

Background: Multiple functions have been ascribed to NLRC5 including MHC-I transcription and cytokine responses.
Results: We generated Nlrc5−/− mice and showed that Nlrc5 increases classical and nonclassical MHC-I and causes removal of the gene-silencing H3K27me3 histone modification on MHC-I promoter.
Conclusion: Nlrc5 regulates MHC-I expression.
Significance: NLRC5, with CIITA, constitutes an NLR subfamily that regulates MHC.
Keywords: Cytokines/Interferon, Gene Transcription, Major Histocompatibility Complex (MHC), Mouse Genetics, Nod-like Receptors (NLR), CIITA, H2-K, I-A, NLRC5, Tla
Abstract
Most of the nucleotide-binding domain, leucine-rich repeat (NLR) proteins regulate responses to microbial and damage-associated products. Class II transactivator (CIITA) has a distinct function as the master regulator of class II major histocompatibility complex (MHC-II) transcription. Recently, human NLRC5 was found to regulate MHC-I in cell lines; however, a host of conflicting positive and negative functions has been attributed to this protein. To address the function of NLRC5 in a physiologic setting, we generated an Nlrc5−/− strain that contains a deletion in the exon that encodes the nucleotide-binding domain. We have not detected a role for this protein in cytokine induction by pathogen-associated molecular patterns and viruses. However, Nlrc5−/− cells showed a dramatic decrease of classical (H-2K) and nonclassical (Tla) MHC-I expression by T/B lymphocytes, natural killer (NK) cells, and myeloid-monocytic lineages. As a comparison, CIITA did not affect mouse MHC-I expression. Nlrc5−/− splenocytes and bone marrow-derived macrophages were able to up-regulate MHC-I in response to IFN-γ; however, the absolute levels of MHC-I expression were significantly lower than WT controls. Chromatin immunoprecipitation of IFN-γ-treated cells indicates that Nlrc5 reduced the silencing H3K27me3 histone modification, but did not affect the activating AcH3 modification on a MHC-I promoter. In summary, we conclude that Nlrc5 is important in the regulation of MHC-I expression by reducing H3K27me3 on MHC-I promoter and joins CIITA as an NLR subfamily that controls MHC gene transcription.
Introduction
NLR3 proteins conduct a variety of functions, including inflammasome-associated function, NF-κB/MAPK activation, interferon (IFN) regulation, cell death, and autophagy (1). These functions are primarily elicited in response to pathogen-associated molecular patterns (PAMPs) or damaged-associated molecular patterns (DAMPs). In contrast, the class II transactivator (CIITA) has remained the only NLR that has been verified in both human and mice as a master transactivator of all major histocompatibility II (MHC-II) genes as well as genes encoding their accessory proteins, such as the invariant chain and H-2DM proteins (2). CIITA is shown to conduct its function by association with the DNA-binding proteins, NF-Y and Regulatory Factor X (RFX), which recognize their cognate binding motifs, the X and Y elements that are conserved in all MHC-II promoters (3, 4). The assembly of this enhanceosome is necessary for the modification of MHC-II promoters via histone acetylation and methylation. Additionally, CIITA interacts with insulator factor, CCCTC binding factor (CTCF), to assure against gene silencing (5).
In addition to the regulation of MHC-II, CIITA was simultaneously found by our group and another laboratory to regulate human MHC-I (6, 7). Both groups showed that mutant cell lines with a defective CIITA protein have defective MHC-I expression and that wild type CIITA complemented this defect. Additionally, CIITA mediates MHC-I gene transactivation through a regulatory DNA motif known as the α site in its promoter. Subsequently, others have shown that CIITA activates MHC-I uniquely in a TATA box binding protein (TBP)-associated factor-1-independent fashion (8).
Recently, human NLRC5 was found to positively regulate MHC-I in cell lines (9, 10), although another group obtained the opposite finding and concluded that NLRC5 reduced MHC-I and CD40 expression (11). The former group overexpressed NLRC5 in Jurkat T cells and found that the expression of class I MHC as well as B2M, Transporter associated with antigen processing (TAP), and Large multifunctional protease (LMP) was enhanced, whereas siRNA targeting NLRC5 caused the reduction of MHC-I. Similar to CIITA, they found that the Walker A and B motifs common among ATP-binding proteins are critical for both the MHC-I-inducing function of NLRC5 as well as its nuclear translocation. Furthermore, overexpressed NLRC5 was detected on MHC-I promoter by chromatin immunoprecipitation (ChIP), suggesting a direct binding of NLRC5 to the MHC-I promoter.
NLRC5 is highly expressed by cells in the immune system, in particular by cells of the lymphocytic and myeloid-monocytic cell lineages (11–13). It can be found in the nucleus (9–11), although cytoplasmic localization (10, 14) is also reported, and an overexpressed NLRC5 has been shown to shuttle between the nucleus and cytoplasm. Its expression is highly inducible by IFN-γ and LPS (9, 11, 14), and IFN-γ and CMV induced NLRC5 promoter activation via the Jak/Stat pathway (14).
However, a host of additional functional roles has been attributed to this protein, although a caveat is that many of the functional studies on NLRC5 have been limited to cell lines. Two groups showed a negative regulatory function for NLRC5. Cui et al. (15) demonstrated the interaction of endogenous NLRC5 with endogenous IκB kinases (IKK) α/β to reduce the phosphorylation state of IKKs. They further showed that siRNA directed at NLRC5 resulted in elevated TNF and IL-6 in response to LPS in both human and mouse macrophage cell lines. This group also found that gene knockdown resulted in elevated antivesicular stomatitis virus response including enhanced expression of type-I interferons. In agreement with this, Benko et al. (11) showed an inhibitory effect of NLRC5 overexpression on NF-κB-, AP-1-, and IFN-sensitive response element reporter assays in the mouse macrophage line RAW264, whereas knocking down the gene resulted in an enhancement of IL-1β, IL-6, TNF, CD40, and class I MHC in response to LPS or IFN-γ. By contrast, two other groups showed that NLRC5 positively regulates cytokine responses to nucleic acids and viruses. Kuenzel et al. (14) used a forced dimerization system involving the caspase recruitment domain (CARD) domain of NLRC5 and found elevated gamma interferon activation site (GAS)- and IFN-sensitive response element-driven reporter activation. These authors further found that NLRC5 overexpression moderately enhanced IFN-α and its downstream targets, OAS1 and P58 repressor protein kinase (PRKRIR), whereas siRNA targeting NLRC5 reduced the IFN-α pathway in response to CMV. In agreement with these authors, Neerincx et al. (13) found that siRNA targeting NLRC5 in primary human dermal fibroblasts caused a drop in IFN-γ and Rantes (regulated on activation normal T cell expressed and secreted) in response to poly(I:C) or Sendai virus.
Our group used a gene knockdown approach to show that NLRC5 reduction resulted in the lack of inflammasome activation in THP-1 cells and in primary human peripheral blood monocytic cells and further that NLRC5 can reconstitute an inflammasome system (16). This result is supported by another group that also found that overexpressed NLRC5 can reconstitute an inflammasome (17). However, this same group produced an Nlrc5−/− mouse strain and found that inflammasome activation, cytokine response to LPS and IFN-γ, type-I IFN and cytokine responses to poly(I:C), RNA virus, stimulatory DNA, and DNA viruses were all normal in this strain. Furthermore, in vivo response to poly(I:C) was also normal in Nlrc5−/− mice.
The composite results revealed a rather confusing picture regarding the function of NLRC5, and the study of Nlrc5−/− mice did not identify a positive function (17). To readdress the function of NLRC5, we produced an Nlrc5 gene deletion mouse targeting the NBD-encoding exon. We observed a profound effect of Nlrc5 deletion on MHC-I expression in lymphocytes and NK cells and explored the effect of Nlrc5 on the histone modification of an MHC-I promoter. The deletion of Nlrc5 resulted in a less dramatic reduction of MHC-I on B cells and myeloid-monocytic cells, leading us to ask whether residual CIITA might account for the differential effect of Nlrc5 on T/NK, which do not express CIITA versus B cells that do express CIITA. In addition, we also explored possible explanations for the discrepant finding where others describe a role for Nlrc5 in regulating cytokine production.
EXPERIMENTAL PROCEDURES
Animal Care and Use
C57BL/6J, Ciita−/− (18), and Nlrc5−/− mice were bred in-house in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Generation of Nlrc5−/− Mice
The Nlrc5−/− NBD was ablated as described in the legend for Fig. 1. Exons 3 and 4 were replaced with the neomycin cassette, and the targeting vector was confirmed by restriction digest analysis and sequencing. The linearized vector was electroporated in IC1 (C57BL6/N) embryonic stem cells (InGenious Targeting Laboratories). Homologous recombinants were verified by Southern blot analysis before injection into BALB/c blastocysts. The resulting chimeras were mated to female C57BL/6J mice, and the resulting heterozygotes were crossed to yield homozygous Nlrc5−/− mice, which were screened using duplex PCR with each reaction containing the following primers: P1 WT NBD primer, 5′-TAGAGTTGTATCACTGTGTGGCTGAGACCC-3′; P2 Neo primer, 5′-GGAACTTCGCTAGACTAGTACGCGTG-3′;P3 common primer, 5′-TTTAGCTGATAGATACAGAGCCTTGGACC-3′, yielding bands of 650 bp for WT and 330 bp for Nlrc5 knock-out.
FIGURE 1.
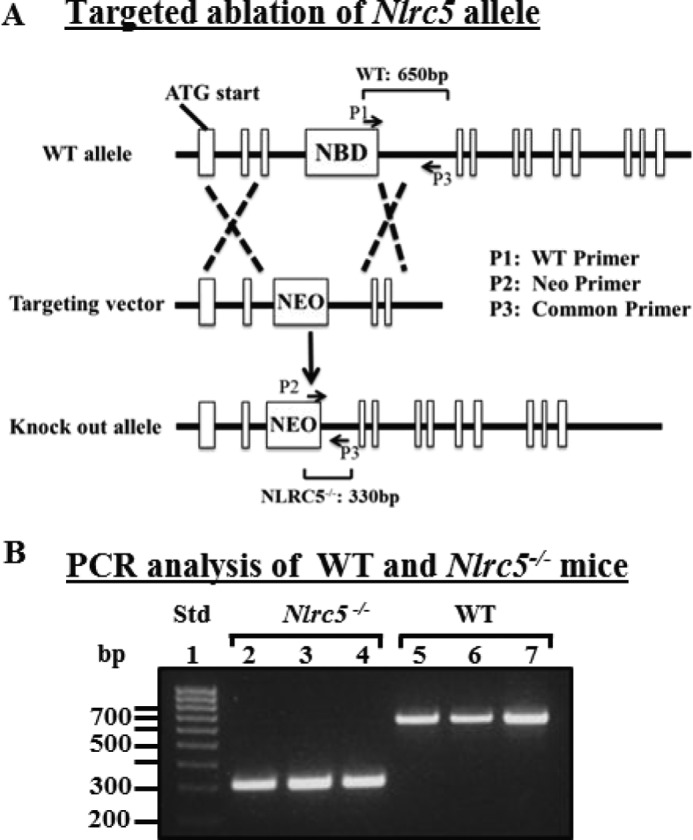
Targeted ablation of the Nlrc5 NBD coding region. A, targeting construct with ∼5-kb region of homology in the 5′ end (targeting the NBD coding region and upstream exon) and ∼2.3-kb homology 3′ to the NBD coding region. Primers for duplex PCR genotype analysis are indicated: P1, WT Nlrc5 NBD-specific primer; P2, neomycin-specific primer; P3, common primer. B, PCR products from WT and Nlrc5−/− genomic DNA using primers described in A. Lane 1, DNA standard (Std); lanes 2–4, tail genomic DNA from three Nlrc5−/− mice; lanes 5–7, tail genomic DNA from three WT mice.
Primary Cell Isolation
Single cell splenocyte and thymocyte suspensions were made by gentle dissociation of tissues with a 1-ml syringe plunger over a 70-μm strainer followed by red blood cell lysis using ACK buffer (Invitrogen), and viable cells were counted using Trypan blue staining and stimulated as indicated. Peritoneal macrophages were isolated by seeding peritoneal lavage cells in DMEM (Invitrogen)+10% FCS(HyClone®) at a density of 5 × 105 cells/well in 96-well plates. After 16 h, the nonadherent cells were removed by washing three times with PBS (Invitrogen), and the remaining adherent cells were used for stimulation. Bone marrow-derived macrophages (BMDM) were generated as described previously (19). All cultures were supplemented with 1× penicillin/streptomycin (Sigma).
Viral Infection
BMDM were seeded at 2 × 105 cells/well in flat bottom 96-well plates and infected with herpes simplex virus (HSV) (multiplicity of infection of 0.2, 1, and 10) or SeV (80 HA unit/ml). Supernatant was collected from triplicate wells of virus or mock-infected cells and tested for IFN-β secretion by ELISA.
PAMP, DAMP, and Cytokine Stimulation
BMDM were seeded at 2 × 105 cells/well in flat bottom 96-well plates and were left untreated or stimulated for 8 h with 200 ng/ml Escherichia coli K12 LPS (InvivoGen), 4 μm CpG (InvivoGen), or 50 μg/ml poly(I:C) (InvivoGen). Supernatant was collected for ELISA, and cells were harvested for mRNA transcript analysis. Inflammasome induction was mediated using an LPS prime of 200 ng/ml for 3 h followed by the addition of 400 μg/ml Imject® alum (Pierce) for 6 h or 2 mm ATP (InvivoGen) for 40 min. IFN-γ stimulation of BMDM and splenocytes used 500 units/ml of recombinant murine IFN-γ (PeproTech) at the indicated time points.
Flow Cytometry
The following reagents were used for flow cytometric staining: CD3-FitC, CD19-eFluor®-450, biotin-H2-Kb, biotin-H2-Db, Avidin-APC, CD11c-PE-Cy5, and CD4-PE (eBioscience); NK1.1-PE-Cy7 and CD8-Pacific Blue (BioLegend); I-Ab-PE (Pharmingen); Live/Dead® Aqua and Violet (Molecular Probes); CD45-Pacific Orange (Invitrogen); CD11b-PE (Beckman Coulter). All data were collected on CyAnTM (Beckman Coulter) or LSRII (BD Biosciences), and sorting was performed using MoFloTM or MoFloTM XDP (Beckman Coulter). All data were analyzed using FlowJo (TreeStar).
ELISA
Detection of murine TNF-α, IL-6, and IL-1β was performed using BD OptEIATM kits (BD Biosciences) according to the manufacturer's instructions. IFN-β ELISA utilizes the following reagents: 4 μg/ml rat anti-mouse IFN-β capture antibody(Yamasa Corp.); polyclonal rabbit anti-mouse IFN-β (1:2000) (R&D Systems); goat anti-rabbit IgG HRP (Cell Signaling Technology), which was used at 1:2000. Standard curve for IFN-β was generated using purified mouse IFN-β (PBL, Inc.).
Quantitative Real-time PCR
Total RNA was harvested using TRIzol® (Invitrogen) followed by reverse transcription using M-MLV (Invitrogen) and oligo(dT) according to manufacturer's instructions. Q-RT-PCR for murine Tnfa, Il6, and Il1b was normalized to Gapdh using SYBR®-Green (Applied Biosystems) and the following primer sets: osTnfa, 5′-CCCTCACACTCAGATCATCTTCT-3′, oaTnf, 5′-GCTACGACGTGGGCTACAG-3′; osIl6, 5′-CAACCACGGCCTTCCCTACTT-3′, oaIl6, 5′-CACGATTTCCCAGAGAACATGTG-3′; osIl1b, 5′-CAACCAACAAGTGATATTCTCCATG-3′, oaIl1b, 5′-GATCCACACTCTCCAGCTGCA-3′; osGapdh, 5′-CCTCGTCCCGTAGACAAAATG-3′, oaGapdh, 5′-TCTCCACTTTGCCACTGCAA-3′. Q-RT-PCR for murine H2-k1, B2m, Tla, Cd1d, I-Ab, and Nlrc5 was normalized to 18 S RNA using TaqMan® primer/probe sets (Applied Biosystems).
ChIP Assay
Total splenocytes were plated at a cell density 10 × 106, stimulated with IFN-γ as indicated (500 units/ml), but only nonadherent lymphocytes were used for ChIP analyses as described previously (20, 21). Following cytokine stimulation, cells were cross-linked with 1% formaldehyde for 10 min. Cross-linking was stopped by the addition of 0.125 m glycine for 5 min. Cells were lysed in SDS lysis buffer and were sonicated to generate an average of 500 bp of sheared DNA. Sonicated lysates were precleared with salmon sperm-coated agarose beads (Millipore), and then lysates were immunoprecipitated overnight with 5 μg of indicated antibody for H3k27me3 (Abcam), acetylated H3 (Millipore), H3 (Millipore), and isotype control antibody (Millipore). Following a 2-h immunoprecipitation with 50 μl of salmon sperm-coated agarose beads, samples were washed with each of the following buffers: low salt buffer, high salt buffer, LiCl, and Tris-EDTA, and samples were eluted with SDS elution buffer. Following elution, cross-links were reversed overnight with 5 m NaCl at 65 °C, and immunoprecipitated DNA was isolated using phenol:chloroform:isopropyl alcohol mix (Invitrogen) as per the manufacturer's instructions. Isolated DNA was analyzed by real-time PCR using primers spanning the proximal promoter region of H2K1, forward, 5′-CCGCGGACGCTGGATA-3′, reverse, 5′-GGCGATTCGCGACTTCTG-3′, and probe, 5′-FAM-AGTCCACGCAGCCCGCAGAACT-TAM-3′; I-Ab forward, 5′-CTGGATGCTTCCTGAGTTTGG-3′, reverse, 5′-TGTGTTTTACTACAGCTATGTTTTGCA-3′, and probe, 5′-FAM-CAATTGGCAAGCTTTGACCCCCAA-TAM-3′; and GAPDH forward, 5′-GAGCGGCCCGGAGTCT-3′, reverse, 5′-GGATTACGGGATGGGTCTGA-3′, and probe, 5′-FAM-AAGTATTAGGAACAACCCCACGCGCC-TAM-3′. Values generated from the Q-PCR reaction were normalized to the total DNA levels (input), and the control IgG levels were subtracted from each immunoprecipitation sample analyzed.
Statistical Analysis and Graphing
Statistical analysis was done using an on-line program through Vassar College, VassarStats: Website for Statistical Computation. Graphing was done using GraphPad Prism®.
RESULTS
Production of an Nlrc5−/− Mouse Strain by Targeted Deletion
We produced an Nlrc5−/− strain by homologous targeted replacement of these residues (AKRPFQSYGSSPRRKNSKKQQL) from exons 3 and the entire exon 4 with the Neo cassette (Fig. 1A). Exons 3 and 4 respectively encode a portion of the CARD-like domain and the entire NBD domain. The targeting construct was generated with an ∼5-kb region of the 5′ end flanking the targeted deletion and an ∼2.3-kb region of the 3′ end flanking sequence. The targeting vector was confirmed by restriction digestion and direct sequencing. The linearized vector was electroporated into IC1 C57BL/6N embryonic stem cells, and homologous recombinants were verified by Southern blot analysis before injection into BALB/c blastocysts. The resulting chimeras were mated to female C57BL/6J mice, and the resulting heterozygotes were crossed to yield homozygous Nlrc5−/− mice. As indicated in Fig. 1B, a common reverse primer, a NEO primer, and an NBD primer generated the expected bands of 650 bp for the WT samples and 330 bp for the gene-targeted samples.
Deletion of Nlrc5 Does Not Affect Cytokine Production in Response to PAMPs and Viruses
As described earlier, previous work has indicated both positive and negative roles for Nlrc5 in cytokine production to PAMPs and viruses (11, 13–15). Additionally, studies in human cells have indicated a role for this protein in inflammasome activation (16, 17). To determine the contribution of mouse Nlrc5 to these processes, we first assessed the production of Tnfa, IL6, and IL-1b mRNA in response to LPS and CpG by BMDM. Cytokine mRNA levels were generally similar between WT and Nlrc5−/− cells with the only statistically significant differences being less than 1.5-fold (Fig. 2A). This is reflected at the protein level for TNF and IL6. For IL-1β production, we additionally tested response to LPS + ATP, or LPS+alum, because IL-1β secretion requires two signals including the activation of pro-IL1β production by LPS and inflammasome activation by ATP or alum to cause the post-translational processing of pro-IL1β to mature IL-1β via caspase-1. Again, deletion of Nlrc5 did not cause a difference. Parallel experiments were performed with LPS-activated resting peritoneal macrophages because BMDM have been expanded in vitro and might generate artificial findings (Fig. 2B). However, no difference was found between WT and Nlrc5−/− peritoneal macrophages. As an additional evaluation, we also tested for the production of interferon β (IFN-β) protein in response to a DNA virus, HSV, and an RNA virus, Sendai virus. No significant difference in cytokine production was observed between WT and Nlrc5−/− BMDM with the exception of the high multiplicity of infection of HSV, which showed a statistically significant, but less than 25% decrease in IFN-β production by Nlrc5−/− cells at 24 h (Fig. 2C). Ifnb mRNA induction by PAMPs including LPS, CpG, and poly(I:C) was similar between WT and Nlrc5−/− BMDM (Fig. 2D). These results confirm those from a previous study, which did not find a detectable role for Nlrc5 in cytokine induction by various PAMPs and microbes (17).
FIGURE 2.

Cytokine expression by WT and Nlrc5−/− macrophages in response to PAMP and DAMP stimulation and viral infection. A and B, Q-RT-PCR for mRNA (top) and ELISA for protein secretion (bottom) of indicated cytokines by BMDM (A) and peritoneal macrophages (B) 8 h after stimulation; data are graphed as -fold change over WT untreated control (UT). Rel. expression, relative expression. C, ELISA for IFN-β secretion by BMDM infected with HSV (left) or Sendai Virus (SeV) (right) at 8 and 24 h after infection. D, Q-RT-PCR for Ifnb mRNA expression in BMDM 8 h after PAMP stimulation. Data in A, B, and D are representative of three independent experiments. Data in C are representative of two independent experiments. Q-RT-PCR data are normalized to 18 S RNA expression. Error bars = S.E.; statistical analysis by two-tailed Student's t test, *, p < 0.05, **, p < 0.01.
Deletion of Nlrc5 Affects Classical and Nonclassical MHC-I Expression but Not CD1 and MHC-II Expression
The work by Meissner et al. (9) showed that in human cell lines, the gene knockdown of NLRC5 caused a reduction of MHC-I expression. Previously, we and others have noted the high levels of NLRC5 gene expression in immune cells, particularly on T and B lymphocytes and NK cells (13, 16). To test this in a physiologic setting, we used CD45 as a marker for all hematopoietic cells and assessed MHC-I (H2-Kb and H2-Db) expression by splenocytes. C57BL/6 lacks the H2-L allele, and thus expression of this gene cannot be tested in this strain. CD45+ hematopoietic cells from Nlrc5−/− mice showed a significant reduction of both H-2K and H-2D expression. Among CD45+ cells, one population has a more dramatic loss of MHC-I than another, as revealed by the two peaks of MHC-I staining in the Nlrc5−/− sample (Fig. 3A). A further separation of the cells shows that splenic T cells showed the most dramatic loss of MHC-I followed by NK cells, B lymphocytes, dendritic cells (CD11bint/CD11c+), and macrophages (CD11b+, CD11clo). To further assess whether this is reflected at the transcriptional level, we sorted different populations, isolated RNA, and tested for the expression of a classical MHC-I gene (H-2k1), the common partner of MHC-I (B2m), and a nonclassical MHC-I gene (Tla). All three were largely reduced in the lymphoid lineage (Fig. 3B). In contrast, Cd1d2, which encodes the CD1 protein that presents lipids and is not generally co-regulated with MHC-I, is not altered in Nlrc5−/− cells. Finally, Nlrc5 deletion did not affect MHC-II gene expression represented by I-A in B cells, but it did cause a slight, but significant, decrease in MHC-II expression in CD11b+ cells. Decreased MHC-I expression in the absence of Nlrc5 is not restricted to the spleen as CD4−CD8− (double negative), as well as CD4 and CD8 single positive thymocytes, in Nlrc5−/− mice all have reduced MHC-I (Fig. 3C). We observed similar MHC-I expression between WT and Nlrc5−/− DP thymocytes, which may reflect low Nlrc5 mRNA expression in the WT population (Fig. 3, C and D).
FIGURE 3.

Flow cytometric and Q-RT-PCR analysis of MHC expression in WT and Nlrc5−/− mice. A, surface H2-Kb and H2-Kd expression in WT (black) and Nlrc5−/− (blue) splenocyte populations. B, Q-RT-PCR for H2-k1, B2m, Tla, Cd1d2, and I-Ab mRNA expression in sorted splenic B, T, and NK populations. Rel. expression, relative expression. C, flow cytometric analysis of H2-Kb expression in WT (black) and Nlrc5−/− (blue) thymocyte populations. % of max, percentage of maximum; DN, double negative; DP, double positive. D, Q-RT-PCR for Nlrc5 mRNA expression in sorted WT thymocyte populations. Flow cytometric data in A and C are representative of five experiments; Q-RT-PCR data are combined from least two independent experiments and normalized to 18 S RNA expression. Error bars = S.E. Statistical analysis by two-tailed Student's t test, *, p < 0.05, **, p < 0.01.
NLRC5 Does Not Alter the Extent of IFN-induced MHC-I Expression
Prior work with CIITA indicates that this protein regulates both basal and inducible expression of MHC-II (2, 22). To determine whether Nlrc5 has a parallel function, we assessed the induced expression of MHC-I by IFN-γ, a known inducer of MHC-I. An analysis of T and B cells indicates that although the basal level of MHC-I is reduced in Nlrc5−/− cells, the induction of MHC-I by IFN-γ in both populations remained intact (Fig. 4A). These data are consistent with MHC-I mRNA analysis showing similar relative increases in H2-k1 message with IFN-γ treatment; however, the MHC-I levels in Nlrc5−/− cells are consistently reduced when compared with WT controls (Fig. 4B). Additionally, IFN-γ-induced MHC-I expression occurs on Nlrc5−/− and Ciita−/− BMDM (Fig. 4C). IFN-γ-induced MHC-II surface expression was normal on Nlrc5−/− BMDM but reduced on Ciita−/− cells as expected (Fig. 4D). The decreased expression of MHC-I but not MHC-II in response to IFN-γ treatment is also reflected at the mRNA level in Nlrc5−/− cells (Fig. 4E). These results indicate that unlike CIITA, which is crucial for both basal and IFN-γ-induced MHC-II expression, Nlrc5 does not affect the extent of IFN-γ-induced up-regulation of MHC-I, but rather is necessary for basal constitutive MHC-I expression, which then affects the final amount of MHC-I expressed by cells after IFN-γ stimulation.
FIGURE 4.

Flow cytometric and Q-RT-PCR analysis of H2-Kb expression by WT and Nlrc5−/− splenocytes or BMDM in response to INF-γ treatment. A, surface H2-Kb expression on in vitro cultured splenocytes, untreated (gray filled) or INF-γ-treated for 24 h (black). % of max, percentage of maximum; UT, untreated control. B, Q-RT-PCR for H2-k1 mRNA expression by in vitro cultured splenocytes from WT or Nlrc5−/− mice at the indicated time points after IFN-γ treatment. C, surface H2-Kb expression on BMDM, untreated (gray filled) or INF-γ-treated at 24 h (black). D, surface I-Ab expression on BMDM, untreated (gray filled) or INF-γ-treated at 24 h (black). Numbers under the histogram in C and D are mean fluorescence intensities. E, Q-RT-PCR for H2-k1 and I-Ab mRNA expression in WT or Nlrc5−/− BMDM treated with IFN-γ. Q-RT-PCR data are combined from at least two independent experiments and normalized to 18 S RNA expression. Error bars = S.E. Data are representative of two independent experiments. Statistical analysis by two-tailed Student's t test, *, p < 0.05, **, p < 0.01. Rel. expression, relative expression.
CIITA Does Not Regulate MHC-I Expression in Mouse B Lymphocytes
The earlier experiments indicate that the effect of Nlrc5 deletion on classical H-2K and B2m expression is most dramatic in T cells and less so in B cells (Fig. 3, A and B). Prior work has shown that CIITA can regulate MHC-I in human cells, and furthermore, CIITA is highly expressed by B cells (6, 7). Thus one possibility is that CIITA and Nlrc5 both regulate MHC-I expression in B cells, whereas CIITA expression does not affect MHC-I expression by T cells because the level of CIITA is negligible in mouse T cells. An assessment of Nlrc5−/−, Ciita−/−, and WT cells indicates that although Nlrc5−/− T and B cells express less H-2K antigen, H-2K expression by Ciita−/− T and B cells is similar to wild type (Fig. 5A). This indicates that Ciita deletion does not result in a detectable loss of MHC-I expression by B cells in the murine system. We also do not detect a decrease in MHC-II expression Nlrc5−/− B cells (Fig. 5B), suggesting that Nlrc5 control of MHC expression is restricted to MHC-I.
FIGURE 5.

Flow cytometric analysis of MHC-I and MHC-II surface expression by WT, Nlrc5−/−, and Ciita−/− mice. A, surface H2-Kb expression in WT (red), Nlrc5−/− (blue), and Ciita−/− (green dashed) splenic B and T cells. Gray, isotype control. % of max, percentage of maximum. B, flow cytometric analysis of I-Ab expression in WT (red), Nlrc5−/− (blue), and Ciita−/− (green dashed) B cells. Data are representative of three independent experiments.
Nlrc5 Is Required for the Removal of the Silencing Mark H3K27me3 from the MHC-I Proximal Promoter
Although others have noted the regulation of MHC-I by Nlrc5, the mechanism has not been explored. Gene expression occurs within the highly ordered context of chromatin, and open chromatin structure is required for transcription initiation. To determine the histone modification of the MHC-I promoter, levels of acetylation and trimethylation of lysine 27 on histone H3 were assessed in lymphocyte-enriched splenocytic populations and BMDM. After IFN-γ treatment, WT cells progressively lost the gene silencing H3K27me3 modification at the MHC-I promoter, H-2K1, which agrees with IFN-γ induction of this gene. By contrast, Nlrc5−/− cells did not show reduced H3K27me3 modification and even exhibited increased methylated state at the 4-h time point (Fig. 6A). Both WT and Nlrc5−/− cells showed similar acetylation of H3 at the MHC-I promoter upon IFN-γ treatment, which is associated with increased chromatin accessibility and gene transcription (Fig. 6B). The unabated and increased H3K27me3 in Nlrc5−/− cells was specific to the MHC-I promoter as normal demethylation and acetylation were generally observed for the MHC-II (Fig. 6, C and D) and GAPDH (Fig. 6, E and F) proximal promoters upon IFN-γ treatment. An exception is the increased H2K27me3 at I-Ab promoter in the BMDM + IFN-γ group at the 4-h time point, which needs to be explored further in the future. Increased H3K27me3 at the MHC-I promoter region is not due to increased histone levels as control assays demonstrated no significant changes in total histone H3 associated with MHC-I or MHC-II (Fig. 6, G and H). These data are consistent with a mechanism in which the MHC-I promoter in Nlrc5−/− cells becomes partially accessible in response to IFN-γ through increased H3 acetylation; however, the inability to reach WT levels of MHC-I expression after IFN-γ stimulation is likely due to an inability of Nlrc5−/− cells to remove H3K27me3 at the MHC-I promoter.
FIGURE 6.

Levels of H3K27me3 and acetylated H3 at MHC-I (H-2K1) and MHC-II (I-Ab) proximal promoters in WT and Nlrc5−/− lymphocyte-enriched splenocytes and BMDM. ChIP assays were carried out in cells stimulated as indicated with IFN-γ. Lysates were immunoprecipitated with control antibody, antibody to H3K27me3 (A, C, E, and F), acetylated H3 (B, D, E, and F), or total H3 (G and H), and associated DNA was isolated and analyzed via Q-PCR using primers spanning the proximal promoter region of H2-K1 (A, B, and G), I-Ab (C, D, and H), and Gapdh (E and F). Values for immunoprecipitations represent the mean ± S.E. of three independent experiments. UT, untreated control.
Detection of a 3′-End Transcript That Originates from Nlrc5 in WT and Nlrc5−/− Mice
While this manuscript was under review, Tong et al. (23) reported that deletion of Nlrc5 by targeting the C-terminal LRR1 also decreased MHC-I expression, but additionally increased NF-κB-dependent cytokine and type-I interferon production in response to various stimuli. Our studies did not indicate an NF-κB or a type-I interferon phenotype, and we considered the possibility that our Nlrc5 knock-out strategy (which targeted the nucleotide-binding domain) may allow for expression of Nlrc5 variant proteins that fail to induce normal MHC-I expression, but are sufficient to dampen NF-κB and type-I interferon responses. We examined Nlrc5 mRNA expression in several resting and stimulated cell subsets using a primer/probe set that anneals to the 3′ end of the mRNA (Fig. 7A, Primer/Probe). We found equivalent, if not increased, levels of Nlrc5 mRNA transcripts containing this end of the mRNA in the Nlrc5−/− splenic populations when compared with WT (Fig. 7B). Stimulation of total splenocytes or BMDM with IFN-γ (Fig. 7, C and D) or stimulation of BMDM with Toll-like receptor (TLR) agonists (Fig. 7E) resulted in up-regulation of this 3′ end transcript equal to, or greater than, WT control samples. These data suggest that Nlrc5 may be autoinhibitory at the mRNA level and leaves open the possibility that alternative splice forms of Nlrc5 that contain the 3′ end, although unable to maintain normal MHC-I expression, could be sufficient for normal regulation of NF-κB and type-I interferon responses.
FIGURE 7.

Q-RT-PCR for 3′ end of Nlrc5 transcript in splenocytes and BMDM from WT or Nlrc5−/− mice. A, schematic depicting the full-length Nlrc5 transcript including the AUG start, the NBD encoded by exon 4, and the 3′ Q-RT-PCR primer/probe set spanning exons 48–49 (dashed line). B–D, Q-RT-PCR with primer/probe set depicted in A. B, sorted resting splenocytes. C, total splenocytes cultured in vitro with IFN-γ for the indicated times. D, BMDM treated with IFN-γ for the indicated times. E. BMDM 8 h after PAMP stimulation. Data in B are combined from at least two independent experiments, and data in C–E are representative of at least two independent experiments. Q-RT-PCR data are normalized to 18 S RNA expression. Error bars = S.E. Rel. expression, relative expression; UT, untreated control.
DISCUSSION
This study describes an assessment of the physiologic function of NLRs by studying a targeted deletion of the NBD domain of Nlrc5. The NBD domain for most NLRs contains Walker A and B motifs, which are conserved among ATP-binding proteins. A study of Nlrc5 has shown that mutation of the Walker A and B motifs destroyed the ability of this protein to induce MHC-I transcription and nuclear translocation (9). Interestingly, previous data similarly showed the critical role of the Walker A and B motifs within CIITA in mediating nuclear translocation as well as MHC-II transactivation function (24–26). Others have shown that bacterially expressed partially purified NBD domain of NLRC4/ipaf binds ATP (27). Nucleotide binding by purified, full-length NLRP3 and NLRP12 was previously reported by our group with studies showing that both NLRs bind ATP and dATP but not CTP, GTP, or UTP and exhibit ATPase activity (28, 29). Others have shown that similar mutations of NOD2 also led to the loss of function (30). Thus the NBD domain plays important functions among many NLRs examined.
The gene expression pattern of Nlrc5 indicates that it is highest in T lymphocytes, NK cells, and B cells. Indeed it has the most profound effect on MHC-I expression by T lymphocytes and NK cells, whereas its effect on MHC-I expression by B cells is less than the former two cell types. One can speculate that perhaps B cells express other MHC-I-regulating NLRs. A highly probable candidate is CIITA, which has been found by many laboratories to update MHC-I in human cells (6, 7). The initial studies documenting the role of CIITA in MHC-I regulation were considered physiologically relevant as they relied on mutant cell lines with defective CIITA, as opposed to artificial overexpression systems. The physiologic relevance of CIITA in murine MHC-I is readdressed in this study, but we failed to find its role in the regulation of mouse MHC-I. In addition, our study also shows that although constitutive MHC-I expression is reduced in Nlrc5−/− cells, the extent of IFN-γ-induced MHC-I expression remained unaltered despite the overall reduction in MHC-I expression when compared with WT cells. Finally, our study indicates that Nlrc5 regulates the transcription of both classical MHC-I as well as the nonclassical Tla gene. Thus Nlrc5 likely controls MHC-I through conserved motifs found in classical and nonclassical MHC-I promoter. Additionally, our data hint at the possibility that Nlrc5 has a slight effect on MHC-II expression. For example, I-Ab mRNA by CD11b+ cells is slightly reduced in the absence of Nlrc5, and H3K27me3 modification of I-Ab promoter revealed a different kinetics in control versus Nlrc5−/− cells. A recent study showed that NLRC5 can associate with RFX, a DNA-binding protein that regulates MHC-II (31). Hence additional work is necessary to assess whether Nlrc5 cross-regulates MHC-II under some conditions.
The above data suggest that unlike CIITA, which is necessary for promoter loading of multiple transcription factors and histone modification factors including histone acetylases and methylases at MHC-II promoters (32), Nlrc5 might play a more defined role in its regulation of MHC-I. Consistent with this working model, the data show that although Nlrc5 is an important regulator of proper chromatin dynamics at MHC-I promoter, the effect is rather specific. Our study demonstrates that decreased expression of MHC-I in Nlrc5−/− knock-out cells is associated with increased levels of H3K27me3 but normal histone 3 acetylation. These observations underscore the dominance of H3K27me3 in down-regulating the transcriptional status of MHC-I chromatin in the absence of Nlrc5.
Aside from the regulation of MHC-I, our study has not been able to identify other functions of Nlrc5 that have been identified by previous studies, including our own, which showed that two interference RNAs targeting Nlrc5 caused a drastic reduction of inflammasome activation by an array of stimuli in human cell lines and primary peripheral blood mononuclear cells (16). Although it is difficult to reconcile the opposing functions found for Nlrc5 in cytokine induction by PAMPs reported in the literature, there are a number of explanations for the inability of Nlrc5−/− cells to confirm previous findings. 1) For the previous studies performed in human cells, it is possible that there are species-specific differences, which are ample among the other NLRs. For example, NLRP1 from humans and mice serve similar functions in inflammasome activation, but the former is responsive to muramyl dipeptide (MDP), whereas the latter is activated by Bacillus anthracis lethal toxin (33). 2) Along this same line, it is possible that there are gene redundancies in mice that are not found in humans. Mice have 38 NLR genes, whereas humans only have 22 NLR genes. As an example, whereas humans have one copy of the NAIP gene, mice have six copies of the gene (34, 35). 3) It is possible that the function of the Nlrc5 is cell type- and stimulus-dependent, and we have not identified the correct stimulus in the appropriate cell types. However, the reduction of MHC-I in Nlrc5−/− cells was observed in all cell types surveyed, so the ability of Nlrc5 to regulate MHC-I is not cell-specific, although the degree in which Nlrc5 controls MHC-I does vary with the cell type. 4) It is possible that deleting the NBD domain is not sufficient to eliminate all functions of Nlrc5 and that the gene deletion might have resulted in a functional protein without NBD. At the time of this submission, another group used a different gene-targeting strategy to show that Nlrc5 deletion resulted in a loss of MHC-I gene expression (36). During the review of this manuscript, two other groups have also shown a role for Nlrc5 in MHC-I expression, but one found an additional role for Nlrc5 in dampening cytokine response (23), whereas another found a role for this gene in inflammasome activation (37). As the various groups used different gene deletion strategies, it is possible that residual transcripts in the different gene deletion strains might be able to regulate functions other than MHC-I expression. The detection of a 3′ end transcript that originates from Nlrc5 in the strain that we produced suggests that this possibility deserves a further investigation.
The above caveats do not take away from the definitive results that support the findings of Meissner et al. (9) observed in cell lines. Together, these results indicate that Nlrc5 is a bona fide MHC-I gene transcriptional regulator. It is interesting that CIITA and Nlrc5 serve similar functions, the human CIITA and Nlrc5 are on the same chromosome (38), and their NBD and leucine-rich repeat (LRR) are most closely related on a phylogenetic tree (11). Since the discovery of the NLR family, some have argued that CIITA does not belong to this family because it is the only NLR protein with a transcriptional function. The discovery of Nlrc5 in MHC-I gene transcription clearly negates this concern and indicates that CIITA has at least one closely related relative with similar function. Thus CIITA and Nlrc5 represent bridges between innate immune genes and regulators of MHC that play pivotal roles in adaptive immunity.
Acknowledgment
The University of North Carolina (UNC) Flow Cytometry Core Facility is supported in part by a National Institutes of Health Center Core Support Grant (P30CA06086) through the NCI to the UNC Lineberger Comprehensive Cancer Center.
This work was supported, in whole or in part, by National Institutes of Health Grants NIAID R37-AI029564 (to J. P. Y. T.), NCI T32 CA009156 (to G. R. R.), and NCI CA131645 (to B. K. D.).
- NLR
- nucleotide-binding domain, leucine-rich repeat
- NBD
- nucleotide-binding domain
- CIITA
- class II transactivator
- PAMP
- pathogen-associated molecular pattern
- DAMP
- damaged-associated molecular pattern
- BMDM
- bone marrow-derived macrophages
- NK
- natural killer
- Q-PCR
- quantitative PCR
- Q-RT-PCR
- quantitative RT-PCR
- FAM
- 6-carboxyfluorescein
- TAM
- N,N,N,N-tetramethyl-6-carboxyrhodamine.
REFERENCES
- 1. Ting J. P., Duncan J. A., Lei Y. (2010) How the noninflammasome NLRs function in the innate immune system. Science 327, 286–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Choi N. M., Majumder P., Boss J. M. (2011) Regulation of major histocompatibility complex class II genes. Curr. Opin. Immunol. 23, 81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu X. S., Linhoff M. W., Li G., Chin K. C., Maity S. N., Ting J. P. (2000) Transcriptional scaffold: CIITA interacts with NF-Y, RFX, and CREB to cause stereospecific regulation of the class II major histocompatibility complex promoter. Mol. Cell Biol. 20, 6051–6061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Masternak K., Muhlethaler-Mottet A., Villard J., Zufferey M., Steimle V., Reith W. (2000) CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 14, 1156–1166 [PMC free article] [PubMed] [Google Scholar]
- 5. Majumder P., Gomez J. A., Chadwick B. P., Boss J. M. (2008) The insulator factor CTCF controls MHC class II gene expression and is required for the formation of long-distance chromatin interactions. J. Exp. Med. 205, 785–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martin B. K., Chin K. C., Olsen J. C., Skinner C. A., Dey A., Ozato K., Ting J. P. (1997) Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity 6, 591–600 [DOI] [PubMed] [Google Scholar]
- 7. Gobin S. J., Peijnenburg A., Keijsers V., van den Elsen P. J. (1997) Site α is crucial for two routes of IFN-γ-induced MHC class I transactivation: the ISRE-mediated route and a novel pathway involving CIITA. Immunity 6, 601–611 [DOI] [PubMed] [Google Scholar]
- 8. Howcroft T. K., Raval A., Weissman J. D., Gegonne A., Singer D. S. (2003) Distinct transcriptional pathways regulate basal and activated major histocompatibility complex class I expression. Mol. Cell Biol. 23, 3377–3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meissner T. B., Li A., Biswas A., Lee K. H., Liu Y. J., Bayir E., Iliopoulos D., van den Elsen P. J., Kobayashi K. S. (2010) NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl. Acad. Sci. U.S.A. 107, 13794–13799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meissner T. B., Li A., Liu Y. J., Gagnon E., Kobayashi K. S. (2012) The nucleotide-binding domain of NLRC5 is critical for nuclear import and transactivation activity. Biochem. Biophys. Res. Commun. 418, 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Benko S., Magalhaes J. G., Philpott D. J., Girardin S. E. (2010) NLRC5 limits the activation of inflammatory pathways. J. Immunol. 185, 1681–1691 [DOI] [PubMed] [Google Scholar]
- 12. Davis B. K., Wen H., Ting J. P. (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neerincx A., Lautz K., Menning M., Kremmer E., Zigrino P., Hösel M., Büning H., Schwarzenbacher R., Kufer T. A. (2010) A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J. Biol. Chem. 285, 26223–26232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuenzel S., Till A., Winkler M., Häsler R., Lipinski S., Jung S., Grötzinger J., Fickenscher H., Schreiber S., Rosenstiel P. (2010) The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J. Immunol. 184, 1990–2000 [DOI] [PubMed] [Google Scholar]
- 15. Cui J., Zhu L., Xia X., Wang H. Y., Legras X., Hong J., Ji J., Shen P., Zheng S., Chen Z. J., Wang R. F. (2010) NLRC5 negatively regulates the NF-κB and type I interferon signaling pathways. Cell 141, 483–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davis B. K., Roberts R. A., Huang M. T., Willingham S. B., Conti B. J., Brickey W. J., Barker B. R., Kwan M., Taxman D. J., Accavitti-Loper M. A., Duncan J. A., Ting J. P. (2011) Cutting edge: NLRC5-dependent activation of the inflammasome. J. Immunol. 186, 1333–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kumar H., Pandey S., Zou J., Kumagai Y., Takahashi K., Akira S., Kawai T. (2011) NLRC5 deficiency does not influence cytokine induction by virus and bacteria infections. J. Immunol. 186, 994–1000 [DOI] [PubMed] [Google Scholar]
- 18. Itoh-Lindstrom Y., Piskurich J. F., Felix N. J., Wang Y., Brickey W. J., Platt J. L., Koller B. H., Ting J. P. (1999) Reduced IL-4-, lipopolysaccharide-, and IFN-γ-induced MHC class II expression in mice lacking class II transactivator due to targeted deletion of the GTP-binding domain. J. Immunol. 163, 2425–2431 [PubMed] [Google Scholar]
- 19. Wen H., Gris D., Lei Y., Jha S., Zhang L., Huang M. T., Brickey W. J., Ting J. P. (2011) Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Truax A. D., Greer S. F. (2012) ChIP and Re-ChIP assays: investigating interactions between regulatory proteins, histone modifications, and the DNA sequences to which they bind. Methods Mol. Biol. 809, 175–188 [DOI] [PubMed] [Google Scholar]
- 21. Truax A. D., Thakkar M., Greer S. F. (2012) Dysregulated recruitment of the histone methyltransferase EZH2 to the class II transactivator (CIITA) promoter IV in breast cancer cells. PloS one 7, e36013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krawczyk M., Reith W. (2006) Regulation of MHC class II expression, a unique regulatory system identified by the study of a primary immunodeficiency disease. Tissue Antigens 67, 183–197 [DOI] [PubMed] [Google Scholar]
- 23. Tong Y., Cui J., Li Q., Zou J., Wang H. Y., Wang R. F. (2012) Enhanced TLR-induced NF-κB signaling and type I interferon responses in NLRC5-deficient mice. Cell Res. 22, 822–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chin K. C., Li G. G., Ting J. P. (1997) Importance of acidic, proline/serine/threonine-rich, and GTP-binding regions in the major histocompatibility complex class II transactivator: generation of transdominant-negative mutants. Proc. Natl. Acad. Sci. U.S.A. 94, 2501–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harton J. A., Cressman D. E., Chin K. C., Der C. J., Ting J. P. (1999) GTP binding by class II transactivator: role in nuclear import. Science 285, 1402–1405 [DOI] [PubMed] [Google Scholar]
- 26. Raval A., Weissman J. D., Howcroft T. K., Singer D. S. (2003) The GTP-binding domain of class II transactivator regulates its nuclear export. J. Immunol. 170, 922–930 [DOI] [PubMed] [Google Scholar]
- 27. Lu C., Wang A., Wang L., Dorsch M., Ocain T. D., Xu Y. (2005) Nucleotide binding to CARD12 and its role in CARD12-mediated caspase-1 activation. Biochem. Biophys. Res. Commun. 331, 1114–1119 [DOI] [PubMed] [Google Scholar]
- 28. Duncan J. A., Bergstralh D. T., Wang Y., Willingham S. B., Ye Z., Zimmermann A. G., Ting J. P. (2007) Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. U.S.A. 104, 8041–8046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ye Z., Lich J. D., Moore C. B., Duncan J. A., Williams K. L., Ting J. P. (2008) ATP binding by monarch-1/NLRP12 is critical for its inhibitory function. Mol. Cell Biol. 28, 1841–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tanabe T., Chamaillard M., Ogura Y., Zhu L., Qiu S., Masumoto J., Ghosh P., Moran A., Predergast M. M., Tromp G., Williams C. J., Inohara N., Núñez G. (2004) Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J. 23, 1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meissner T. B., Liu Y. J., Lee K. H., Li A., Biswas A., van Eggermond M. C., van den Elsen P. J., Kobayashi K. S. (2012) NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J. Immunol. 188, 4951–4958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wright K. L., Ting J. P. (2006) Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol. 27, 405–412 [DOI] [PubMed] [Google Scholar]
- 33. Moayeri M., Sastalla I., Leppla S. H. (2011) Anthrax and the inflammasome. Microbes Infect. 14, 392–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kofoed E. M., Vance R. E. (2011) Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao Y., Yang J., Shi J., Gong Y. N., Lu Q., Xu H., Liu L., Shao F. (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600 [DOI] [PubMed] [Google Scholar]
- 36. Staehli F., Ludigs K., Heinz L. X., Seguín-Estévez Q., Ferrero I., Braun M., Schroder K., Rebsamen M., Tardivel A., Mattmann C., Macdonald H. R., Romero P., Reith W., Guarda G., Tschopp J. (2012) NLRC5 deficiency selectively impairs MHC class I-dependent lymphocyte killing by cytotoxic T cells. J. Immunol. 188, 3820–3828 [DOI] [PubMed] [Google Scholar]
- 37. Yao Y., Wang Y., Chen F., Huang Y., Zhu S., Leng Q., Wang H., Shi Y., Qian Y. (2012) NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res. 22, 836–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harton J. A., Linhoff M. W., Zhang J., Ting J. P. (2002) Cutting edge: CATERPILLER: a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J. Immunol. 169, 4088–4093 [DOI] [PubMed] [Google Scholar]