

Abstract
Protein glycosylation is a complex process that depends not only on the activities of several enzymes and transporters but also on a subtle balance between vesicular Golgi trafficking, compartmental pH, and ion homeostasis. Through a combination of autozygosity mapping and expression analysis in two siblings with an abnormal serum-transferrin isoelectric focusing test (type 2) and a peculiar skeletal phenotype with epiphyseal, metaphyseal, and diaphyseal dysplasia, we identified TMEM165 (also named TPARL) as a gene involved in congenital disorders of glycosylation (CDG). The affected individuals are homozygous for a deep intronic splice mutation in TMEM165. In our cohort of unsolved CDG-II cases, we found another individual with the same mutation and two unrelated individuals with missense mutations in TMEM165. TMEM165 encodes a putative transmembrane 324 amino acid protein whose cellular functions are unknown. Using a siRNA strategy, we showed that TMEM165 deficiency causes Golgi glycosylation defects in HEK cells.
Introduction
Congenital disorders of glycosylation (CDG) are a rapidly growing family of inborn errors of metabolism. To date, some 50 different CDG types have been identified, and most of them affect protein glycosylation (defects in N-glycosylation, O-glycosylation, and both N- and O-glycosylation).1–4 Because glycosylation is a ubiquitous cellular function, it is not surprising that CDG encompasses an extremely broad range of clinical phenotypes and affect every organ system.
Initially, mutations were found in genes encoding glycosyltransferases, glycosidases, and nucleotide-sugar transporters. However, CDG types that are caused by deficiencies of vesicular Golgi trafficking5–10 and Golgi pH homeostasis11 have recently been identified.
For the affected individuals described in this study, the N-glycan analysis did not allow us to pinpoint the underlying molecular defect. In addition, a candidate-gene approach would have been a matter of mere luck because of the huge number of enzymes and proteins involved in N-glycosylation as well as in the structure and function of the Golgi apparatus.
We decided to exploit the possibility of identifying the candidate gene by a mapping approach, combined with expression analysis, in a family with two affected siblings presenting with a new, specific phenotype and a type 2 pattern of serum-transferrin isoelectric focusing (sTf-IEF). This strategy led us to identify CDG-II individuals with mutations in TMEM165.
Material and Methods
Cell Culture
Primary skin fibroblasts from healthy controls and affected individuals and HEK cells were cultured at 37°C under 5% CO2 in DMEM/F12 (Life Technologies) supplemented with 10% fetal bovine serum (Clone III, HyClones). Where appropriate, microtubule (MT)-depolymerizing drugs nocodazole (5 μM, Calbiochem) and puromycin (200 μg/ml, Sigma) were added to the cultures. Research on CDG-affected cells was approved by the ethics committee of the University of Leuven, and proper informed consent was obtained for affected individuals and family members.
DNA, RNA Extraction, and cDNA Synthesis
Blood DNA was isolated via a classical salt-based extraction procedure. Total RNA was isolated from 2 × 107 fibroblasts with the TRIzol LS reagent according to the manufacturer's (Invitrogen) instructions. TMEM165 cDNA was prepared with oligo-dT priming (Amersham) and Superscript III RNase (Invitrogen) according to the manufacturers' protocol, and the protein-coding region was amplified by PCR. Primers were designed by Primer3 to amplify the entire cDNA as well as the exons on genomic DNA, including the intron-exon boundaries. Primer sequences, based on the sequence described in ENSEMBL (ENSG00000134851), are available upon request. After purification of the PCR products with EXO-SAP-IT (Roche), sequencing was performed with the Big Dye Terminator v.3.1 cycle sequencing kit on ABI 3130xl (Applied Biosystems). DNA-mutation numbering is based on the cDNA reference sequence (RefSeq accession number NM_018475, version NM_018475.3 GI:195539338).
The sequence variants identified in this study were not found in 100 controls.
Microarray Analyses
Molecular karyotyping was performed in all six family members with Affymetrix Human Mapping 50K XbaI SNP chips according to the manufacturer's (Affymetrix) instructions. In brief, total genomic DNA was digested with XbaI, and the 50K fragments were ligated to adaptors and amplified with a single primer. After purification of the PCR products, amplicons were quantified, fragmented, labeled, and hybridized on the array. Signal intensities were measured with Affymetrix GeneChip Scanner 3000 7G. Genotypes were generated with the Dynamic model.
Copy-Number and Loss-of-Heterozygosity Analyses
Intensity of hybridization was quantified for the estimation of relative copy numbers for each chromosome. The signal-intensity ratios of tested samples were divided by the mean of a set of 200 internal reference samples for the calculation of copy-number status with the use of the Copy-Number Analyzer for GeneChip (CNAG)12,13 software version 3.3.0.1. Intensity of hybridization was estimated with the hidden Markov model.
Autozygosity Mapping
Autozygosity refers to the state of a genetic variation in which the two alleles in an individual are homozygous as a result of being inherited from a common ancestor carrying the same allele. Autozygous alleles are commonly described as being identical by descent (IBD). Autozygosity mapping consists of the following two principal steps. (1) A LOD score is computed for each variation per affected individual. This score indicates how likely the observed value for a variation results from autozygosity as opposed to being drawn from a known population. (2) LOD scores are computed for regions. LOD scores are summed for contiguous variations (on the same chromosome). Regions are made from the contiguous variations that have high LOD scores.
Linkage Analysis and Haplotype Analysis
Genome-wide parametric and nonparametric multipoint linkage analyses were performed in all families with the use of Genspring GT software (Agilent Technologies). The parametric analyses were run under an autosomal-recessive mode. The penetrance was set to 90%, and the phenocopy rate for nongene carriers was set to 1%. DECODE map was selected for LOD-score calculation, and the Caucasian population was taken as a reference for allele frequencies.
Expression Profiling by Microarray Hybridization
RNA extracted from fibroblasts of each family member was used for gene-expression analysis. The quality of total RNA was checked on the basis of A260/A280 ratios (>1.9) and 28S/18S rRNA peak-height ratios (>1.6), which were determined with a high-resolution electrophoresis system (Agilent 2100 Bioanalyzer, Agilent Technologies). All microarray procedures followed Human Genome U133 Plus 2.0 Array (Affymetrix). In brief, 20 μg of biotin-labeled cRNA was fragmented according to Affymetrix's instructions. All labeled samples were hybridized to Human Genome U133 Plus 2.0 Array (Affymetrix). GeneChip data were normalized with MAS 5.0 algorithms, and global normalization and scaling were set to a signal intensity of 100. Isolation of total RNA, synthesis of cRNA, microarray processing, and data normalization were carried out according to standard Affymetrix protocols.
Data Analysis
A number of quality-control checks were conducted for the evaluation of GeneChip quality. GAPDH was used for the assessment of the quality of the RNA sample and assay specifically for the GeneChip array. The signal values for the 3′ probe set for GAPDH were compared to those for the 5′ probe set. The ratio of the 3′ to the 5′ probe sets was expected to be no more than 3. Hybridization controls on the GeneChip array (four E. Coli genes, bioB, bioC, bioD, and cre) were spiked into each sample independent of RNA-sample preparation for the evaluation of hybridization efficiency. Raw noise (Q value), a measure of how pixel-to-pixel variation of probe cells on a GeneChip array is due to operation-associated electrical noise of the GeneArray scanner, was evaluated. To serve as internal controls, PolyA control genes, dap, lys, phe, thr, and trp from B. subtilis, were amplified and spiked into the RNA samples prior to amplification. Microarray data were normalized, and “present,” “marginal,” and “absent” calls were made with the microarray suite v.5 software (MAS5) algorithm in the Affymetrix package. A probe was removed if it was not expressed in all family members. In fact, the MAS5 detection call (present or absent) was used for the removal of 23,914 probe sets (from a total of 54,675 probe sets) that were not reliably detected in all samples.
Principal component analysis (PCA) and hierarchical clustering (HCL) were applied for the comparison of the whole-genome gene-expression profiles among the different samples; median intensity, HCL and K-Medians Support (KMS) were used for the generation of clusters of genes that frequently group together across multiple runs and that have a threshold of 80% of occurrence in the same cluster.
t Test
Gene expressions were measured with the GeneChip Human Genome U133 Plus 2.0 Array. A Welch's t test was performed with 500 permutations. We set the false discovery rate (FDR) to 5 or the p value to 0.01, and we used standard Bonferonni corrections. The HCL between two affected individuals and controls was based on Pearson correlation and complete linkage clustering.
Significance Analysis of Microarrays
Differential expression between affected members and controls was investigated with the use of significance analysis of microarrays (SAM14). Dynamic tuning of delta parameters and the FDR give the proportion of genes likely to have been identified by chance as being significant. We performed two types of testing: (1) two-class unpaired testing, in which samples fell in one of two groups and the subjects were different between the two groups; and (2) two-class paired testing, in which samples were paired on a one-to-one basis.
Integration of Mapping and Expression Data
On the remaining dataset, we used two filtering methods. First, we looked for probe sets with a signal more than 1.5× lower in affected members than in controls; we selected 862 significant genes, of which 76 were located in the autozygous and linked regions on chromosomes 4 and 13. Second, 3,000 probe sets were selected by a maximal standard-deviation filter. Five highly significant genes came out in all statistical analyses.
Real-Time PCR
Real-time PCR (RT-PCR) in cDNA isolated from control and TMEM165-deficient cell lines was performed in triplicate with SYBR green for the detection and quantification of PCR products on a 7500 RT-PCR system (Applied Biosystems). Hypoxanthine-guanine phophoribosyltransferase (HPRT) was used as a reference for the calculation of 2-ΔΔCt.
Mass-Spectrometry Analysis of N- and O-Linked Glycans Released from Total Plasma Proteins
Analysis of N- and O-linked glycans on plasma glycoproteins was carried out by surface-enhanced laser desorption/ionization time-of-flight (TOF) mass spectrometry (Ciphergen) essentially as described by Foulquier et al., 2007 for matrix-assisted laser desorption/ionization TOF mass spectrometry (MALDI-TOF-MS). Analysis of acidic N-linked glycans was performed in linear negative-ion mode. The total N-linked glycans from plasma proteins were released enzymatically by PNGase. For O-linked glycans, the permethylated derivatives were analyzed in positive-ion reflective mode as [M+Na]+.The reference samples were anonymous plasma samples from individuals without CDG or a lysosomal-storage disease.
Antibodies
Anti-GM130 (mab) and anti-TGN46 (pab) were purchased from BD Biosciences. Anti-TMEM165 (pab) was from Sigma Aldrich. Anti-Lamp-2 was purchased from Santa Cruz Biotechnology. Anti-B4GALT1 (GalT) was a kind gift from E. G. Berger (University of Zürich, Zürich, Switzerland).
Immunoblotting
For TMEM165 immunoblotting, fibroblasts cells (8 × 106 cells per sample) were lysed directly in lithium dodecyl sulfate (LDS) loading buffer (Invitrogen). Samples (20 μl) were separated on 4%–12% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose membranes (GE Healthcare). The membrane was blocked in blocking buffer (5% milk powder in TBS-T [1X TBS with 0.05% Tween20]) for 1 hr at room temperature, incubated for the same amount of time with the primary rabbit anti-TMEM165 antibody (HPA038299, Sigma-Aldrich; used at a dilution of 1:2,000) in blocking buffer, and washed three times for 5 min in TBS-T. The membranes were then incubated with the peroxidase-conjugated secondary goat anti-rabbit antibody (P0448, Dako; used at a dilution of 1:10,000) in blocking buffer for 1 hr at room temperature and later washed three times for 5 min in TBS-T. Signal was detected with chemiluminescence reagent (ECL, PerkinElmer) on imaging film (GE Healthcare).
Immunofluorescence Microscopy
Cells were grown on glass coverslips for 12–24 hr, washed once with PBS, and fixed by being incubated for 25 min with 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.2) at room temperature. The coverslips were rinsed twice with 0.1 M glycine in PBS for 15 min. The fixed cells were incubated with primary antibodies for 1 hr.7 After being washed with PBS, Alexa 488- or Alexa 568-conjugated secondary antibodies (Molecular Probes) diluted in blocking solution were applied for 1 hr. Immunostaining was detected through an inverted Leica TCS-SP5 confocal microscope. Data were collected with Adobe Photoshop 7.0 (Adobe Systems).
Lectin Binding and Flow-Cytometry Assay
Control and siTMEM165-transfected HEK cells were harvested via pipetting, washed in PBS, fixed with 4% paraformaldehyde in PBS on ice for 1 hr, and then incubated in 50 mM NH4Cl on ice for 15 min. After two washes with PBS, cells were permeabilized with 0.5% triton and 0.1% BSA for 10 min at room temperature. Cells were then washed once in PBS and resuspended to a final concentration of 1.106 cells/ml in PBS containing 0.1% BSA. All washing and incubation steps were carried out in the buffered BSA solution. Forty micrograms of fluorescein isothiocyanate (FITC)-labeled Sambucus nigra (SNA) was added to 100 μl cell-suspension aliquots of 5 × 104 cells. SNA lectin was purchased from Vector Laboratories. Incubations were carried out in the dark for 30 min with occasional agitation. In addition, the degree of specificity of SNA-lectin binding was determined by incubation with 200 mM lactose. This adequate competitive sugar was incubated with SNA lectin during the incubation time. Flow-cytometry analysis was performed on a Becton Dickinson FACSCalibur flow cytometer. Immediately after incubation with lectin, cells were resuspended in 500 μl of Isoton solution (Becton Dickinson) and submitted to cytometry analysis. Each analysis was performed on 20,000 events gated on the region of the HEK cell population on the basis of light-scatter properties (forward scatter [FSC] versus side scatter [SSC]). Fluorescence intensity data from the FL1 (green fluorescence) channel were collected and analyzed with the Cell Quest Pro software.
RNA Interfering Experiment
Control and TMEM165 siRNA (Dharmacon Smart Pools) were transfected into HEK cells at a final concentration of 50 nM with the use of Lipofectamine 2000 according to the manufacturer's (Invitrogen) protocol with slight modifications. Cells were tranfected in OptiMEM medium in the absence of fetal bovine serum. Three cycles of transfections were done every 48 hr for the achievement of maximal knockdown. Cells were transfected with corresponding siRNAs and collected for analysis 7 days later.
Results
Clinical Presentation
The index case (case 1), a boy, is the first child of Georgian Jewish parents (Figure 1A). Parental consanguinity was denied. The boy was mainly affected by growth retardation (he was unresponsive to growth hormone, which eventually led to severe dwarfism; he is now 19 years old), psychomotor retardation (he began independently walking at 2 years of age), midface hypoplasia, muscular weakness, fat excess, joint laxity, and hepatosplenomegaly. He showed increased serum transaminases (the level of aspartate transaminase [AST] [195–357 U/l; nl < 32] was higher than that of alanine transaminase [ALT] [43–54 U/l; nl < 31] and creatine kinase [1,376–1915 U/l; nl < 145) and partial growth-hormone deficiency. Upon radiological examination, osteoporosis (MIM 166710) and significant epiphyseal, metaphyseal, and diaphyseal dysplasia were evident (Figure 1A). A brain magnetic resonance image [MRI] revealed white-matter abnormalities and relative hypoplasia of the pituitary gland.
Figure 1.
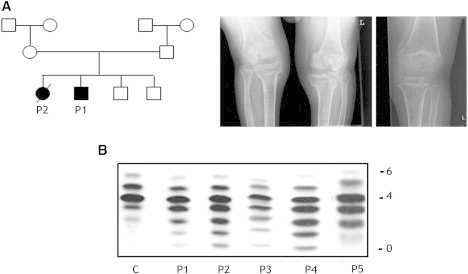
Family tree, Presentation of Radiological Findings, and sTf-IEF Pattern
(A) The family of cases 1 and 2 is the index family in this study. Radiological image of a knee in case 1 at the age of 17 years (left panel) and of case 3 at the age of 3.5 years (right panel). Note severe osteopenia, a very thin bone cortex, broad metaphyses, and significant dysplasia of the epiphyses.
(B) sTf-IEF pattern from a control and from the individuals under investigation. The number of negative charges is indicated on the right.
Case 2, the sister of case 1, presented with similar clinical features. In addition, she had recurrent, unexplained fever episodes and died at 14 months of age from an acute infectious shock.
Case 3, the fourth child of an unrelated Georgian Jewish family, showed the same clinical, biochemical, and radiological features as cases 1 and 2. He had unexplained fever episodes and transient epilepsy that responded well to antiepileptic medication, and he needed artificial ventilation (via tracheostomy) up to the age of 4 years because of an unexplained restrictive lung pathology. Growth-hormone treatment had no effect on his growth, and now, at the age of 4 years, he has severe dwarfism.
Case 4 is a Turkish boy. Only limited information is available on this person. At the age of 9 years, his psychomotor level is that of a 4-year-old. His growth is normal. There is no dysmorphy except for mild rhizomelia, no hepatosplenomegaly, and no epilepsy. He has no clear skeletal anomalies.
Case 5 is an American girl. She presented with a short stature, facial dysmorphy, wrinkled skin (MIM 278250), abnormal fat distribution, and dysplastic toenails. She had amelogenesis imperfecta and skeletal abnormalities, including osteoporosis, anterior beaking of lumbal vertebrae, dysplastic vertebrae and ribs, dysplastic fourth metacarpals and metatarsals, hypoplasia of femoral heads, and kyphoscoliosis (a comparison of the main clinical phenotypes is given in Table 1).
Table 1.
Comparison of Clinical Phenotypes
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | |
|---|---|---|---|---|---|
| Ethnicity | Jewish | Jewish | Jewish | Turkish | American |
| Consanguinity | − | − | − | + | ND |
| Lethality | − | 14 months | − | − | − |
| Gestational age | 40 weeks | 38 weeks | 40 weeks | at term | ND |
| Length at birth | P25 | P3 | P3–P25 | P3–P25 | ND |
| Head circumference at birth | P50 | P50 | ND | P3–P25 | ND |
| Acquired microcephaly | + | + | + | + | ND |
| Growth retardation | +++ | ++ | +++ | + | + |
| Dysmorphy | + | + | + | + | + |
| Hypotonia | + | + | + | + | ND |
| Convulsions | ND | EEG abnormal | + | ND | ND |
| Brain MRI | abnormal | abnormal | abnormal | ND | ND |
| Eye abnormalities | + | + | ND | ND | + |
| Hepatomegaly | + | + | + | + | ND |
| Failure to thrive | + | + | + | + | + |
| Skeletal dysplasia | + | + | + | ND | + |
| Renal abnormalities | − | − | − | HUS | ND |
| Periods with hyperthermia | − | + | + | − | − |
| Feeding problems/ pseudo-obstruction | + | + | − | ND | ND |
| Thrombopenia | borderline | + | + | + | ND |
| CK elevations in blood | + | + | + | + | ND |
| Transaminases (AST/ALT) | +/− | +/− | +/+ | +/+ | ND |
The presence (+) or absence (−) of each feature is reported for each individual. The following abbreviations are used: +++, severe; ++, moderate; +, mild; ND, not done; P, percentile; EEG, electroencephalography; MRI, magnetic resonance image; HUS, haemolytic uremic syndrome (MIM 235400); CK, creatine kinase; AST, aspartate transaminase; and ALT, alanine transaminase.
The diagnosis of CDG-II in these cases was established by standard sTf-IEF showing a marked increase of asialotransferrin, monosialotransferrin, disialotransferrin, and trisialotransferrin in all cases (Figure 1B).
Structural Analysis of N- and O-Glycans and Golgi Morphology
Because of the type 2 sTf-IEF pattern, we investigated the Golgi glycosylation in more detail; we determined the structures of the N- and O-linked glycans on serum glycoproteins from controls and affected individuals by using mass spectrometry (Figure 2). In the affected individuals, there is a relative increase in the undersialylated and undergalactosylated glycans (NeuAc1Hex4HexNAc4 was at m/z 2,227, Hex5HexNAc4 was at m/z 2,040, and Hex4HexNAc4 was at m/z 1,836). The presence of these abnormal ions points to a slight defect in both sialylation and galactosylation and corroborates the IEF data. Similar results have been obtained for case 5.15 To test for an O-glycan deficit, we permethylated, purified, and analyzed the O-glycans by MALDI-TOF-MS (Figure S1, available online, represents O-glycans from affected individuals and control serum). A comparison of the glycan profiles for the affected individuals and controls revealed no differences in the relative amounts of glycans, especially in Hex1HexNAcitol (m/z 534). These results are consistent with the normal ApoC-III profile and prove the absence of an O-glycan defect.
Figure 2.
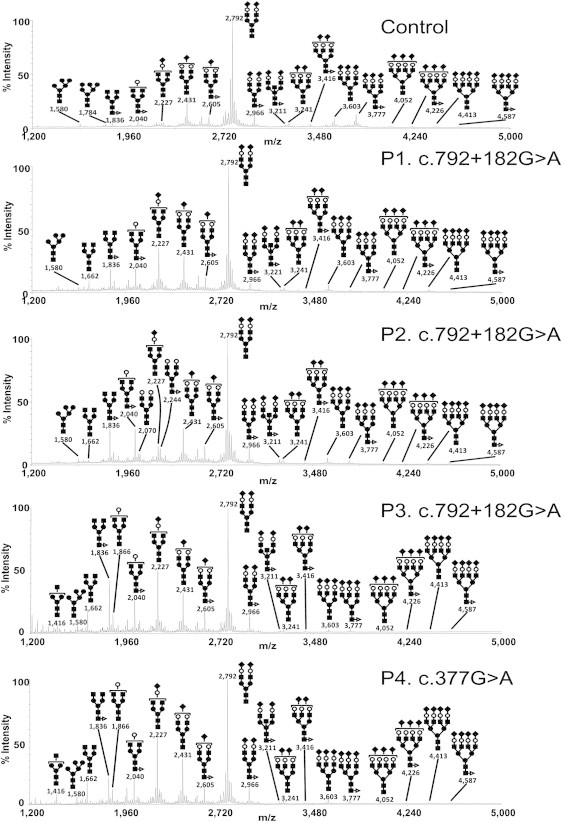
Protein N-Glycosylation Deficiencies
MALDI-TOF-MS spectra of the permethylated N-glycans from sera of control and TMEM165-deficient individuals. The symbols representing sugar residues are as follows: closed square, N-acetylglucosamine; open circle, mannose; closed circle, galactose; open diamond, sialic acid; and closed triangle, fucose. Linkages between sugar residues have been removed for simplicity.
Because the N-glycan results point to a Golgi glycosylation defect, the Golgi morphology was investigated. Staining with Golgi markers TGN46 and GM130 detected significant alterations in Golgi morphology in fibroblasts of all affected individuals. Compared to that in controls, the Golgi apparatus in all affected individuals was found dilated, and the trans-Golgi network was markedly fragmented (Figure 3, see arrows).
Figure 3.
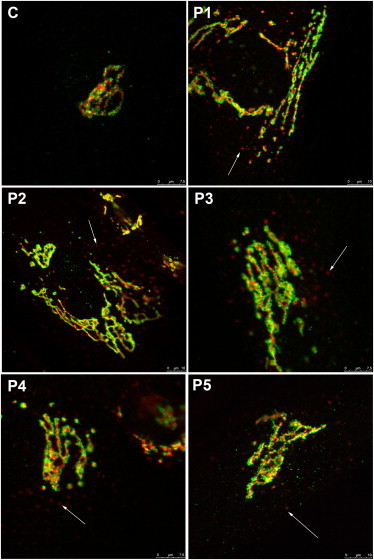
Alterations in Golgi Structure
Golgi localization of GM130 (green) and TGN46 (red) in control and TMEM165-deficient fibroblasts. The cells were double labeled with antibodies against GM130 and TGN46 and visualized by confocal microscopy. The arrows point to fragmented Golgi.
Mapping and Gene Identification
Autozygosity mapping in the first family (cases 1 and 2; Figure 1A) identified two homozygous regions on chromosomes 4 and 13 (Figure S2A). According to the haplotype data from the affected individuals, the interval on chromosome 4 was delineated by the SNP markers rs6828553 and rs1505663 and comprised 18 Mb of genomic DNA, and the interval on chromosome 13 was delimited by the SNP markers rs10507629 and rs9318228 and comprised 14.8 Mb of genomic DNA. Both autozygous loci were confirmed by multipoint linkage analysis and had a Zmax score of 3.6 (Figure S3). The two regions together contain approximately 140 genes.
To reduce the number of candidate genes, we analyzed their expression in two affected members, two unaffected members, and one unrelated control by using total RNA from primary fibroblasts. We assayed the differential expression between affected members and the three controls by using Affymetrix arrays, and we evaluated them by using a Student's t test and SAM,14 KMS, and HCL with two-class unpaired and two-class paired testing (Figures S2B, S2C, and S4). Five highly significant genes that came out in all statistical analyses were sequenced at the cDNA level, and the result revealed the absence of exon 4 in the predicted TMEM165 transcript and the insertion of an aberrant 117 bp fragment derived from intron 4. Sequencing of the coding sequence and the flanking intronic regions of TMEM165 and of the alternative exon at the genomic level revealed a homozygous mutation, c.792+182G>A, in both affected siblings (Figure 4A, left panel). The mutation activates a cryptic splice donor site, which leads to two different transcripts: the wild-type one and an additional one resulting in the replacement of exon 4 with a 117 bp intronic sequence (Figure 4B). The splice prediction program of fruit flies showed that the normal splice donor has a strength of 0.99, whereas the cryptic splice donor site has a strength of 0.5 without mutation and 0.94 with mutation c.792+182G>A. If translated, this aberrant transcript would lead to a protein shortened by 94 amino acids and have 27 amino acids changed at the C-terminal part (Figure 4C).
Figure 4.
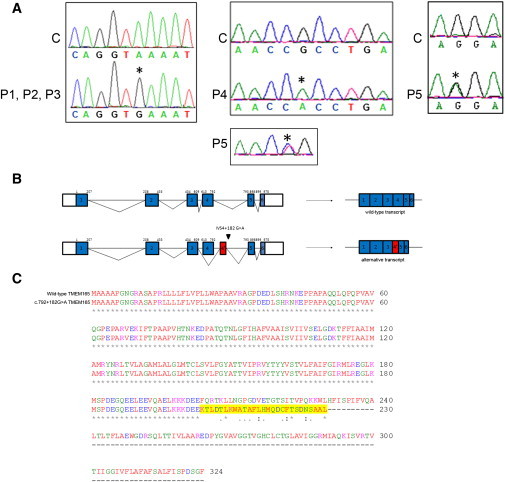
TMEM165 Mutations
(A) In the left panel is the sequence alignment of the genomic DNA fragment from a control C and cases P1, P2, and P3 homozygous for the G>A transition at position c.792+182. In the middle and right panels is the sequence alignment of the cDNA fragment from a control C and cases P4 and P5. P4 is homozygous for the G>A transition at position c.377, and P5 is compound heterozygous for the C>T transition at position 376 and the G>A transition at position 910. Nucleotides affected by the mutations and the predicted effects on protein length are indicated above each sequence.
(B) Schematic representation of the predicted genomic (top) and cDNA (bottom) structure of TMEM165 in the control and cases P1, P2, and P3. Coding exons are shown in blue boxes, and introns (not drawn to scale) are represented by lines. The beginning and end of the open reading frame are labeled; the numbering above each exon is based on coding nucleotides. The red box represents the alternative exon 4 (4′) used in cases P1, P2, and P3.
(C) Predicted effect of the c.792+182G>A mutation on TMEM165. Sequence alignment was compared between wild-type TMEM165 and mutant TMEM165. The amino acids that changed as a result of the mutation are highlighted in yellow. The mutated TMEM165 is truncated and shortened by 94 amino acids.
Sequencing of TMEM165 in a series of 30 individuals with unsolved CDG-II allowed us to identify three other individuals with a TMEM165 mutation. Case 3 (P3) is a Jewish boy whose parents originated from the same region as the index case, and we detected the same homozygous intronic c.792+182G>A mutation in his DNA. Case 4 (P4), who is from a consanguineous Turkish family, is homozygous for a missense mutation (c.377G>A [p.Arg126His]) (Figure 4A, middle panel). Case 5 (P5) is compound heterozygous for a distinct missense mutation (c.376C>T [p.Arg126Cys]) in the same codon and another missense mutation (c.910G>A [p.Gly304Arg]) (Figure 4A, right panel). The arginine at position 126 and the glycine at position 304 are phylogenetically strictly conserved and are likely to be pathogenic (Figure S5B).
Topography of TMEM165
TMEM165 is predicted to encode a 324 amino acid protein that seems to be ubiquitously expressed (Figure S6). SignalIP predicts that this protein contains a signal-peptide cleavage site at position 33 (Figure S5A). If the signal peptide is removed, ConPredII methods predict six transmembrane domains and a luminal N-tail. Sequence comparison of TMEM165 revealed a high conservation of at least ∼230 residues throughout evolution, and putative orthologs could be identified in virtually all eukaryotes and in many bacteria (>300), including cyanobacteria, actinobacteria (e.g., Mycobacteria), and betaproteobacteria (e.g., Burkholderia), indicating that TMEM165 has a very ancient function. All of these putative proteins belong to the UPF0016 family of integral membrane proteins of unknown function; this family usually contains two copies of a region that includes an EXGDK/R motif (see alignment in Figure S5B) and that is flanked by two hydrophobic regions containing 16–20 residues on the N-terminal side and 8–9 residues on the C-terminal side. Both repeats also comprise additional hydrophobic regions of approximately 25–28 and 16–19 residues.
Functional Analysis of TMEM165 Mutations
We used quantitative RT-PCR (qRT-PCR) to investigate the effects of the mutations on TMEM165 expression by comparing TMEM165 cDNA levels amplified from mRNA isolated from control and TMEM165-deficient fibroblasts cultured in the presence or absence of the translation inhibitor puromycin. Compared with controls, case 4, who harbors the missense mutation, had a normal expression level, whereas affected individuals with the splice mutation c.792+182G>A and affected individual P5 with the compound heterozygote mutations showed a clear reduction in expression of the wild-type transcript (Figure 5A, left panel). The aberrant transcript is detectable only in small amounts in controls, whereas its expression level is high in all affected individuals harboring the splice mutation c.792+182G>A (Figure 5A, right panel). Culturing control and TMEM165-deficient fibroblasts in the presence of puromycin did not drastically change the expression level except for in individual P5, in whom the amount of wild-type transcript was restored. However, in the three affected individuals with the splice mutation, a high increase in the amount of alternative transcript was observed, suggesting that the aberrant splice variant is subject to nonsense-mediated decay.
Figure 5.
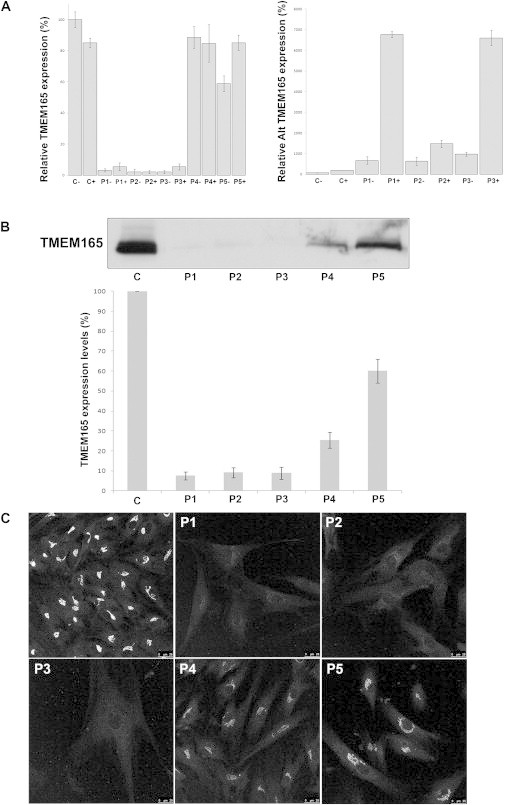
Functional Analyses of TMEM165 Mutations
(A) Quantification of the wild-type (left panel) and alternative (alt) TMEM165 transcripts (right panel) in controls and affected individuals by qPCR with (+) and without (−) puromycin. Expression is measured relative to a housekeeping gene. Values plotted with a wild-type control group untreated with puromycin are set to 1. Error bars represent ± SEM. Expression of TMEM165 in control and TMEM165-deficient fibroblasts: steady-state levels of expression (B) and intracellular distribution (C).
(B) Immunoblots of whole-cell lysates from control (C) and TMEM165-deficient (P) fibroblasts. Actin levels were used as a loading control.
(C) Control and TMEM165-deficient fibroblasts were processed for immunofluorescence microscopy with TMEM165 antibodies. For comparison, images were collected under identical settings.
To investigate the effect of the mutations on TMEM165, we performed immunoblotting in control and TMEM165-deficient fibroblasts. As shown in Figure 5B, an extremely low level of full-length TMEM165 was detected in affected individuals with the truncating mutation. However, no shortened protein was observed, suggesting that the truncated protein, if translated, was unstable and prone to degradation. Moreover, only 25% of the immunoreactivity was recovered in case 4 with the missense mutation, suggesting that the mutant protein is also unstable (Figure 5B). In case 5, only a slight decrease in TMEM165 stability was observed.
We next compared the subcellular localizations of wild-type and mutated TMEM165 in fibroblasts by confocal microscopy. At the steady state, TMEM165 shows a clear perinuclear Golgi-like distribution (Figure 5C). As expected from immunoblot analysis, the TMEM165-deficient individuals showed a much weaker signal than did the controls. However, a faint Golgi localization could be seen in all affected individuals (Figure 5C).
Subcellular TMEM165 Localization
As depicted above, TMEM165 displays a perinuclear Golgi-like distribution. In order to gain insight into its precise Golgi localization, we used the microtubule-depolymerizing agent nocodazole, which has been shown to disrupt the Golgi complex. Nocodazole treatment is known to disperse Golgi markers into discrete puncta and has therefore been used for the localization of proteins to distinct Golgi subcompartments.16,17 Double labeling of nocodazole-treated cells with cis-Golgi (GM130) and trans-Golgi (GalT) markers showed clear, although incomplete, segregation of the two markers (Figure 6). After nocodazole treatment, TMEM165 showed a relatively low degree of colocalization with GM130 but a rather complete colocalization with GalT. The level of colocalization was quantified as the area occupied by the colocalizing spots in the image as a percentage of the total labeled spots. Consistent with the qualitative data, these results showed a much higher degree of colocalization for TMEM165 versus GalT (73 ± 4%) than for TMEM165 versus GM130 (37 ± 8%). These results suggest that TMEM165 is present in the late Golgi (trans) rather than in the early Golgi (cis and median).
Figure 6.
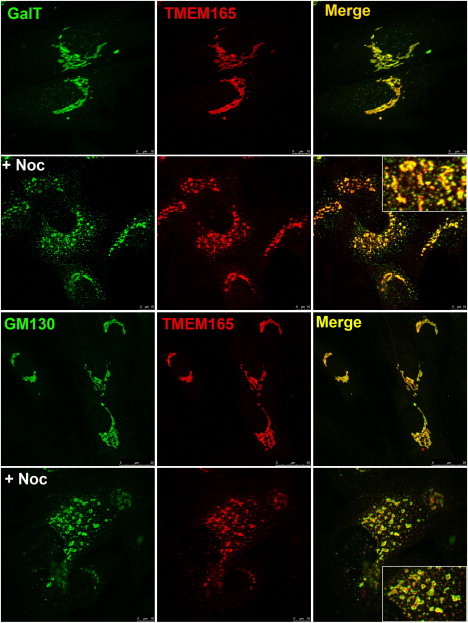
Subcellular Localization of Wild-Type TMEM165
Indirect double immunofluorescence staining of control fibroblasts treated in the absence and presence of nocodazole (+ Noc). The cells were then double labeled with antibodies against TMEM165 and with antibodies against either GalT or GM130. The cells were then examined by confocal microscopy. In the presence of nocodazole, close examination of the overlays shows that the colocalization of TMEM165 with GalT is more complete than it is with GM130.
Analysis of Glycosylation Defects
The impact of TMEM165 on Golgi glycosylation was studied in TMEM165-deficient fibroblasts. Surprisingly, we did not find a general glycosylation deficiency (data not shown). To further investigate this link, we used siRNA to study whether defective TMEM165 was sufficient to cause a Golgi glycosylation deficiency. To this goal, HEK cells were treated with TMEM165 siRNA for 7 days. As shown in Figure 7A, compared with controls, HEK cells treated with siRNA showed a strong (>90%) decrease in the level of TMEM165 after 7 days, whereas actin protein levels remained unchanged. This has been confirmed by immunofluorescence microscopy in which only a few cells showed a Golgi staining (Figure 7B) and by fluorescence-activated cell sorting (FACS) analysis (data not shown).
Figure 7.
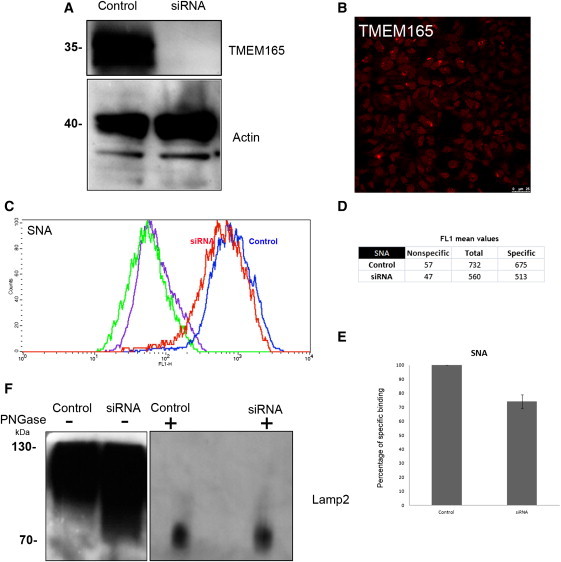
Depletion of TMEM165 Slightly Affects Golgi Glycosylation
(A) HEK cells were transfected with nontarget siRNA (“Control”) or siRNA targeted to TMEM165 (“siRNA”). Seven days after transfection, cells were lysed and equivalent amounts of homogenate were subjected to SDS PAGE and immunoblotted with antibodies against TMEM165 and actin.
(B) Indirect immunofluorescence staining of the siRNA TMEM165 HEK cells with the use of anti-TMEM165.
(C) HEK cells were transfected with nontarget siRNA (“Control”) or siRNA targeted to TMEM165 (“siRNA”). Seven days after transfection, cells were harvested, permeabilized, and incubated with FITC-labeled SNA for flow-cytometry analysis. Histogram plots of control (blue line) and siRNA-TMEM165-transfected (red line) HEK cells analyzed for SNA binding and incubated with FITC-labeled SNA are shown. For each condition, the fluorescence peak corresponding to SNA binding in the presence of 200 mM lactose is represented (nonspecific binding: green line for control and violet line for siRNA). Specific binding was calculated as the difference between the mean fluorescence intensities of total and nonspecific binding peaks (see D).
(E) The results were expressed as the percentages of specific lectin binding to cells. In the calculation, specific lectin binding to control cells, which corresponds to the difference between the total and the nonspecific-binding peaks, was considered as 100%.
(F) HEK cells were transfected with nontarget siRNA (“Control”) or siRNA targeted to TMEM165 (“siRNA”). Seven days after transfection, cells were lysed (incubated or not with PNGase [1,000 U per sample]), and equivalent amounts of homogenate were subjected to SDS and immunoblotted with antibodies against Lamp2.
To investigate the Golgi glycosylation status in siTMEM165 HEK cells, we used SNA-FITC-conjugated lectin for flow-cytometry analysis with and without 200 mM lactose. For each histogram, the fluorescence peak corresponding to SNA binding in the presence of 200 mM lactose (nonspecific binding) is represented (Figure 7C). Specific binding was calculated as the difference between the mean fluorescence intensities (Figure 7D) of total and nonspecific peaks. Weak fluorescence intensity shifts for specific SNA binding were observed for TMEM165-knockdown HEK cells (compared to controls, they showed a 25% decrease in fluorescence) (Figure 7E).
The observed decrease in sialylation indicates disturbed terminal Golgi glycosylation in TMEM165-depleted cells. To confirm this finding, we determined the steady-state glycosylation status of the lysosomal resident protein Lamp2 (Figure 7F). After 7 days of TMEM165 knockdown, the fact that the gel mobility of Lamp2 was altered indicated the presence of underglycosylated protein species.
To confirm that the increase in gel mobility was caused by defective glycosylation, we subjected both control and TMEM165-knockdown cell lysates to PNGase F treatment (Figure 7F). We found that deglycosylation of all forms of Lamp2 produced a single polypeptide of the same relative molecular mass, indicating that N-glycosylation was affected.
Discussion
We have identified TMEM165 (also called TPARL) as a gene encoding a protein of unknown function. Mutations in this gene are responsible for a previously undescribed type of CDG-II. The identification of the gene was achieved by a combination of homozygosity mapping and expression profiling. In total, four different mutations in TMEM165 were identified. The results point to an important role for TMEM165 in Golgi glycosylation and Golgi morphology maintenance.
The precise role of TMEM165 needs to be further determined, but on the basis of the predicted topology and the comparative phylogeny, some predictions regarding its function can be made. TMEM165 includes seven transmembrane domains—or six if one assumes that a signal peptide is cleaved off. The protein is extremely well conserved during evolution. Almost all orthologs that we could identify are integral membrane proteins containing two copies of an EXGDK/R motif flanked by two hydrophobic regions. Iterated Blast searches indicated that TMEM165 is distantly related to bacterial sodium-phosphate transporters, which, however, do not contain the typical EXGDK/R motif that is present in TMEM165. This finding, as well as an analysis with the ProtFun software, points to TMEM165 as being a transporter.18
The fact that the highly conserved motifs, which are presumably critical for the function of TMEM165, comprise more negative than positive charges suggests that the transported species is a cation, possibly a proton. For comparison, it has been shown that protonatable carboxylic groups of several aspartates and a glutamate and a prosthetic group bound to a lysine via a Schiff base are important for the proton-pumping action for bacteriorhodopsin, the best characterized proton-pump protein. A nearby positively charged arginine residue could be involved in controlling pKa of the asparagine by acting as a central proton donor and acceptor.19
Another argument supporting TMEM165 as a proton pump stems from its expression pattern. It is highly expressed in the lactating mammary gland (BioGPS database). The synthesis of lactose involves the generation of two protons inside the Golgi apparatus; one is generated during the transfer of galactose from UDP-Gal onto glucose, and the other is created during the subsequent hydrolysis of UDP to UMP and inorganic phosphate. Because the lactose concentration is around 100 mM in milk, this represents a significant proton load. A good way of getting rid of this proton load would be a passive transport to the cytosol, where the protons will be “reused” in the course of UDP-Gal synthesis.
A third argument is derived from the genomic organization of the TMEM165 ortholog in certain bacteria. Indeed, bacterial genes are frequently organized in operons and often encode proteins that are functionally related. Inspection of the genomic environment of the bacterial orthologs of TMEM165 indicated that, in most cases, these orthologs were isolated open reading frames. However, the TMEM165 orthologs appeared to belong to the same operon as a putative guanosine deaminase and adenosine deaminase in Ralstonia solanacearum, and it was just downstream of operons encoding these enzymes in several other species (Cupriavidus metallidurans, Burkholderia cepacia, Burkholderia cenocepacia, and Burkholderia pseudomallei). Adenosine deaminase and guanosine deaminase, which are readily protonated at a neutral pH, presumably cause alkalinization of the cytosol. We speculate that the TMEM165 ortholog allows protons to flow in from the extracellular medium and thereby neutralizes the alkaline load. The optimal pH in secretory and endocytic organelles has to be maintained by the balance between active protein pumping and passive proton efflux. It is therefore possible that TMEM165 is responsible for passive proton leakage within the secretory pathway.
The link between the TMEM165 defect and the Golgi glycosylation deficiency is far from solved, and we found some discrepancies. Indeed, although a severe glycosylation deficiency was observed from sTf-IEF, only a slight deficiency was found from a MALDI-TOF analysis of total serum N-glycoproteins. Moreover, a glycosylation analysis of TMEM165-deficient fibroblasts was normal. This could be the result of a functional compensation in (slowly growing) cells in culture, although it should be mentioned that fibroblasts are not good models for the study of glycosylation. We showed that a depletion of TMEM165 in HEK cells does lead to a mild deficiency of glycosylation. Still, the observed hypoglycosylation could be secondary, i.e., the primary molecular defect could reside outside the glycosylation machinery.
The major clinical findings in the individuals with a homozygous splice mutation are severe psychomotor retardation, major skeletal dysplasia, and, in the surviving individuals, pronounced dwarfism. Interestingly, the individuals harboring missense mutations have a milder disease and lack the skeletal phenotype. The possibility that the skeletal phenotype is not due to the mutations in TMEM165 but to a concomitant defect in a neighboring gene cannot be completely excluded. The answer to this question will have to await the development of specific animal knockout models.
Finally, it is not without importance that, in view of the present hype around exome sequencing, the causal mutation in the index family would never have been identified if the affected individuals' samples had been directly subjected to exome sequencing. Hence, we wish to underline that the combination of autozygosity mapping and expression analysis is still useful. Of course, it has limitations, e.g., we were fortunate to deal with a mutation that affects the expression level of the mRNA. It is remarkable that the proposed strategy has rarely been used or reported in the literature.
In conclusion, we have identified a protein that is associated with protein hypoglycosylation and that seems to be of prime importance for Golgi structure, maintenance, and/or pH.
Acknowledgments
We are indebted to Ragna Sannerud for providing advice and tools for the imaging experiments and to Professor Eric Berger, Professor Thierry Hennet, Ellen Reynders, Professor Jack Rohrer, and many others for helpful discussions. The work was supported by grant G.0553.08 from the FWO (Fonds voor Wetenschappelijk Onderzoek, Vlaanderen, Belgium) and grant 12540 from the AFM (Association Française contre les Myopathies, France) to G.M.; a PEPS-CNRS (Projets Exploratoires/Premier Soutien du Centre National de la Recherche Scientifique, France), a Marie Curie Reintegration Grant (VTG-CDG), and an Agence Nationale Recherche Jeune Chercheur to F.F.; grants from the F.R.S.-FNRS (Fonds de la Recherche Scientifique, Wallonie), la Communauté Française de Wallonie-Bruxelles, and la Loterie Nationale (Belgium) to M.V.; the Interuniversity Attraction Poles Program (network 6/05) of the Belgian Science Policy (Belgium) to E.V.S., G.M., and M.V.; grant R01DK55615 and The Rocket Fund to H.F.; the European Commission (Sixth Framework Program, contract LSHM-CT.2005-512131 to EUROGLYCANET) to G.M. and E.V.S.; and the Körber Stiftung to J.J., E.V.S., and G.M. M.A. is a “Scientific Logistics Manager” of the F.R.S.-FNRS. The authors thank the Genomics/Transcriptomics platform of Université catholique de Louvain for performing the Affymetrix chip experiments and the Bioimaging Core Facility of Lille (Institut de Recherche Interdisciplinaire, CNRS).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/
GenBank, http://www.ncbi.nlm.nih.gov/GenBank/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Primer 3, http://frodo.wi.mit.edu
Symatlas, http://symatlas.gnf.org/Symatlas/
References
- 1.Jaeken J., Matthijs G. Congenital disorders of glycosylation: A rapidly expanding disease family. Annu. Rev. Genomics Hum. Genet. 2007;8:261–278. doi: 10.1146/annurev.genom.8.080706.092327. [DOI] [PubMed] [Google Scholar]
- 2.Jaeken J., Hennet T., Matthijs G., Freeze H.H. CDG nomenclature: Time for a change! Biochim. Biophys. Acta. 2009;1792:825–826. doi: 10.1016/j.bbadis.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schachter H., Freeze H.H. Glycosylation diseases: Quo vadis? Biochim. Biophys. Acta. 2009;1792:925–930. doi: 10.1016/j.bbadis.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foulquier F. COG defects, birth and rise! Biochim. Biophys. Acta. 2009;1792:896–902. doi: 10.1016/j.bbadis.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 5.Wu X., Steet R.A., Bohorov O., Bakker J., Newell J., Krieger M., Spaapen L., Kornfeld S., Freeze H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004;10:518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]
- 6.Foulquier F., Ungar D., Reynders E., Zeevaert R., Mills P., García-Silva M.T., Briones P., Winchester B., Morelle W., Krieger M. A new inborn error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1-Cog8 interaction in COG complex formation. Hum. Mol. Genet. 2007;16:717–730. doi: 10.1093/hmg/ddl476. [DOI] [PubMed] [Google Scholar]
- 7.Foulquier F., Vasile E., Schollen E., Callewaert N., Raemaekers T., Quelhas D., Jaeken J., Mills P., Winchester B., Krieger M. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc. Natl. Acad. Sci. USA. 2006;103:3764–3769. doi: 10.1073/pnas.0507685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kranz C., Ng B.G., Sun L., Sharma V., Eklund E.A., Miura Y., Ungar D., Lupashin V., Winkel R.D., Cipollo J.F. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum. Mol. Genet. 2007;16:731–741. doi: 10.1093/hmg/ddm028. [DOI] [PubMed] [Google Scholar]
- 9.Reynders E., Foulquier F., Leão Teles E., Quelhas D., Morelle W., Rabouille C., Annaert W., Matthijs G. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum. Mol. Genet. 2009;18:3244–3256. doi: 10.1093/hmg/ddp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paesold-Burda P., Maag C., Troxler H., Foulquier F., Kleinert P., Schnabel S., Baumgartner M., Hennet T. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum. Mol. Genet. 2009;18:4350–4356. doi: 10.1093/hmg/ddp389. [DOI] [PubMed] [Google Scholar]
- 11.Kornak U., Reynders E., Dimopoulou A., van Reeuwijk J., Fischer B., Rajab A., Budde B., Nürnberg P., Foulquier F., Lefeber D., ARCL Debré-type Study Group Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat. Genet. 2008;40:32–34. doi: 10.1038/ng.2007.45. [DOI] [PubMed] [Google Scholar]
- 12.Nannya Y., Sanada M., Nakazaki K., Hosoya N., Wang L., Hangaishi A., Kurokawa M., Chiba S., Bailey D.K., Kennedy G.C., Ogawa S. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto G., Nannya Y., Kato M., Sanada M., Levine R.L., Kawamata N., Hangaishi A., Kurokawa M., Chiba S., Gilliland D.G. Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of Affymetrix single-nucleotide-polymorphism genotyping microarrays. Am. J. Hum. Genet. 2007;81:114–126. doi: 10.1086/518809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tusher V.G., Tibshirani R., Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miura Y., Hato M., Shinohara Y., Kuramoto H., Furukawa J., Kurogochi M., Shimaoka H., Tada M., Nakanishi K., Ozaki M. BlotGlycoABCTM, an integrated glycoblotting technique for rapid and large scale clinical glycomics. Mol. Cell. Proteomics. 2008;7:370–377. doi: 10.1074/mcp.M700377-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Chavrier P., Parton R.G., Hauri H.P., Simons K., Zerial M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62:317–329. doi: 10.1016/0092-8674(90)90369-p. [DOI] [PubMed] [Google Scholar]
- 17.Ullrich O., Reinsch S., Urbé S., Zerial M., Parton R.G. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demaurex N. pH Homeostasis of cellular organelles. News Physiol. Sci. 2002;17:1–5. doi: 10.1152/physiologyonline.2002.17.1.1. [DOI] [PubMed] [Google Scholar]
- 19.Krogh A., Larsson B., von Heijne G., Sonnhammer E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.