540-7. [DOI] [PubMed] [Google Scholar]
- Sun W, Kang KS, Morita I, Trosko JE, Chang CC. High susceptibility of a human breast epithelial cell type with stem cell characteristics to telomerase activation and immortalization. Cancer Res. 1999;59:6118–6123. [PubMed] [Google Scholar]
- Tabata Y, Iizuka Y, Masuda NT, Shinei R, Kurihara K, Okonogi T, et al. In vitro and in vivo characterization of novel nonsteroidal progesterone receptor antagonists derived from the fungal metabolite PF1092C. J Steroid Biochem Mol Biol. 2002;82:217–223. doi: 10.1016/s0960-0760(02)00157-7. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Kyo S, Takakura M, Kanaya T, Sagawa T, Yamashita K, et al. Expression of telomerase activity in human endometrium is localized to epithelial glandular cells and regulated in a menstrual phase-dependent manner correlated with cell proliferation. Am J Pathol. 1998;153:1985–1991. doi: 10.1016/S0002-9440(10)65712-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trask D, Band V, Zajchowski DA, Yaswen P, Suh T, Sager R. Keratins as markers that distinguish normal and tumor-derived mammary epithelial cells. Proc Natl Acad Sci USA. 1990;87:2319–2323. doi: 10.1073/pnas.87.6.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kyo S, Takakura M, Tanaka M, Yatabe N, Maida Y, et al. Progesterone regulates human telomerase reverse transcriptase gene expression via activation of mitogen-activated protein kinase signaling pathway. Cancer Res. 2000;60:5376–5381. [PubMed] [Google Scholar]
- Williams CD, Boggess JF, LaMarque LR, Meyer WR, Murray MJ, Fritz MA, et al. A prospective, randomized study ofendometrial telomerase during the menstrual cycle. J Clin Endocrinol Metab. 2001;86:3912–3917. doi: 10.1210/jcem.86.8.7729. [DOI] [PubMed] [Google Scholar]
- Yaswen P, Stampfer M. Molecular changes accompanying senescence and immortalization of cultured human mammary epithelial cells. Int J Biochem Cell Biol. 2002;34:1382–1394. doi: 10.1016/s1357-2725(02)00047-x. [DOI] [PubMed] [Google Scholar]
- Yu CC, Lo SC, Wang TC. Telomerase is regulated by protein kinase C-zeta in human nasopharyngeal cancer cells. Biochem J. 2001;355:459–464. doi: 10.1042/0264-6021:3550459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky B, Kroemer G. Apoptosis and genomic instability. Nature Rev Mol Cell Biol. 2004;5:752–762. doi: 10.1038/nrm1443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.