

Abstract
Pityriasis rubra pilaris (PRP) is a papulosquamous disorder phenotypically related to psoriasis. The disease has been occasionally shown to be inherited in an autosomal-dominant fashion. To identify the genetic cause of familial PRP, we ascertained four unrelated families affected by autosomal-dominant PRP. We initially mapped PRP to 17q25.3, a region overlapping with psoriasis susceptibility locus 2 (PSORS2 [MIM 602723]). Using a combination of linkage analysis followed by targeted whole-exome sequencing and candidate-gene screening, we identified three different heterozygous mutations in CARD14, which encodes caspase recruitment domain family, member 14. CARD14 was found to be specifically expressed in the skin. CARD14 is a known activator of nuclear factor kappa B signaling, which has been implicated in inflammatory disorders. Accordingly, CARD14 levels were increased, and p65 was found to be activated in the skin of PRP-affected individuals. The present data demonstrate that autosomal-dominant PRP is allelic to familial psoriasis, which was recently shown to also be caused by mutations in CARD14.
Main Text
Pityriasis rubra pilaris (PRP [MIM 173200]) is an uncommon skin disorder of unknown etiology and is characterized by the appearance of keratotic follicular papules, well-demarcated salmon-colored erythematous plaques covered with fine powdery scales interspersed with distinct islands of uninvolved skin, and palmoplantar keratoderma (thickening of the skin of the palms and soles). PRP occurs equally in men and women and is seen in about 1 in 5,000 individuals diagnosed with skin disease in Great Britain.1 Although most cases of PRP are simplex cases, up to 6.5% of all PRP-affected individuals report a positive family history.1–3 Those rare familial cases demonstrate autosomal-dominant inheritance with an early age of onset, incomplete penetrance, and variable expression.3–8
Griffiths classified PRP into five types on the basis of age of onset, clinical features, and prognosis; these types are classic adult type I, atypical adult type II, classic juvenile type III, circumscribed juvenile type IV, and atypical juvenile type V.9,10 More recently, a type VI PRP, or human immunodeficiency virus (HIV)-associated PRP, has been described.11–14 Familial PRP belongs to the type V group, which accounts for about 5% of all cases.1,8,9 It is usually present at birth or appears during the first years of life and runs a chronic course. It is characterized by prominent follicular hyperkeratosis, diffuse palmoplantar keratoderma, and erythema8,9 Response to treatment is disappointing. Modest results have been reported for retinoids, cyclosporine, and, lately, TNF-alpha-blocking agents.
The pathogenesis of PRP remains elusive. An infective etiology was proposed on the basis of clinical observations15 and the known association between PRP and HIV infection.11–14 PRP has also been reported to be associated with defective vitamin A metabolism or vitamin A deficiency.1 However, on the basis of clinical and pathological features, it is now classified as a papulosquamous disease, which, like psoriasis, is thought to result from abnormal activation of inflammatory pathways.16 Although PRP and psoriasis might manifest with overlapping features, they are distinguishable on clinical and histopathological grounds. PRP is characterized by prominent involvement of the face, classical islands of sparing over the trunk, follicular hyperkeratosis, distinctive diffuse red-orange palmoplantar keratoderma, and the absence of psoriasis-associated nail changes. Although psoriasis is typically associated with hypogranulosis, intraepidermal neutrophilic infiltration culminating with the formation of Munro microabscesses, elongation of the rete ridges, and marked vascular dilation in the papillary dermis,1,17–19 PRP features include alternating parakeratosis and orthokeratosis, hypergranulosis, broad thickening of the rete ridges, thick suprapapillary plates, follicular hyperkeratosis, lack of neutrophilic infiltration, and limited vascular dilatation.20
In the present study, we demonstrate that familial PRP results from mutations in CARD14 (MIM 607211), which encodes caspase recruitment domain family, member 14, a known activator of nuclear factor kappa B (NF-κB) signaling.21,22
We assessed four families affected by dominant PRP. In family D, we only had access to DNA from two affected individuals. All participants provided their written informed consent to participate to this study in accordance with protocols reviewed and approved by institutional review boards at each participating center. A diagnosis of PRP was based on the presence of characteristic clinical and histopathological features (Figure 1 and Table 1). All affected individuals showed well-demarcated erythematous plaques coalescing into large areas interspersed with islands of normal skin, follicular papules or accentuation, palmoplantar keratoderma, and a lack of psoriasis-associated nail changes. Histopathology of skin lesions showed alternating orthokeratosis and parakeratosis, acanthosis with broadening of the rete ridges, follicular plugging, lymphocytic infiltrate in the dermis, and a lack of neutrophils in the epidermis. Age of onset varied from 4 to 36 months of age. DNA from members of family A was extracted from saliva samples with the use of an Oragene DNA kit (DNA Genotek, Kanata, Ontario, Canada), DNA from members of families B and C was extracted from lymphoblast cell lines, and DNA from two affected individuals of family D was extracted from peripheral-blood lymphocytes.
Figure 1.
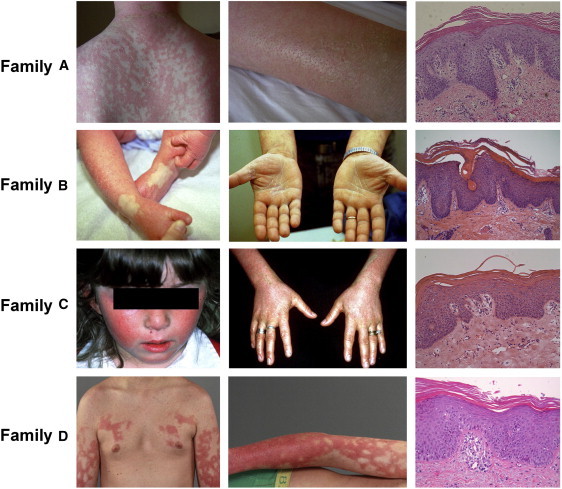
Clinical and Histopathological Features of PRP-Affected Families
Note the conspicuous presence of widespread erythematous plaques coalescing into large areas containing islands of normal-appearing skin, follicular plugging, and palmoplantar keratoderma. In all cases, histopathology demonstrates alternating parakeratosis and orthokeratosis, acanthosis, and a mononuclear dermal infiltrate.
Table 1.
Clinical and Pathological Features
| Family A | Family B | Family C | Family D | |
|---|---|---|---|---|
| Age of onset (months) | 4 | 9 | 8 | 36 |
| Face involvement | + | + | + | + |
| Scalp involvement | + | − | − | − |
| Islands of normal skin | + | + | + | + |
| PPK | mild | erythematous with scale but no thickening | accentuate skin markings with a beaded appearance but not thickened | mild |
| Nail changes | none | none | thickened with onychorrhexis | +/− |
| Alternating orthokeratosis and parakeratosis | + | + | + | + |
| Lymphocytic infiltrate on pathology | mild dermal lymphocytic | mild dermal lymphocytic | mild dermal lymphocytic | mild dermal lymphocytic |
| Follicular plugging | + | + | + | + |
We genotyped all available members of family A by using the Illumina Human Linkage 12 chip comprising 6,000 tagged SNPs distributed across the genome. Two hundred nanograms of DNA was hybridized according to the Infinium II assay (Illumina, San Diego, CA) and scanned with an Illumina BeadArray reader. The scanned images were imported into BeadStudio 3.1.3.0 (Illumina) for extraction and quality control and produced an average call rate of 99.9%. Multipoint linkage analysis with the Allegro software23 implemented in the easyLinkage package24 generated a LOD score of 2.79 at marker rs11150586 in the centrosomal region of chromosome 16 and a LOD score of 2.50 at marker rs938283 on chromosome 17 (Figure 2A). Fine mapping of the two regions of interest in families A, B, and C was performed with polymorphic microsatellite markers that were selected from the National Center for Biotechnology Information (NCBI) database. Microsatellite analysis was performed on an automated sequencer (ABI 3100 Genetic Analyzer; PE Applied Biosystems, Foster City, CA, USA) with sequence-specific forward and reverse primers and a universal fluorescently labeled M13 primer as previously reported.25 Allele sizes were determined with GeneMapper v.4.0 software. Haplotype analysis ruled out linkage to the chromosome 16 region (Figure S1, available online). All three families were found to map to 17q25.3, generating a combined LOD score of 3.73 (analysis was performed with the Superlink software;26 the LOD scores calculated for families A, B, and C were, respectively, 1.9366, 1.1950, and 0.5977). Critical recombination events in family A members II-2 and III-3 set the disease interval between markers D17S722 and D17S668 (Figure 2B). Of note, two obligatory carriers (I-3 and II-4) were completely asymptomatic despite the fact that they were carriers of the disease-associated haplotype.
Figure 2.
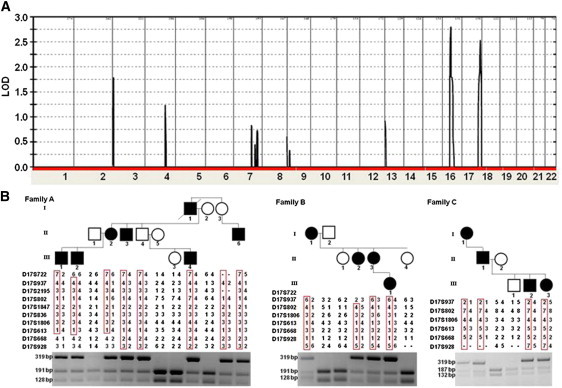
Linkage, Haplotype, and PCR-RFLP Analysis
(A) Multipoint LOD-score analysis was performed with the Allegro software23 implemented in the easyLinkage package24 and produced an estimated penetrance of 90%. LOD scores are plotted against all SNP markers distributed across the genome.
(B) Haplotype analysis with polymorphic markers across chromosomal region 17q25.3 reveals a heterozygous 5 Mb interval between markers D17S722 and D17S668; this interval is uniquely shared by affected individuals or obligatory carriers. PCR restriction fragment-length polymorphism (PCR-RFLP) analysis, shown below each pedigree, confirmed segregation of the mutations in the families. Mutation c.467T>C in CARD14 abolishes a recognition site for AlwnNI; thus, healthy individuals display 191 bp and 128 bp fragments, whereas affected heterozygous individuals also show a 319 bp fragment (families A and B). Mutation c.412_414delGAG in CARD14 disrupts a recognition site for EcoNI; thus, healthy individuals display 187 bp and 132 bp fragments, whereas affected heterozygous individuals also show a 319 bp fragment (family C).
The 5 Mb candidate region was found to contain 118 genes. To identify the gene harboring PRP-causing mutations, we performed targeted capture (for individuals II-2, II-4, II-5, and III-4 from family A) and exome capture (for individuals II-2 and III-4 from family A). The DNA samples used for targeted capture were prepared with paired-end adapters according to the manufacturer's (Illumina) guidelines. After quality control, the library was captured on a custom repeat-masked 244K Agilent SureSelect solid array (Agilent, Santa Clara, CA). Next-generation sequencing was performed on an Illumina Genome Analyzer IIx. The resulting reads were aligned to the UCSC human reference sequence (hg19/GRCh37) with the use of the Burrows Wheeler aligner (BWA)27 and SoapSNP for SNP calling. Because of the low coverage obtained by the targeted capture (Table S1), we used two different approaches for exome sequencing. First, a genomic library of the DNA sample from III-4 was prepared according to the Agilent standard protocol for SOLiD library preparation. The library was captured with the SureSelect Human All Exon kit (Agilent). Next-generation sequencing was carried out on the SOLiD V3 system (Applied Biosystems). The resulting reads were aligned to the hg19 reference sequence with Bioscope 1.2 (Applied Biosystems) and the BWA. SNP calling was performed with Bioscope 1.2 and SAMTOOLS.28 Second, a genomic library of the DNA sample from II-2 was prepared with paired-end adapters according to Illumina's protocol, and exome enrichment was achieved with a NimbleGen SeqCap EZ Human Exome Library v.2.0 (Roche NimbleGen, Madison, WI, USA). The sample was sequenced on an Illumina HiSeq2000 system. We analyzed all data for quality, exome coverage, and exome-wide SNPs and indels by using the platform provided by DNAnexus (DNAnexus, Mountain View, CA, USA).
A total of 436 sequence variants were identified in the candidate region. After having filtered out 409 variants that were found to correspond to common variants in public databases (NCBI, UCSC, and the 1000 Genomes Project), we verified all putative mutations by Sanger sequencing. Only one of the sequence variants was found to cosegregate with the disease phenotype in family A; this variant was c.290G>C (p.Gly97Ala) in TMEM105 (RefSeq accession number NM_178520.3), the gene encoding the transmembrane protein 105. Direct sequencing of this gene in families B–D revealed additional cosegregating sequence alterations in TMEM105. In family B, we identified a 7 bp deletion (c.58delGGGCCCA) in the 5′ UTR of the gene, whereas in family D, we identified c.2726C>G in the 3′ UTR of the gene. These variations have recently been reported to represent very rare polymorphisms (rs147929646, rs137864500, and rs182343738, respectively).
Because next-generation-sequencing efforts did not lead to the identification of the proximal cause of PRP in the affected four families, we scrutinized all exons that were within the disease interval and that had insufficient coverage (<10×) by either exome or targeted sequencing. All together, 17.4% and 6% of exons from individuals III-4 and II-2, respectively, were defined as candidates for resequencing by Sanger technology (Table S1). We prioritized those exons by functional relevance to the pathogenesis of PRP, and we initially sequenced exons of genes encoding regulators of epithelial differentiation or mediators of inflammation. This strategy finally revealed heterozygous mutations in CARD14 (RefSeq NM_024110.4) in all families (Figure 3A). Mutations c.467T>C (p.Leu156Pro) and c.412_414delGAG (p.Glu138del) were found in exon 4 and alter highly conserved amino acids (Figure 3B). Mutation c.349+1G>A abolishes the consensus donor splice site in intron 3 of CARD14 and generates an aberrant splice variant as a result of the use of a cryptic splice site, causing the insertion of an extra 66 bp originating from intron 3 (Figure 3C). We confirmed segregation of the mutation with the disease phenotype throughout families A, B, and C by using PCR restriction fragment-length polymorphism (PCR-RFLP). PCR fragments, encompassing exons 3 or 4, were PCR amplified and digested with AlwnNI, EcoNI, or AciI (New England Biolabs, Ipswich, MA) at 37°C for 16 hr. We also excluded all mutations from a panel of 100 healthy population-matched control individuals (200 alleles). In addition, none of the mutations were found in major public databases (NCBI or UCSC), including in a total of more than 8,200 individual sequences deposited in the 1000 Genomes Project and in the National Heart, Lung, and Blood Institute (NHLBI) Grand Opportunity Exome Sequencing Project. Of note, mutation c.467T>C was detected in two families (families A and B) on the background of distinct haplotypes (not shown), suggesting spontaneously recurrent mutational events rather than a founder effect.
Figure 3.
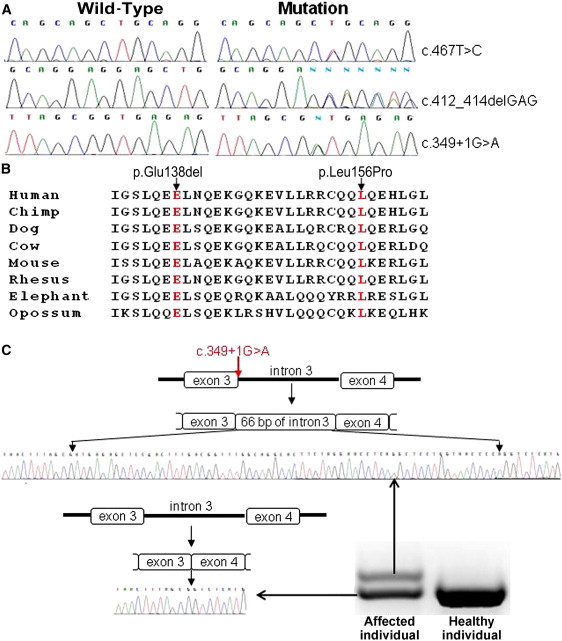
Mutation Analysis
(A) Direct sequencing of CARD14 revealed three heterozygous mutations: c.467T>C in families A and B, c.412_414delGAG in family C, and c.349+1G>A in family D.
(B) Analysis of the CARD14 region encompassing mutant residues p.Glu138del and p.Leu156 (both in red) demonstrates conservation across species.
(C) Sequence analysis of RT-PCR products generated with RNA extracted from a skin biopsy of an affected individual of family D revealed two splice variants, which, through sequencing, were found to correspond to the wild-type cDNA sequence and to an aberrant splice variant containing an extra 66 bp originating from intron 3.
To assess the relevance of our findings to the skin pathology seen in PRP, we assessed CARD14 tissue-specific expression. cDNA was synthesized from 500 ng of Human Total RNA Survey Panel (Ambion, Austin, TX). CARD14 cDNA PCR amplification was carried out with the Fast SYBR Green Master Mix on a StepOne Real-Time PCR System (Applied Biosystems) with gene-specific intron-crossing oligonucleotide pairs (forward primer 5′-AGGCAGGTGTTCGAGCTG-3′ and reverse primer 5′-GGTCCTGGCTTCCTGCTT-3′). In line with previous semiquantitative RT-PCR experiments21 and microarray data,29 we detected a high level of CARD14 expression in mucosal tissue. Comparative quantitative analysis of CARD14 expression in 22 different tissues showed that CARD14 RNA levels are five times higher in the skin than in any other human tissue (Figure 4).
Figure 4.
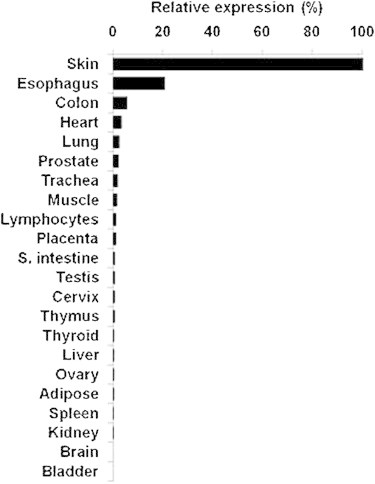
CARD14 Expression Analysis
Results are provided as a percentage of CARD14 expression relative to its level of expression in skin after normalization to ACTB.
To assess the functional consequences of the mutations detected in PRP-affected individuals, we stained skin biopsies from these individuals with a CARD14 antibody (Novus Biologicals, Littleton, CO). As shown in Figure 5B, CARD14 immunostaining was found to be stronger in PRP-affected skin than in normal skin. In addition, whereas CARD14 staining was mostly restricted to the lower layers of the normal epidermis, CARD14 was localized up to the granular layers in PRP-affected skin. In addition, because CARD14 has been shown to modulate the activity of NF-κB, which was previously shown to be involved in inflammatory skin diseases,30 we stained PRP-affected and normal control-derived skin biopsies with a monoclonal antibody that recognizes an epitope overlapping the nuclear localization signal of the p65 subunit (Millipore, Billerica, MA). In addition to the stronger intensity of the staining found in PRP-affected skin (Figure 5D), we also observed a significant difference in the percentage of activated p65-positive suprabasal cells in PRP-affected skin as compared with normal skin (Figure 5E). These results are in agreement with in vitro data showing that highly similar CARD14 mutations recently implicated in the pathogenesis of familial psoriasis result in NF-κB activation.31 To further assess the effect of the CARD14 mutations on NF-κB activity, we studied the expression of three known NF-κB target genes in PRP-affected skin (Figure 5F). We performed PCR amplification of CCL20 (MIM 601960), IL1B (MIM 147720), and NOS2 (MIM 163731) cDNA on a StepOne Real-Time PCR System (Applied Biosystems) by using the Fast SYBR Green Master Mix, cDNA derived from skin biopsies obtained from an affected member of family D and from a healthy individual, and gene-specific intron-crossing oligonucleotide pairs (primer pairs for CCL20, IL1B, and NOS2 were 5′-CGAATCAGAAGCAAGCAA-3′ and 5′-AGCATTGATGTCACAGCCTTCAT-3′, 5′-CATTGCTCAAGTGTCTGAAGCA-3′ and 5′-CTGGAAGGAGCACTTCATCTGTT-3, and 5′-CTGCAGACACGTGCGTTACTC-3′ and 5′- GAAGTCGTGCTTGCCATCACT-3′, respectively). Compared with normal skin, the PRP-affected skin showed upregulation of all three genes (Figure 5F).
Figure 5.
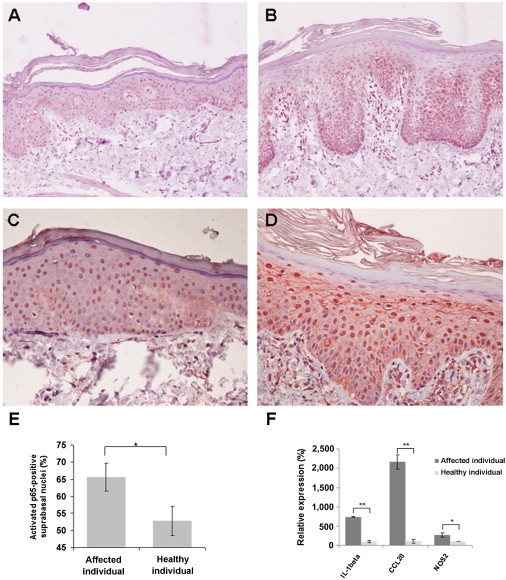
Elevated CARD14 Levels and NF-κB Hyperactivation in the Epidermis of PRP-Affected Individuals
Skin-biopsy sections obtained from a person with PRP (B and D) and from a healthy individual (A and C) were stained with antibodies directed against CARD14 (A and B) or an activated p65 subunit of NF-κB (C and D).
(E) The percentage of activated-p65-positive nuclei was determined in the suprabasal layers of the epidermis. The percentage of positive nuclei was significantly higher in the affected individual (χ = 33.2, p = 8.5 × 10−9).
(F) Expression levels of three NFkB target genes were determined with quantitative RT-PCR and cDNA derived from a skin biopsy obtained from a member of family D. The results were normalized to ACTB (MIM 102630) expression and are expressed as a percentage of the control. Data shown represent mean values of triplicates ± SD (∗p < 0.05,∗∗p < 0.01 by a Student's t test).
In the present study, we provide evidence indicating that mutations in CARD14 cause familial PRP. The known role of CARD14 in the regulation of inflammatory processes,29,32 the high expression of CARD14 in the skin, and the fact that NF-κB is activated in PRP-affected skin are also supportive of a role for this protein in the pathogenesis of PRP. Of note, the fact that penetrance was incomplete in some of the families we studied suggests a possible role for additional modifying traits, epigenetic factors, or environmental elements in determining the phenotypic expression of the causative mutations.
CARD14, CARD10, and CARD11, also known as CARD- and membrane-associated guanylate kinase-like domain-containing proteins, share a high degree of sequence and structural homology.21,22,29 All three proteins contain a CARD domain, coiled-coil (C-C) domain, SH3 domain, PDZ domain, and GuK domain;21,22 however, they exhibit a distinct tissue-distribution pattern. CARD10 is found in a broad range of tissues and not in hematopoietic cells, whereas CARD11 is present in hematopoietic tissues.29 CARD14 was previously found to be abundant in placental and mucosal tissues.22,29 Here, we demonstrate its high level of expression in the skin.
Recently, pathogenic mutations located in the vicinity of the mutations reported in the present study were found to underlie both familial and nonfamilial cases of psoriasis.31,33 Psoriasis and PRP have been considered over the years as two separate entities both clinically and histologically.18 PRP and psoriasis have traditionally been differentiated on the basis of the distinctive clinical and histopathological features reviewed above as well as the inconsistent response of PRP to UVB phototherapy. The present data suggest that the two diseases, at least in their familial forms, share a common pathophysiology, which in turn might bear important implications for the therapeutic approach to PRP.
CARD14 binds to B cell lymphoma 10 (BCL10) through a CARD-CARD interaction and also functions as an upstream regulator of NF-κB signaling and thereby protects cells from apoptosis induced by different stimuli.21,22 All three PRP-causing mutations identified in this study alter the structure of the coiled-coil domain of CARD14, which was found to be associated with NF-κB activation. The function of the coiled-coil domain of CARD14 remains to be fully delineated. Of note, mice carrying an ethylnitrosourea-induced point mutation in the coiled-coil domain of CARD11 showed defects in humoral immune responses, defective B cell activation by antigen receptors, and T cell costimulation by CD28 and developed a skin disorder similar to atopic dermatitis with elevated levels of IgE.34 This suggests that the coiled-coil domain is important for the regulation of CARD activation.
NF-κB has been shown to promote keratinocyte viability during differentiation.35 Mice displaying constitutive activation of NF-κB develop a generalized skin disorder histologically featuring epidermal hyperplasia, hyperkeratosis, parakeratosis, hypogranulosis, T cell infiltration, and the formation of microabscesses. These data are in line with the possibility that the CARD14 mutations identified in our study modulate the NF-κB signaling pathway within keratinocytes and ultimately lead to aberrant immunological activation. It is therefore of interest that anti-TNF-alpha treatment has been shown to be of therapeutic benefit for PRP, including familial PRP,8 and that TNF-alpha antagonists are known to target NF-κB signaling,36 and response to these drugs is even in part determined by polymorphisms in genes belonging to the NF-κB pathway36
Moreover, an abnormal response to microbial agents has been proposed to contribute to the pathogenesis of both psoriasis and PRP.20,37 Other CARD domain proteins, notably NOD2 (CARD15), are involved in the response to bacteria. Colitis-associated CARD15 mutations cause a decreased response to bacterial cell-wall components. It is tempting to speculate that PRP might be akin to colitis in that respect and might, in part, result from altered host-microbiome interactions.
In conclusion, a century after familial PRP was originally described,38 we demonstrate in the present study that this disorder results from mutations in CARD14, which regulates NF-κB signaling and which is strongly expressed in the skin.
Acknowledgments
We would like to acknowledge the participation of all family members in this study. We are grateful to Temima Schnitzer-Perlman for her help with exome sequencing and to Jason Lee and Mark Pawlowski for their assistance with the histopathological analysis.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
Berkeley Drosophila Genome Project, http://www.fruitfly.org/
The ConSurf Server, http://consurftest.tau.ac.il/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
NHLBI Grand Opportunity Exome Sequencing Project, https://esp.gs.washington.edu/drupal/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Superlink, http://bioinfo.cs.technion.ac.il/superlink-online-twoloci/makeped/TwoLociMultiPoint.html
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Albert M.R., Mackool B.T. Pityriasis rubra pilaris. Int. J. Dermatol. 1999;38:1–11. doi: 10.1046/j.1365-4362.1999.00513.x. [DOI] [PubMed] [Google Scholar]
- 2.Sehgal V.N., Srivastava G. (Juvenile) Pityriasis rubra pilaris. Int. J. Dermatol. 2006;45:438–446. doi: 10.1111/j.1365-4632.2006.02666.x. [DOI] [PubMed] [Google Scholar]
- 3.Thomson M.A., Moss C. Pityriasis rubra pilaris in a mother and two daughters. Br. J. Dermatol. 2007;157:202–204. doi: 10.1111/j.1365-2133.2007.07938.x. [DOI] [PubMed] [Google Scholar]
- 4.Sehgal V.N., Bajaj P., Jain S. Pityriasis rubra pilaris (PRP): Report of four cases. J. Dermatol. 2000;27:174–177. doi: 10.1111/j.1346-8138.2000.tb02146.x. [DOI] [PubMed] [Google Scholar]
- 5.Kint A., De Bie S., Geerts M.L., T'Kint R. Pityriasis rubra pilaris, a familial condition. Arch. Belg. Dermatol. Syphiligr. 1972;28:371–376. [PubMed] [Google Scholar]
- 6.Sehgal V.N., Jain S., Kumar S., Bhattacharya S.N., Sardana K., Bajaj P. Familial pityriasis rubra pilaris (adult classic-I): A report of three cases in a single family. Skinmed. 2002;1:161–164. doi: 10.1111/j.1540-9740.2002.01663.x. [DOI] [PubMed] [Google Scholar]
- 7.Vanderhooft S.L., Francis J.S., Holbrook K.A., Dale B.A., Fleckman P. Familial pityriasis rubra pilaris. Arch. Dermatol. 1995;131:448–453. [PubMed] [Google Scholar]
- 8.Vasher M., Smithberger E., Lien M.H., Fenske N.A. Familial pityriasis rubra pilaris: Report of a family and therapeutic response to etanercept. J. Drugs Dermatol. 2010;9:844–850. [PubMed] [Google Scholar]
- 9.Griffiths W.A. Pityriasis rubra pilaris. Clin. Exp. Dermatol. 1980;5:105–112. doi: 10.1111/j.1365-2230.1980.tb01676.x. [DOI] [PubMed] [Google Scholar]
- 10.Griffiths W.A. Pityriasis rubra pilaris: The problem of its classification. J. Am. Acad. Dermatol. 1992;26:140–142. doi: 10.1016/s0190-9622(08)80543-9. [DOI] [PubMed] [Google Scholar]
- 11.Misery I., Faure M., Claidy A. Pityriasis rubra pilaris and human immunodeficiency virus infection—type 6 pityriasis rubra pilaris? Br. J. Dermatol. 1996;135:1008–1009. doi: 10.1046/j.1365-2133.1996.d01-1114.x. [DOI] [PubMed] [Google Scholar]
- 12.Miralles E.S., Núñez M., De Las Heras M.E., Pérez B., Moreno R., Ledo A. Pityriasis rubra pilaris and human immunodeficiency virus infection. Br. J. Dermatol. 1995;133:990–993. doi: 10.1111/j.1365-2133.1995.tb06939.x. [DOI] [PubMed] [Google Scholar]
- 13.Menni S., Brancaleone W., Grimalt R. Pityriasis rubra pilaris in a child seropositive for the human immunodeficiency virus. J. Am. Acad. Dermatol. 1992;27:1009. doi: 10.1016/s0190-9622(08)80267-8. [DOI] [PubMed] [Google Scholar]
- 14.Auffret N., Quint L., Domart P., Dubertret L., Lecam J.Y., Binet O. Pityriasis rubra pilaris in a patient with human immunodeficiency virus infection. J. Am. Acad. Dermatol. 1992;27:260–261. doi: 10.1016/s0190-9622(08)80734-7. [DOI] [PubMed] [Google Scholar]
- 15.Betlloch I., Ramón R., Silvestre J.F., Carnero L., Albares M.P., Bañuls J. Acute juvenile pityriasis rubra pilaris: A superantigen mediated disease? Pediatr. Dermatol. 2001;18:411–414. doi: 10.1046/j.1525-1470.2001.01967.x. [DOI] [PubMed] [Google Scholar]
- 16.Klein A., Landthaler M., Karrer S. Pityriasis rubra pilaris: A review of diagnosis and treatment. Am. J. Clin. Dermatol. 2010;11:157–170. doi: 10.2165/11530070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 17.Braun-Falco O., Ryckmanns F., Schmoeckel C., Landthaler M. Pityriasis rubra pilaris: A clinico-pathological and therapeutic study with special reference to histochemistry, autoradiography, and electron microscopy. Arch. Dermatol. Res. 1983;275:287–295. doi: 10.1007/BF00417199. [DOI] [PubMed] [Google Scholar]
- 18.Soeprono F.F. Histologic criteria for the diagnosis of pityriasis rubra pilaris. Am. J. Dermatopathol. 1986;8:277–283. doi: 10.1097/00000372-198608000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Schön M.P., Boehncke W.H. Psoriasis. N. Engl. J. Med. 2005;352:1899–1912. doi: 10.1056/NEJMra041320. [DOI] [PubMed] [Google Scholar]
- 20.Magro C.M., Crowson A.N. The clinical and histomorphological features of pityriasis rubra pilaris. A comparative analysis with psoriasis. J. Cutan. Pathol. 1997;24:416–424. doi: 10.1111/j.1600-0560.1997.tb00816.x. [DOI] [PubMed] [Google Scholar]
- 21.Scudiero I., Zotti T., Ferravante A., Vessichelli M., Vito P., Stilo R. Alternative splicing of CARMA2/CARD14 transcripts generates protein variants with differential effect on NF-κB activation and endoplasmic reticulum stress-induced cell death. J. Cell. Physiol. 2011;226:3121–3131. doi: 10.1002/jcp.22667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertin J., Wang L., Guo Y., Jacobson M.D., Poyet J.L., Srinivasula S.M., Merriam S., DiStefano P.S., Alnemri E.S. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J. Biol. Chem. 2001;276:11877–11882. doi: 10.1074/jbc.M010512200. [DOI] [PubMed] [Google Scholar]
- 23.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann K., Lindner T.H. easyLINKAGE-Plus—automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- 25.Schuelke M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000;18:233–234. doi: 10.1038/72708. [DOI] [PubMed] [Google Scholar]
- 26.Fishelson M., Geiger D. Exact genetic linkage computations for general pedigrees. Bioinformatics. 2002;18(Suppl 1):S189–S198. doi: 10.1093/bioinformatics/18.suppl_1.s189. [DOI] [PubMed] [Google Scholar]
- 27.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blonska M., Lin X. NF-κB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res. 2011;21:55–70. doi: 10.1038/cr.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wullaert A., Bonnet M.C., Pasparakis M. NF-κB in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011;21:146–158. doi: 10.1038/cr.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jordan C.T., Cao L., Roberson E.D., Pierson K.C., Yang C.F., Joyce C.E., Ryan C., Duan S., Helms C.A., Liu Y. PSORS2 Is Due to Mutations in CARD14. Am. J. Hum. Genet. 2012;90:784–795. doi: 10.1016/j.ajhg.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seibl R., Kyburz D., Lauener R.P., Gay S. Pattern recognition receptors and their involvement in the pathogenesis of arthritis. Curr. Opin. Rheumatol. 2004;16:411–418. doi: 10.1097/01.bor.0000127108.08398.34. [DOI] [PubMed] [Google Scholar]
- 33.Jordan C.T., Cao L., Roberson E.D., Duan S., Helms C.A., Nair R.P., Duffin K.C., Stuart P.E., Goldgar D., Hayashi G. Rare and Common Variants in CARD14, Encoding an Epidermal Regulator of NF-kappaB, in Psoriasis. Am. J. Hum. Genet. 2012;90:796–808. doi: 10.1016/j.ajhg.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jun J.E., Wilson L.E., Vinuesa C.G., Lesage S., Blery M., Miosge L.A., Cook M.C., Kucharska E.M., Hara H., Penninger J.M. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 2003;18:751–762. doi: 10.1016/s1074-7613(03)00141-9. [DOI] [PubMed] [Google Scholar]
- 35.Seitz C.S., Freiberg R.A., Hinata K., Khavari P.A. NF-kappaB determines localization and features of cell death in epidermis. J. Clin. Invest. 2000;105:253–260. doi: 10.1172/JCI7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lories R.J., Derese I., Luyten F.P., de Vlam K. Activation of nuclear factor kappa B and mitogen activated protein kinases in psoriatic arthritis before and after etanercept treatment. Clin. Exp. Rheumatol. 2008;26:96–102. [PubMed] [Google Scholar]
- 37.Karin M., Lawrence T., Nizet V. Innate immunity gone awry: Linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 38.De Beurmann, B., Heuyer. (1910). Pityriasis rubra pilaire familial. Ann Dermatol Syphiligr (Paris) 1(ser 5), 609–619.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.