

Background: EPR/55Mn ENDOR spectroscopy of the oxygen-evolving complex (OEC) and Mn2+ in Ca2+-depleted photosystem II.
Results: Electronic model of the Ca2+-depleted OEC; characterization of Mn2+ binding.
Conclusion: Ca2+ is not critical for maintaining the electronic and spatial structure of the OEC. Its removal exposes a Mn2+ binding site supposedly in an extrinsic subunit.
Significance: Mechanistic implications for water oxidation; Mn2+ in photoassembly/D1 protein repair.
Keywords: Calcium, Electron Paramagnetic Resonance (EPR), ENDOR Spectroscopy, Manganese, Metalloproteins, Photosystem II, Oxygen-evolving Complex (OEC), Photoassembly/Photoactivation, Water-oxidizing Complex (WOC), Zero-field Splitting
Abstract
Ca2+ is an integral component of the Mn4O5Ca cluster of the oxygen-evolving complex in photosystem II (PS II). Its removal leads to the loss of the water oxidizing functionality. The S2′ state of the Ca2+-depleted cluster from spinach is examined by X- and Q-band EPR and 55Mn electron nuclear double resonance (ENDOR) spectroscopy. Spectral simulations demonstrate that upon Ca2+ removal, its electronic structure remains essentially unaltered, i.e. that of a manganese tetramer. No redistribution of the manganese valence states and only minor perturbation of the exchange interactions between the manganese ions were found. Interestingly, the S2′ state in spinach PS II is very similar to the native S2 state of Thermosynechococcus elongatus in terms of spin state energies and insensitivity to methanol addition. These results assign the Ca2+ a functional as opposed to a structural role in water splitting catalysis, such as (i) being essential for efficient proton-coupled electron transfer between YZ and the manganese cluster and/or (ii) providing an initial binding site for substrate water. Additionally, a novel 55Mn2+ signal, detected by Q-band pulse EPR and ENDOR, was observed in Ca2+-depleted PS II. Mn2+ titration, monitored by 55Mn ENDOR, revealed a specific Mn2+ binding site with a submicromolar KD. Ca2+ titration of Mn2+-loaded, Ca2+-depleted PS II demonstrated that the site is reversibly made accessible to Mn2+ by Ca2+ depletion and reconstitution. Mn2+ is proposed to bind at one of the extrinsic subunits. This process is possibly relevant for the formation of the Mn4O5Ca cluster during photoassembly and/or D1 repair.
Introduction
The oxygen-evolving complex (OEC)5 of photosystem II (PS II) catalyzes the light-driven oxidation of water. The OEC contains an inorganic Mn4O5Ca metallocofactor that includes five μ-oxo bridge linkages and is coordinated by a framework of surrounding amino acids (1–6) in a highly defined manner that confers catalytic function. The redox-active tyrosine residue YZ (D1-Tyr-161) enables electron transfer from the Mn4O5Ca cluster to P680+·, the radical cation formed upon photon absorption and charge separation. The Mn4O5Ca cluster undergoes four successive oxidations, cycling through a series of different net valence states, referred to as the Si states (where i = 0–4 denotes the number of oxidizing equivalents stored in the cluster). The transient state S4 spontaneously returns to S0 upon regaining four electrons from the two substrate water molecules, which in the process form molecular oxygen. The release of O2 is followed by the rebinding of at least one H2O molecule (for reviews, see Refs. 7–14).
X-ray crystallographic structures of the PS II protein complex provided an atomic picture of the structure of the OEC (1–6), identifying all amino acids that ligate the Mn4O5Ca cluster. The metallocofactor resembles a distorted chair, consisting of the cuboidal moiety Mn3O3Ca (MnB3MnC2MnD1),6 with the fourth, outer manganese ion (MnA4), connected to the cuboid via an additional μ-oxo bridge (O4) to one of the manganese vertices (MnB3). The reported cluster is likely modified due to photoreduction of the MnIII and MnIV ions, such that the Mn-Mn and Mn-Ca distances seen in the x-ray structure are all elongated as compared with those derived from extended x-ray absorption fine structure (EXAFS) measurements (15). Allowing for this, the basic topology of the x-ray structure is similar to earlier literature models, including the geometry-optimized density functional theory (DFT) models of Kusunoki (16), Siegbahn (17), and the recent model of Ames et al. (18), in which the cuboid exhibits an open conformation with MnA4 connected to MnB3 via a di-μ-oxo bridge (Fig. 1).
FIGURE 1.

Stereo view of a DFT model of the Mn4O5Ca cluster in the S2 state and directly ligating amino acid residues and H2O/OH− molecules (18). Amino acids, except CP43-Glu-354, are from PS II subunit D1. Manganese, calcium, nitrogen, oxygen, carbon, and hydrogen atoms are shown in purple, green, blue, red, gray, and white, respectively. Nonpolar H atoms are omitted for clarity.
The Ca2+ ion of the Mn4O5Ca cluster, which can be removed from and reconstituted into the OEC (19–21), is essential for catalytic function (19–23). The non-catalytic Ca2+-depleted OEC cannot complete the S state cycle, advancing only to a modified S2 state, termed S2′ (24, 25). The reason for this remains unclear. However, four basic explanations exist in the current literature based on the proposed role(s) for the Ca2+ ion during the S state cycle (for reviews, see Refs. 26–28). These include the following: (i) As an integral component of the OEC (6), the Ca2+ ion can be suspected to be of crucial structural importance. However, EXAFS experiments suggest that Ca2+ depletion leads to only a small spatial reorganization of the remnant Mn4O5 cluster (29). (ii) It facilitates fast one-electron transfer from the OEC to YZ+· (for reviews, see Refs. 11 and 30). The formation of the S2′ state requires long visible light illumination at temperatures ≥0 °C. This is in contrast to the native S2 state, which can be generated via visible light illumination at −78 °C. This apparent increase in the activation energy of OEC turnover upon Ca2+ removal may represent a decoupling of the YZ+· from the OEC, such that Ca2+-mediated protein conformational changes and/or H+ translocations associated with physiological S state transitions are blocked. (iii) It is a binding/staging site for substrate water and its deprotonation (26, 31). The kinetics of substrate water binding to the OEC are affected by biochemical exchange of Ca2+ with Sr2+, the only surrogate ion able to confer catalytic activity (19, 23, 32). It can presumably act in place of Ca2+ as it has approximately the same size and a similar Lewis acidity (31). This result has been interpreted as evidence for Ca2+ binding one of the substrate waters. Inhibition due to Ca2+ depletion would then reflect the loss of a substrate binding site. (iv) Although the basic structural arrangement of manganese ions in the cluster is retained upon Ca2+ removal, it is uncertain if their magnetic and/or electronic interactions are perturbed, which could lead to a decoupling of the cluster or a rearrangement of the manganese valence states. Thus, Ca2+ depletion could potentially change the redox properties as well as substrate and/or protein interactions of the complex, inhibiting catalytic function.
The Mn4O5Ca cluster in the S2 state exhibits a characteristic multiline EPR signal centered at g ≈ 2 (33) that arises from an S = 1/2 ground spin state of the cluster. Under certain conditions (illumination, reactants), additional signals are observed at higher g values; in spinach, a second broad signal can be detected at g ≈ 4.1 (34, 35), attributed to an S = 5/2 spin state (36). These signals are affected by the presence of small alcohols, foremost methanol (MeOH) (37–41), which enhance the intensity of the multiline signal at the expense of the g ≈ 4.1 signal (37) (for a full discussion see Ref. 41). The Mn4O5 cluster in the S2′ state also exhibits a multiline signal; however, its hyperfine splitting pattern is perturbed. It contains a larger number of resolved lines as compared with the native S2 multiline signal, with a smaller average line spacing (5.5–6 versus 8.8 mT). The magnetic interaction between YZ• and the OEC is also perturbed in Ca2+-depleted PS II as evidenced by changes in the tyrosine split signal of the S2′YZ• state (24, 25).
A detailed understanding of the electronic structure of the Mn4O5Ca cluster in the S2 state has been developed from pulse EPR data (42–46), in particular 55Mn electron nuclear double resonance (ENDOR). These experiments demonstrated that the four manganese ions contribute about equally to the ground electronic state of the S2 state; i.e. all four manganese ions carry approximately the same spin density. This requirement allows an assessment of the electronic exchange interactions between the four manganese ions and the development of Mn4 coupling schemes. These necessarily reflect the geometric structure of the OEC and allow the assignment of the individual manganese oxidation states. Our recently proposed model for the S2 state (18) is described under “Discussion.” This scheme places the only MnIII ion inside the cuboidal unit (MnD1) (see also Ref. 47) and compares favorably with information from complementary spectroscopic measurements (48–50).
Although it has not been directly observed by EPR spectroscopy, the possibility of another paramagnetic manganese species being able to bind to the Ca2+-depleted PS II has been suggested in an earlier study by Booth et al. (51). The additional species was suggested to be a Mn2+ ion that can bind specifically to a site in the protein complex that is created or becomes accessible via structural changes in the course of Ca2+ removal. This was based on the observation that, after equimolar amounts of Mn2+ ions had been added to Ca2+-depleted PS II, no Mn2+ was observed by X-band continuous wave (CW) EPR. Upon titrating Ca2+ ions back into these samples, Mn2+ was released as seen from the appearance of the six-line Mn2+ EPR signal.
In this work, both the spin system of the Mn4O5 cluster in the S2′ state of Ca2+-depleted PS II and the binding of Mn2+ ions to this protein were studied by EPR and ENDOR spectroscopy at X- and Q-band frequencies. The results provide new insight into the role of the Ca2+ ion in the native OEC.
EXPERIMENTAL PROCEDURES
Sample Preparation
PS II-enriched thylakoid membranes were prepared from spinach based on the procedure of Berthold et al. (52) using detergent treatment by incubation with Triton X-100 for 15 min. All work was performed in the dark or very dim green light, and the PS II was kept at 4 °C before storage in the dark at −80 °C or in liquid N2. Chlorophyll concentrations were determined by assays using aqueous acetone (80%) extracts (53) with updated extinction coefficients (54) using an ATI Unicam UV-visible spectrometer UV2–300.
Ca2+ depletion and reconstitution based on the low pH/citrate treatment method (21) was achieved as described previously (55). The final buffer used was 50 mm MES, 15 mm NaCl, 0.4 m sucrose, 1 mm EDTA, pH 6.5. Ca2+ removal and, as a proof for the integrity of the OEC, Ca2+ rebinding was confirmed both by enzymatic assays and by X-band CW EPR. The O2 evolution rates of native PS II were ∼400 μmol O2/mg of chlorophyll/h (see the following section). O2 evolution rates dropped to 5–10% in Ca2+-depleted and were reactivated to >80% in Ca2+-reconstituted samples. Similar percentages of the S2 multiline signal were observed after white light illumination with a tungsten lamp through an aqueous 5% CuSO4 IR filter of the respective samples at 200 K for 5 min (Table 1, Fig. 2A). These numbers are consistent with previous literature reports (25, 29, 56).
TABLE 1.
Oxygen evolution activities and relative S2 multiline EPR signal intensities of the Ca2+-containing native, the Ca2+-depleted, and the Ca2+-reconstituted PS II membrane preparations from spinach
| Observable | Native | Ca2+-depleted | Ca2+-reconstituted |
|---|---|---|---|
| Enzymatic rates/μmol O2/mg chlorophyll/ha | 390 ± 30 | 27 ± 1 | 330 ± 30 |
| Relative enzymatic rates | 100% | 7 ± 0% | 84 ± 8% |
| Relative S2 state multiline signal intensitiesb | 100% | 8 ± 3% | 105 ± 12% |
a Determined as an average of at least 8 single measurements at a minimum of 2 different chlorophyll concentrations from 5 to 25 μg/ml.
b Determined from the peak-to-trough distances of four prominent derivative peaks in the CW EPR spectrum (100).
FIGURE 2.

EPR and ENDOR experimental spectra (black solid traces) and simulations (red dashed traces). A, X-band CW EPR of PS II isolated from spinach. Shown are the Ca2+-depleted OEC poised in the S2′ state (a) and the native (b), Ca2+-depleted (c), and Ca2+-reconstituted OECs (d) illuminated at 200 K. In the experimental spectrum, the region of the overlapping YD• signal (g ≈ 2) was omitted for clarity. In a, a fourth order polynomial, and in b–d, a background signal of the resonator cavity were subtracted from the raw data. Experimental parameters: MW frequencies, 9.634 GHz (a), 9.44 GHz (b–d); MW power, 0.5 milliwatt (a), 20 milliwatts (b–d); modulation amplitude, 7.5 G (a), 15 G (b–d); time constant, 82 ms; temperature, 8 K (a), 10 K (b–d). B, X-band (a) and Q-band (b–e) Davies ENDOR of the Ca2+-depleted S2′ state from spinach compared with the native and Sr2+-substituted S2 states from spinach and T. elongatus. Shown are Ca2+-depleted Mn4O5 S2′, spinach (a and b), native Mn4O5Ca S2, spinach (taken from Refs. 45 and 46) (c), native Mn4O5Ca S2, T. elongatus (from Ref. 49) (d), and Sr2+-substituted Mn4O5Sr S2, T. elongatus (from Ref. 49) (e). a and b were smoothed using a 9- and 5-point moving average, respectively. b is the difference of an S2′ state spectrum after illumination at 0 °C minus an S1′ state spectrum after dark adaptation (supplemental Fig. S2) to remove an overlapping Mn2+ signal. The Mn2+ signal is attributed to residual Mn2+ ions stemming from a small fraction of damaged manganese clusters. In X-band pulse EPR (not shown) and ENDOR spectra (a), Mn2+ contributions were avoided by optimizing the MW pulse lengths for the S2′ state signal of the Mn4O5 spin system with an S = 1/2 ground spin state. Experimental parameters: a and b, MW frequencies, 9.717 GHz (X-band), 34.033 GHz (Q-band); shot repetition rate, 5 ms; MW pulse lengths π, 12 ns (X-band), 72 ns (Q-band); τ, 200 ns (X-band), 440 ns (Q-band); magnetic fields, 380 mT (X-band), 1208 mT (Q-band); RF pulse length πRF, 4 μs; temperature, 5 K; c–e, see Refs. 45 and 49.
Advancement of dark-adapted S1′ state EPR samples to the S2′ and S2′YZ• states (25) was done by illumination at 0 °C for 3 min, with 125 μm 3-(3,4-dichlorophenyl)-1,1-dimethylurea (10 mm in dimethyl sulfoxide) added to the samples advanced to the S2′ state, which restricts the acceptor site and, thus, YZ to one turnover.
For Ca2+ and Mn2+ titration experiments, dark-adapted Ca2+-depleted PS II membranes were rebuffered in EDTA-free buffer by three cycles of dilution, centrifugation at 39,000 × g for 15 min, and resuspension using 50 mm MES, 15 mm NaCl, 5 mm MgCl2, 0.4 m sucrose, pH 6.5. The final concentration of PS II reaction centers (RCs) in the samples was 28 ± 3 μm based on a chlorophyll concentration of 6.3 ± 0.8 mg ml−1 and assuming 250 chlorophylls/RC (57) after 15 min Triton X-100 treatment. The samples were incubated with known amounts of Mn2+ ranging from 0 to 4 eq per RC for 2 h. For the Ca2+ titration, samples containing 0.8 added eq of Mn2+ were incubated with known amounts of Ca2+ between 0 and 2400 eq for one additional hour. Mn2+ and Ca2+ ions were added from stock solutions of their chlorides.
Oxygen Evolution Measurements
Steady state PS II enzyme activity at 25 °C was determined by polarographic measurement of the O2 concentration in a PS II-containing assay mixture using a Clark-type Hansatech oxygen electrode with a high sensitivity Teflon membrane under continuous illumination with a tungsten lamp through an aqueous 5% CuSO4 IR filter. The assay medium was the buffer of the samples lacking EDTA and with 5 mm MgCl2 and 0.2 mm phenyl-p-benzoquinone (20 mm in dimethyl sulfoxide) added as an electron acceptor.
EPR/ENDOR Spectroscopy
X-band CW EPR spectra were recorded on a Bruker ELEXSYS E500 spectrometer equipped with an ESR900 liquid helium flow cryostat and an ITC503 helium flow temperature controller (Oxford Instruments Ltd.). X-band pulse experiments were performed with a Bruker ESP 380E spectrometer equipped with a dielectric ring resonator, an Oxford ITC liquid helium flow system, and a temperature controller. Q-band pulse experiments were performed using a Bruker ELEXSYS E580 spectrometer equipped with a laboratory-built cylindrical ENDOR resonator (58), a CF935 cryostat, and an ITC5025 temperature controller (Oxford Instruments Ltd.). Field-swept electron spin echo (ESE)-detected experiments were performed at Q-band frequencies using the pulse sequence π/2-τ- π/2-τ-echo with π = 72 ns and τ = 440 ns. For 55Mn Davies ENDOR, the pulse sequence was π-πRF-T-π/2-τ- π/2-τ-echo, with π = 12 ns (X-band), 72 ns (Q-band), or 16 ns (Q-band Mn2+ titration/quantification), πRF = 4 μs (X-, Q-band) or 4.5 μs (Q-band Mn2+ titration/quantification), T = 3.4 μs (X-, Q-band) or 1 μs (Q-band Mn2+ titration/quantification), and τ = 200 ns (X-band), 440 ns (Q-Band), or 320 ns (Q-band Mn2+ titration/quantification). The radio frequency (RF) was varied randomly in the desired range, and the RF pulses were amplified by an ENI 5100L amplifier. Except for Mn2+ titration/quantification, 55Mn Davies ENDOR spectra were collected using a home-built computer console with SpecMan control software (59) coupled to an SMT02 external RF pulse generator.
EPR/ENDOR Spectral Simulations
Simulations of EPR and 55Mn ENDOR spectra were performed numerically using the EasySpin software package (60). The fitting procedures employed a least squares minimization routine. All tensors were set to be collinear. The Ca2+-depleted Mn4O5 cluster in the S2′ state was treated as an effective electronic spin S = 1/2 ground state coupled to the four 55Mn nuclei, described by the following spin Hamiltonians for the EPR (Equation 1) and 55Mn ENDOR (Equation 2) spectra.
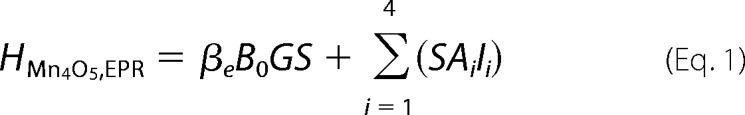 |
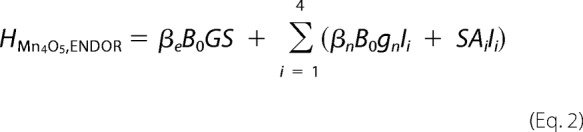 |
The EPR spectrum was calculated using second order perturbation theory, neglecting nuclear Zeeman terms and forbidden transitions. The 55Mn ENDOR spectra were calculated exactly, including nuclear Zeeman terms and considering all transitions. For the monomeric Mn2+ ion (S = 5/2, I = 5/2) bound to the Ca2+-depleted PS II, the following spin Hamiltonian was solved exactly both for the ESE and ENDOR spectra:
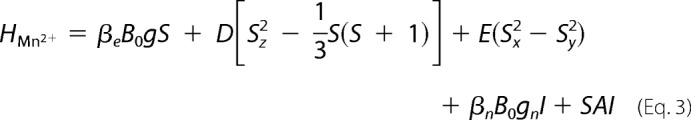 |
For details on the simulation procedure and the theoretical background, see Refs. 46, 49, and 61.
Temperature-dependent CW EPR Signal Intensity
The temperature was calibrated using a thermometer in place of the sample in the EPR tube. To guarantee that the actual unsaturated intensity I1 of the S2′ state modified multiline, as the ground state signal, was measured at all temperatures, the saturation behavior was studied at the lowest temperature employed. As a result, the non-saturating microwave (MW) power of 0.1 milliwatt was used throughout. The intensities I1 of the derivative signals were measured by means of the heights of 19 peaks throughout the spectral range, thereby minimizing statistical errors and contributions of underlying broader signals, such as from cytochrome b559 and the semiquinone-iron complex. How the ground-to-first excited state energy difference Δ is determined from the temperature dependence of I1 is outlined in the supplemental data.
Quantification of the Relative Concentrations of PS II-bound Mn2+ and Hexaquo-Mn2+
The Mn2+ species in Ca2+ and Mn2+ titration samples were quantified by means of their Q-band 55Mn Davies ENDOR spectra in two ways, and the results were averaged. (i) The relative contributions of the spectra from the pure Mn2+ species needed to reproduce the spectra from the various titration points were determined. The spectra from Mn2+ already present in the Ca2+-depleted PS II samples without the addition of Mn2+ ions and from 40 μm MnCl2 dissolved in the titration buffer represented PS II-bound and hexaquo-Mn2+, respectively. (ii) The relative amplitudes of the 55Mn ENDOR mS = −3/2 transitions, which appear in different RF ranges characteristic for the two Mn2+ species, were quantified in the regions of 353–376 MHz for PS II-bound Mn2+ and 390–395 MHz for hexaquo-Mn2+.
RESULTS
EPR and 55Mn ENDOR of the Ca2+-depleted Mn4O5 Cluster in the S2′ State
The characteristic modified multiline CW EPR signal (24, 25) was observed for Ca2+-depleted PS II samples poised in the S2′ state. It is centered at g ≈ 2 and spans the magnetic field range from ∼260 to ∼430 mT, resolving at least 27 hyperfine interaction (HFI) lines with an average peak-to-peak spacing of ∼6 mT (Fig. 2A). The central HFI lines are superimposed by the signal of the stable tyrosyl radical YD• centered at g ≈ 2, which is not depicted for clarity of presentation. The broad underlying signal of the reduced QA−·Fe2+ complex (62) contributes in the 350–375-mT region (24, 25, 29).
Traces a and b in Fig. 2B show the X- and Q-band Davies ENDOR spectra of the S2′ state recorded at 5 K and magnetic fields of 380 and 1208 mT, respectively. The 55Mn ENDOR spectrum of the Mn4O5 cluster in the S2′ state is essentially invariant across the corresponding EPR signal envelope (supplemental Fig. S1). It is ∼130 MHz wide, extending over a range from ∼60 to ∼190 MHz. As compared with the 55Mn ENDOR spectrum of the native S2 state (Fig. 2Bc, supplemental Fig. S1), the Ca2+-depleted S2′ state spectrum is broader. The edges of the spectrum change up to 10 MHz, resulting in a ∼20 and ∼10 MHz increase in the width of the X- and Q-band 55Mn ENDOR spectra, respectively, as compared with the Ca2+-containing S2 state of spinach PS II (42, 45, 46). The Q-band spectrum of the S2′ state exhibits five clearly resolved peaks, as also seen for the native S2 state spectrum from Thermosynechococcus elongatus (Fig. 2Bd); however, their positions differ slightly.
The X-band CW EPR and X- and Q-band 55Mn Davies ENDOR spectra were simultaneously simulated using the spin Hamiltonian formalism (for details see “Experimental Procedures” and Refs. 46 and 49). In these simulations, the S2′ state Mn4O5 cluster is treated as an effective S = 1/2 electronic spin state coupled to the four 55Mn nuclei, the same as for the native S2 state (41, 42, 46, 48, 49, 63–66). This approach requires the ground electronic spin state to be well separated from higher states, as is experimentally observed (see the following section). The simulations reproduce all the major spectral features of the EPR and 55Mn ENDOR spectra (Fig. 2, dashed red traces).
The isotropic and anisotropic values of the fitted effective 55Mn HFI tensors Ai (i = 1–4) are given in Table 2. For means of comparison, the numbers for the native S2 state from spinach (46) and the native and Sr2+-substituted S2 states from T. elongatus (49) are also listed. A full set of G and HFI tensor components is listed in supplemental Table S1. As seen for the Mn4O5Ca/Sr clusters, four effective HFI tensors are required to simulate the Mn4O5 cluster spectra. Their magnitudes are on the order seen for mono- and dimeric Mn3+ and Mn4+ complexes. Hence, their individual spin projection coefficients ρi must be on the order of 1 (see Ref. 49). In contrast to preliminary simulations of the S2′ spectra (67) or others on the S2 state from spinach PS II (46), the HFI tensors were not constrained to axial symmetry. However, as was found previously in simulations of the Mn4O5Ca/Sr clusters in T. elongatus (49), the tensors nevertheless show a considerable degree of axial symmetry. Moreover, these four OEC clusters show the same geometries of their HFI tensors, with larger axial than equatorial tensor components (Aaniso < 0) for A2 − A4 and vice versa for the largest HFI A1 (Aaniso > 0).
TABLE 2.
Isotropic and anisotropic values of the effective 55Mn HFI tensors Ai (i = 1–4) used for the simulations of the X- and Q-band EPR and ENDOR spectra of the Ca2+-depleted PS II from spinach in the S2′ state (Fig. 2) and for the S2 states of native spinach PS II (46) and native and Sr2+-substituted PS II from T. elongatus (49)
| Species | OEC state | Tensor component | A1 | A2 | A3 | A4 |
|---|---|---|---|---|---|---|
| MHz | MHz | MHz | MHz | |||
| Spinach | -Ca2+ S2′ | isoa | 311 | 234 | 202 | 171 |
| anisob | 72 | −84 | −38 | −59 | ||
| Ca2+ S2 | iso | 298 | 248 | 205 | 193 | |
| aniso | 35 | −40 | −60 | −70 | ||
| T. elongatus | Ca2+ S2 | iso | 312 | 251 | 208 | 191 |
| aniso | 55 | −40 | −48 | −108 | ||
| Sr2+ S2 | iso | 332 | 243 | 203 | 173 | |
| aniso | 59 | −37 | −30 | −56 |
a The isotropic Ai,iso (i = 1–4) values are the averages of the principal values: Ai,iso = (Ai,x + Ai,y + Ai,z)/3.
b The anisotropy in the Ai values is expressed as the difference Ai,aniso = A⊥ − A‖ between the equatorial and axial components of the tensor. The equatorial and axial Ai values are defined as Ai,⊥ = (Ai,x + Ai,y)/2, Ai,‖ = Ai,z.
Spin State Energies of the Ca2+-depleted Mn4O5 Cluster in the S2′ State
The energy difference Δ of the paramagnetic ground spin state and the first exited state was estimated from the temperature dependence of the unsaturated X-band CW modified multiline signal of the Ca2+-depleted S2′ state. The measured intensities I1 of the derivative signal at a series of temperatures are depicted in a Curie plot versus 1/T in Fig. 3. This relation is approximately linear over the measured range from 14.4 to 5.5 K and extrapolates to 0 for T → ∞. This Curie behavior of the temperature dependence indicates that the Ca2+-depleted S2′ state features an S = 1/2 ground spin state energetically well separated from states of higher spin multiplicity. The temperature dependence of the S2′ modified multiline signal can be reproduced reasonably well with Δ ≥ 35 cm−1 corresponding to Jeff ≥ 12 cm−1 (see “Experimental Procedures”). This relatively large separation from states of higher spin multiplicity allows the S2′ state Mn4O5 spin system to be treated in the strong exchange limit, i.e. as an effective S = 1/2 spin state, as assumed in the previous section.
FIGURE 3.

Curie plot showing the dependence of the intensity I1 of the modified multiline derivative signal of the Ca2+-depleted S2′ state on the inverse temperature T. The error of the x-values comes from the calibration of the actual temperature at the sample position (see “Experimental Procedures”). The curves are simulations of the Curie temperature dependence over a range of Δ values on the basis of Equation S1 in the supplemental data and the simplified electron 2-spin coupling scheme for the OEC outlined under “Experimental Procedures.” Experimental parameters: MW frequency, 9.437 GHz; MW power, 0.1 milliwatt; modulation amplitude, 0.75 mT; time constant, 82 ms; temperatures, 5.5, 6.3, 7.3, 8.7, and 14.4 K.
EPR and 55Mn ENDOR of a Specifically Bound Mn2+ Ion
The Ca2+-depleted PS II preparations exhibit an additional EPR and ENDOR signal in all accessible S′ states that is not present in native PS II samples. At 5 K, Q-band ESE-detected field sweep EPR spectra of the dark-adapted Ca2+-depleted PS II preparations (S1′ state), in which the Mn4O5 cluster does not show a perpendicular mode EPR signal, displayed a broad EPR signal centered at g = 1.99 with a full width at half-maximum of ∼63 mT (Fig. 4, inset). A corresponding signal was not observed using CW X-band EPR spectroscopy; the signal is probably too broad to be discerned from the base-line drift in the CW EPR experiment (51). Q-band Davies ENDOR spectra were recorded at several magnetic fields in the RF frequency range of 30 to 400 MHz (Fig. 4). The 55Mn ENDOR spectra are dominated by two broad peaks between 100–195 MHz and another line centered at ∼370 MHz. The two lines at 100–195 MHz are dependent on the magnetic field and shift to higher frequencies with increasing magnetic field. The spectra also contain sharp proton signals, one centered at the 1H Larmor frequency (∼50 MHz) and a strongly coupled one at ∼75 MHz with decreasing amplitude at increasing field positions. Its partner at low frequency (∼25 MHz) lies outside the spectral range. No further low frequency signals were detected for this species using either ENDOR or electron spin echo envelope modulation (ESEEM).
FIGURE 4.

Q-band pulse ESE-detected field-swept EPR (inset) and Davies ENDOR experimental spectra (black solid traces) and simulations (Sim.; red dashed traces) of the Mn2+ ion bound to Ca2+-depleted PS II isolated from spinach and poised in the S1′ state. In the EPR spectrum, the region of the overlapping YD• EPR signal (g ≈ 2) is not displayed for clarity and was omitted in the simulations. The arrows indicate the four magnetic fields at which the ENDOR spectra were measured. Experimental parameters: MW frequency, 34.07 GHz; shot repetition rate, 5 ms; MW pulse length π, 72 ns; τ, 440 ns; Davies ENDOR, magnetic fields, 1195, 1208, 1224, 1260 mT (top to bottom); RF pulse length πRF, 4 μs; temperature, 5 K.
These EPR and 55Mn ENDOR signals can be readily assigned to high spin Mn2+ with S = 5/2, although their appearance is different from the spectra typically associated with Mn2+ complexes (see Discussion and Fig. 5A). Simultaneous simulations of the EPR and of four ENDOR spectra at different magnetic fields (Fig. 4, dashed red traces) are consistent with this assignment. They reproduce both the spectral breadth and line shape of the EPR absorption signal and the peaks in the four 55Mn ENDOR spectra. Besides a near-isotropic G tensor (principal values 1.983, 1.996, 2.002), the simulations yielded an almost isotropic HFI tensor with the principal components Ax = 256 MHz, Ay = 260 MHz and Az = 257 MHz, resulting in an isotropic average Aiso of 258 MHz. In addition, the simulations required a large fine structure parameter D = −2355 MHz with a pronounced rhombicity η = E/D = −0.38 of the zero-field splitting (ZFS). It is noted that the predominant contribution to the width of the EPR and ENDOR signals is the large and rhombic ZFS interaction (more information on the effect of the ZFS can be found in the supplemental data and Ref. 61). Hence, considering the good agreement of the measured and calculated EPR and ENDOR signals (Fig. 4), the optimized fine structure parameter D can be considered robust; i.e. a single set of D and E values is sufficient to rationalize the data. The fact that the inclusion of a distribution of the ZFS parameters is not required indicates that there are only small site-to-site inhomogeneities of the Mn2+ ligand sphere. Therefore, we propose that the Mn2+ ion is bound to one specific site in Ca2+-depleted PS II.
FIGURE 5.

Mn2+ and Ca2+ titrations monitored by Q-band 55Mn ENDOR. A, Q-band 55Mn Davies ENDOR spectra of dark-adapted Ca2+-depleted PS II samples (S1′ state) with 0.16, 1.2, and 4.0 eq (black, red, and blue trace) of Mn2+ ions added relative to the number of PS II RCs and of 40 μm MnCl2 (corresponding to 1.6 eq) dissolved in the same buffer used for the PS II titration experiments. For the titration curve, see supplemental Fig. S3. The spectra were smoothed using a 5-point moving average. B, titration of Ca2+-depleted PS II samples containing 1 eq of Mn2+ ions with respect to the PS II RCs with Ca2+. The relative 55Mn ENDOR signal amplitudes of Mn2+ ions bound to the PS II protein complex (black squares) and hexaquo-Mn2+ in solution (red circles), quantified as described under “Experimental Procedures,” are plotted against the equivalents of Ca2+ ions added to the samples. The concentrations of both Mn2+ species as a function of added Ca2+ were reproduced by a sigmoid fit curve (solid lines). The concentration of RCs in the samples was 25 ± 3 μm. Experimental parameters: MW frequency, 34.03 GHz; shot repetition rate, 5 ms; MW pulse length π, 16 ns; τ, 320 ns; magnetic field, 1224 mT; RF pulse length πRF, 4.5 μs; temperature, 5 K.
Mn2+ and Ca2+ Titration Experiments
To further investigate the Mn2+ species described in the previous section, Mn2+ and Ca2+ titration experiments of Ca2+-depleted PS II samples were performed, monitoring the CW EPR and ENDOR signal described above.
Mn2+/Ca2+ Titration Monitored by CW EPR
Mn2+ ions were added to Ca2+-depleted PS II samples and the characteristic S2′YZ• state split signal, S2′ multiline signal, and hexaquo-Mn2+ signal (not shown) were measured. The addition of ≤0.8 eq of Mn2+ ions relative to the number of PS II RCs did not quantitatively alter the three signals. The Mn2+ ions added are CW EPR-silent, as seen in the study of Booth et al. (51), which is consistent with a protein-bound Mn2+ species. In addition, this species does not cause any line broadening or even splitting of the signals from the OEC or the tyrosyl radicals. The addition of ≥0.8 equivalents of Mn2+ ions resulted in the appearance of the hexaquo-Mn2+ signal. The subsequent addition of Ca2+ to Ca2+-depleted, Mn2+-loaded PS II samples led to a loss of the S2′YZ• state split signal and of the multiline signal, as the Ca2+-reconstituted Mn4O5Ca cluster can proceed beyond the S2′ state upon illumination. A concomitant increase of the Mn2+ six-line signal was observed due to the release of the PS II-bound Mn2+ into solution (51).
Mn2+/Ca2+ Titration Monitored by 55Mn ENDOR
Mn2+ binding was also directly monitored by Q-band ENDOR. The concentrations of PS II-bound and solubilized Mn2+ ions in each sample were quantified by means of the relative amplitudes of their characteristic 55Mn ENDOR signals (Fig. 5A; for the titration curve, see supplemental Fig. S3). Without the addition of MnCl2, dark-adapted Ca2+-depleted PS II (S1′ state) always displayed the PS II-bound Mn2+ signal shown in Fig. 4. The addition of ∼0.8 eq of MnCl2 led to a 4–5-fold increase of this signal with only little free hexaquo-Mn2+ (15 ± 4%) present at the same time. This suggests that ∼20% of RCs contain a bound Mn2+ before exogenous addition of MnCl2 so that in the end a total of ∼1 eq Mn2+ is in the sample. The basal Mn2+ is likely derived from centers damaged during the Ca2+ depletion procedure and nominally corresponds to the loss of ∼5% Mn4O5(Ca) clusters. The high occupancy of the Mn2+ site suggests that it is of high affinity, with a dissociation constant KD that is too small to be determined here. From the employed concentrations of the binding partner, KD is expected to be in the submicromolar/nanomolar range. It is also noted that the addition of the chelating agent EDTA did not remove or alter the appearance of the bound Mn2+ signal, consistent with the protein site having a high affinity for Mn2+.
An additional Ca2+ titration was performed on the fully Mn2+-loaded Ca2+-depleted PS II (+0.8 eq of MnCl2, i.e. a final ratio of 1 Mn2+ ion per PS II RC). The Ca2+ concentrations ranged from 0 to 2400 eq Ca2+ per RC (0–60 mm). In Fig. 5B, the relative concentrations of the two Mn2+ species (PS II-bound and solubilized) are plotted against the equivalents of Ca2+ ions added. This behavior could be reproduced by a sigmoid curve with a half-saturation value of 700 Ca2+ ions per RC. This value is similar to 1200 eq of Ca2+ reported in Booth et al. (51). The difference may be due to the Ca2+ depletion method used, the low pH/citrate treatment in this study versus a NaCl salt wash (24) in the study of Booth et al. (51). Their differing effects on the extrinsic PS II subunit composition could alter the Ca2+ binding kinetics (see Refs. 24 and 51).
DISCUSSION
Location of the Mn2+ Binding Site
Based on the observations described above (see “Results”), a preliminary assignment can be made as to where the binding site of the Mn2+ ion is located. No strong magnetic interaction was observed between the Mn2+ ion and the Ca2+-depleted Mn4O5 cluster or the tyrosyl radical YZ• in the form of a broadening or splitting of the corresponding EPR signals. Thus, Mn2+ binding directly to the Ca2+ site of the OEC can be excluded. This Mn2+ ion must be at least 10 Å away not to be detectable via dipolar magnetic interaction. A similar argument holds for YD• (D2-Tyr-160), as it also displays an unperturbed EPR lineshape when Mn2+ is bound. These “exclusion zones” are indicated by green and violet spheres in Fig. 6A. There is, however, a long range dipolar interaction between the Mn2+ ion and YD• as evidenced by the relaxation enhancement of its EPR signal (51). Being smaller than the enhancement resulting from the Mn4O5Ca cluster in the S2 state suggests a weaker Mn2+-YD• interaction and thus a longer distance than the 31 Å measured between the cluster and YD (6).
FIGURE 6.

Ca2+ and potential Mn2+ binding sites in cyanobacterial PS II crystals. A, PS II crystal structure from T. vulcanus (6) (PDB accession number 3ARC) highlighting the Ca2+ ions (black spheres) as well as a Ca2+ binding site found in PS II from T. elongatus (gray sphere) and their distances to the paramagnetic entities Mn4O5Ca cluster, YZ•, and YD•. The 10 Å spheres around the latter indicate the approximate region in which a bound Mn2+ would cause a splitting of their EPR signals and thus can be excluded to contain the Mn2+ binding site. B and C, ligand environments of the Ca2+ ions in the extrinsic PsbO proteins from T. vulcanus and T. elongatus (T.e.) (5), respectively. Oxygen, nitrogen, and carbon atoms are shown in red, blue, and yellow, respectively. Differences between the PsbO proteins of these cyanobacterial species and from higher plant spinach are displayed by a sequence alignment in supplemental Fig. S4. All distances are in Å.
The binding and titration behavior can either be rationalized by a significant allosteric effect of Ca2+ on the Mn2+ site, or Mn2+ binding could take place directly at a depleted Ca2+ site. The recent crystal structure (6) of PS II from Thermosynechococcus vulcanus exhibits three additional Ca2+ sites at distances greater than 30 Å from the paramagnetic species monitored, i.e. the Mn4O5(Ca) cluster, YZ•, and YD• (Fig. 6A). In the structure of PS II from T. elongatus, a different Ca2+ site in PsbO has been identified (4, 5, 68), not found in the T. vulcanus crystals. All these Ca2+ sites are located on the lumenal/donor side of PS II in the subunit CP47, the cytochrome b559 subunit β (PsbF), and the extrinsic protein PsbO, and are solvent-accessible. It is not clear, however, whether Ca2+ binding at these sites is solely a crystallization artifact under the conditions used or of physiological relevance. With the exception of the two sites in PsbO, the Ca2+ sites appear to be of low affinity, as the Ca2+ ions are ligated to a large part by H2O and glycerol. In contrast, the two Ca2+ sites seen in the PsbO possess at least three ligands from amino acid side chains (Fig. 6, B and C) and thus are potentially of high affinity. In the homologous PsbO from spinach, which has also been reported to bind Ca2+ (69–71), Asn-197 and Val-198 of the binding motif in Fig. 6B correspond to the conserved residues Ser-286 and Val-287, whereas there is no equivalent for Thr-135. Glu-81, Glu-140, and His-257 in the other binding motif (Fig. 6C) correspond to Glu-146, Glu-205, and Glu-317 (for a sequence alignment, see supplemental Fig. S4). Mn2+ binding to PsbO has indeed been demonstrated previously in isolated PsbO from higher plants (72–74). As in the present study and Ref. 51, protein-bound Mn2+ did not show a CW EPR signal, but a six-line signal was observed after denaturation of the protein (73). PsbO was reported to show carbonic anhydrase activity, which was maximal in the presence of Mn2+ (74).
The magnetic properties of the Mn2+ ion provide information about the immediate ligand environment in this binding pocket. The D and E values of Mn2+ complexes of higher symmetry, such as Mn2+-EDTA and hexaquo-Mn2+, are significantly smaller than those for the PS II-bound Mn2+ described here (see supplemental data and Ref. 61). The large and highly rhombic ZFS reflects an asymmetric coordination sphere. Both the 7- and the 5-fold coordination geometries of the Ca2+ ions in PsbO from the two cyanobacterial species exhibit considerable asymmetry (Fig. 6, B and C). In addition, the large proton coupling seen also suggests the Mn2+ ion to have at least one water ligand. The absence of any smaller coupling, such as from 14N (not shown), indicates that the Mn2+ ion does not bind to a N-containing ligand residue like histidine. Thus, the absence of a (visible) water and the presence of His-257 as ligands of the Ca2+ ion in T. elongatus PsbO (Fig. 6C) favor Mn2+ binding to the Ca2+ site in PsbO identified in the T. vulcanus crystal structure (Fig. 6B).
PS II from higher plants exhibits an extrinsic subunit composition different from that of the cyanobacterial system. Higher plant lumenal PsbP has been reported to be capable of binding Mn2+ stoichiometrically (75, 76). Similar to Ca2+-depleted PS II in this study and in Ref. 51, isolated PsbP loaded with Mn2+ did not show a Mn2+ X-band CW EPR signal unless it was denatured. A bound Mn2+ could be detected by high field EPR spectroscopy and distinguished from non-specifically attached Mn2+, similar to the present study. It is noted though that the binding constant reported in Bondarava et al. (76) is probably incorrect; for a discussion, see Ref. 77. Moreover, the Mn2+ ion in PsbP could be (partially) replaced by Ca2+, and Zn2+ has been found to bind at one of the two proposed Mn2+ sites in PsbP crystals from spinach (PDB accession number 2VU4) and its cyanobacterial homologue CyanoP (78). Mn2+ bound to the PsbP would be at least 30 Å from either the OEC or YD, again consistent with the distance constraints identified above. Thus, PsbP could also contain the putative site of specific Mn2+ binding in Ca2+-depleted higher plant PS II.
The physiological role of the putative Mn2+ binding site is the delivery of Mn2+ to the OEC during photoassembly and/or the storage of Mn2+ during the damage/repair cycle of the D1 protein (see Refs. 79–82). Ca2+ is essential for photoactivation of the OEC. It was suggested to bind at a site within the PS II complex, which leads to a conformational change of the protein pocket where the OEC is assembled (i.e. the C terminus of D1). Thus, it appears reasonable that in the absence of Ca2+, it is favorable for the PS II supercomplex to sequester in a site Mn2+ that can be rapidly delivered upon an increase in Ca2+ concentration. In this scenario the lumenal Ca2+ concentration would be a signaling mechanism for OEC assembly and repair.
Spectral Properties of the Mn4O5 Cluster in the S2′ State Compared with Other S2 State Systems
The appearance of the 55Mn ENDOR spectra, the fitted 55Mn HFI tensors, and the ground-to-first excited state energy separation of the Ca2+-depleted S2′ state all fall within the natural spectral variations observed for the native S2 states in different species (41). This demonstrates that the basic electronic and thus also spatial structure of the Mn4O5 cluster remains intact upon Ca2+ removal. This confirms and further refines observations on the interatomic distances of the manganese ions from earlier EXAFS experiments (29).
The Ca2+-depleted S2′ state from spinach resembles the native S2 state from T. elongatus with regard to the spin state energies. Upon removal of the Ca2+ ion, Δ increases to ≥35 cm−1, which is much larger than for the native spinach S2 state (Δ = 3–6 cm−1) but more similar to T. elongatus (Δ = 12–25 cm−1) (41, 83, 84). In intact spinach PS II, the energy ladder is sensitive to MeOH addition. The mechanism by which MeOH binding perturbs the electronic structure of the S2 state was recently discussed in Su et al. (41). In the model proposed, MeOH binding to the OEC increases the electronic coupling of the pending manganese (MnA4) to the cuboidal (MnB3,MnC2,MnD1) unit. It is this effective coupling that defines the ground-to-first excited state energy difference Δ of the S2 state. Ca2+ depletion appears to have the same effect. However, the addition of MeOH did not modify the appearance of the S2′ state ESE and ENDOR spectra (not shown). It is emphasized though that this effect is of the same size as that of the variation between species and thus is unlikely to be of physiological significance.
Electronic Structure/Exchange Coupling Scheme of the Ca2+-depleted Mn4O5 Cluster in the S2′ State
To further rationalize the spectral results from the Ca2+-depleted Mn4O5 cluster, a spin coupling scheme for the S2′ state was developed. It was constructed to meet the following requirements: (i) a ground state of spin multiplicity S = 1/2, (ii) a ground-to-first excited state energy difference Δ ≈ 35 cm−1, (iii) spin projection factors |ρi| ≈ 1 for all four manganese electronic spins, and (iv) intrinsic ZFS constants di of the manganese ions that lie within the range found for mono- and dimeric model complexes, i.e. 1 cm−1 < |d| < 5 cm−1 for MnIII and |d| < 0.1 cm−1 for MnIV ions in an octahedral ligand environment (see Refs. 18, 47, and 49). The inferred structural (29) and spectral similarity of the native and the Ca2+-depleted manganese cluster suggest that the spin coupling scheme for the native S2 state (Fig. 7, c = 1) (18), in which MnD1 is the MnIII ion, can be used as a starting point. Calculated on the basis of the refined model of the OEC in the latest crystal structure (6), the basic arrangement of this scheme is in accordance with the spatial organization as described by Siegbahn and our group (17, 18, 47, 85), in which MnB3, MnC2, and MnD1 form a trimeric core unit connected to MnA4 by a di-μ-oxo bridge via MnB3 (Fig. 1). Thus, this scheme represents an extension of the (3 + 1)- or Y-coupling schemes, proposed earlier in EPR spectroscopic studies (42, 46, 47, 49), where JA4-C2 = JA4-D1 = 0.
FIGURE 7.
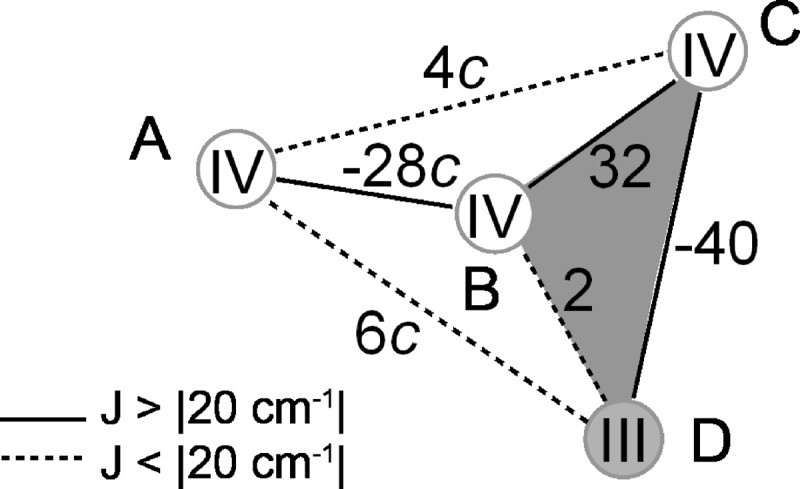
Model for the electronic structure of the OEC in the native S2 and Ca2+-depleted S2′ states calculated based on a refined DFT structure of the OEC (18) in the latest crystal structure (6). A–D label the manganese ions in their respective oxidation state, and the numbers give the pair-wise exchange coupling Jij between the electronic spins of the MnIII and MnIV ions in cm−1. The constant c is 1 in the originally derived model but differs for the various clusters and conditions, such as the presence or absence of MeOH. The S2′ state can be described by the scheme with c = 1.65.
The coupling topology fulfills criteria (i) and (iii) as ground spin state multiplicity and spin projection factors are the same for the two states, S2 and S2′. In contrast, their ground-to-first excited state energy differences Δ and effective 55Mn HFI tensors Ai, relevant for (ii) and (iv), are different. Thus, the Δ = 10.5 cm−1 calculated for the S2 state coupling scheme also differs from the experimental Δ ≥ 35 cm−1 determined for the S2′ state. Correlations between the exchange coupling scheme and this energy difference have been investigated in previous studies (41, 47). One mechanism by which Δ is influenced directly was shown to be the strength and the sign of the exchange coupling between MnA4 and the trimeric unit comprising MnB3, MnC2, and MnD1. An increase or decrease in the magnitudes of the coupling constant JA4-B3 results in a larger or smaller energy gap, respectively. As the monomer-trimer joint is in the vicinity of a possible binding site of a MeOH molecule, this rationalizes the effect of MeOH on the electronic structure of the Mn4O5Ca cluster in the native S2 state (41). For the Ca2+-depleted S2′ state, the coupling of MnA4 to the trimeric unit was varied by multiplying the respective exchange coupling constants JA4-B3, JA4-C2, and JA4-C2 by a factor c (Fig. 7). It can be readily calculated that with c = 1.65 (JA4-B3 = −46 cm−1, JA4-C2 = 7 cm−1, and JA4-D1 = 10 cm−1), Δ is 35 cm−1 and thus in the desired range.
For testing whether the obtained model also reproduces reasonable estimates for the intrinsic ZFS values di of the MnIII and MnIV ions, a brief description on how those can be assessed based on the inferred coupling scheme and the fitted effective HFI tensors is given in the supplemental data. Because of their inherently small ZFSs, the di values of the three MnIV ions can be assumed to be 0 cm−1 for the calculations of the intrinsic HFI tensors ai from the fitted effective Ai and the computed ρi tensors. MnIII ions generally exhibit an absolute isotropic HFI value |aiso| in the range between 165 and 225 MHz and considerable anisotropy defined as the difference ai,aniso = |a‖| − |a⊥| between the absolute values. MnIV ions tend to exhibit slightly larger isotropic HFI values (|aiso| = 187–253 MHz) and only small intrinsic HFI anisotropies (|aiso| < ∼30 MHz) (see Ref. 49). For the Ca2+-depleted S2′ state, a ZFS value dD1 of the MnD1III ion in the range of −2.24 to −2.31 cm−1 yields ai tensors consistent with the valence states of the individual manganese ions. An optimized ZFS value dD1 = −2.27 cm−1 leads to the spin projection and intrinsic HFI tensors ρi and ai listed in Table 3. In terms of the intrinsic isotropic and anisotropic HFI values, the calculated numbers match the prerequisites as found in the literature very well. As the ZFS dD1 = −2.24 to −2.31 cm−1 lies in the range usually found for MnIII ions (1 cm−1 < |d| < 5 cm−1), the developed model fulfills the four essential criteria imposed.
TABLE 3.
Calculated spin projection tensor components ρ⊥ and ρ‖, intrinsic 55Mn HFI tensor components a⊥ and a‖, and isotropic and anisotropic intrinsic HFI values aiso and aaniso for the Mn ions of the OEC in the Ca2+-depleted S2′ state on the basis of the electronic exchange coupling scheme in Fig. 7 with c = 1.65 and intrinsic ZFS values dA4 = dB3 = dC2 = 0 cm−1 for the MnIV ions and dD1 = −2.27 cm−1 for the MnD1III ion
| Manganese ion | ρ⊥ | ρ‖ | a⊥a | a‖a | aisob | aanisoc |
|---|---|---|---|---|---|---|
| MHz | MHz | MHz | MHz | |||
| MnA4 (MnIV) | 1.03 | 1.25 | 197 | 230 | 208 | 33 |
| MnB3 (MnIV) | −0.81 | −1.09 | 187 | 190 | 188 | 3 |
| MnC2 (MnIV) | −0.87 | −1.21 | 220 | 188 | 209 | −31 |
| MnD1 (MnIII) | 1.66 | 2.04 | 202 | 123 | 175 | −79 |
a The equatorial and axial ai values are defined as: a⊥ = (ax + ay)/2, a‖ = az.
b The isotropic aiso values are the averages of the individual components of the tensor aiso = (ax + ay + az)/3.
c The anisotropy of the a tensor is expressed as the difference aaniso = a‖ − a⊥ between the parallel and perpendicular tensor components.
Structural Implications of the Zero-field Splitting dD1 of the MnD1III Ion
The removal of the Ca2+ ion from the spinach OEC is found to result in a significant change of dD1 from −1.2 cm−1 (41) to −2.2 to −2.3 cm−1. This perturbation is larger than for the Ca2+/Sr2+ replacement in PS II from T. elongatus. For these systems, the intrinsic ZFS values of the MnD1III ion are relatively similar (Ca2+, dD1 = −1.3 cm−1; Sr2+, dD1 = −1.2 cm−1) (49). It is, however, noted that the signs of the dD1 and of the HFI anisotropy of the MnIII ion do not change between the Ca2+-depleted S2′ and the Ca2+-containing S2 state. These parameters can be related to the ligand sphere of the MnIII ion (86–88). Negative numbers for dD1 and aD1,aniso correspond to a 5B1g ground state, obtained in the cases of square pyramidal 5-coordinate or tetragonally elongated 6-coordinate ligand geometries. This suggests the coordination sphere of the MnD1III for the S2′ and S2 states to be similar. However, the increase in the magnitude of dD1 upon Ca2+ removal does indicate modifications of the precise binding mode, e.g. altered ligand distances and angles. One possible mechanism for altering dD1 is protonation of one of the μ-oxo bridges ligating the Ca2+ ion (Fig. 1) as a means of overall charge compensation of the cluster upon Ca2+ removal. It is known from model complexes that Mn-Mn distances are elongated upon protonation of Mn-O-Mn bridges (89). However, within the trimeric cuboidal unit, this lengthening could be strongly impaired for the MnC2-MnD1 distance. The fitted averaged distance of the Mn-Mn interactions at 2.7–2.8 Å from EXAFS on Ca2+-depleted PS II samples (29), however, does not allow for a conclusive assessment. Also, glutamate 189 of the D1 protein (D1-Glu-189), which directly coordinates the MnD1III (6, 17, 47) and potentially also the Ca2+ ion (18), could be reoriented upon Ca2+ depletion leading to a distortion of the coordination sphere and thus an altered dD1.
In the latest crystal structure, all four manganese ions are 6-coordinate (6). This, however, requires the O5 μ-oxo bridge to be a ligand of MnA4, MnB3, and MnD1, engendering very long Mn-O5 bond distances well outside the range seen in model complexes (see Ref. 18) and by EXAFS spectroscopy of the Mn4O5Ca cluster in PS II (90, 91). In most geometry-optimized DFT structures, such as those proposed by Siegbahn and our group (17, 18, 47), the position of O5 is significantly altered (Fig. 1). The O5 shifts toward the MnA4, forming a genuine μ-oxo bridge between MnA4 and MnB3, and results in MnD1 having an open coordination site. In this case, in the Ca2+-depleted S2′ state, Glu-189 might function as a bidentate ligand in a then tetragonally elongated 6-coordinate MnD1III ligand sphere, leading to the observed change of dD1.
Alternatively, the absence of Ca2+ may allow this open site to be occupied by a water molecule in the S2′ state (Fig. 8) forming a sixth ligand to MnD1. The MnD1-bound water molecule is the second substrate water in the mechanism proposed by Siegbahn (17), which potentially binds during the S2-to-S3 transition. Thus, within this model, one of the roles of Ca2+ in the active cluster would be to prevent the second substrate from binding too early in the reaction cycle (25, 92). This activity would presumably avoid detrimental side product formation (reactive oxygen species) and lead to single product (O2) formation. Consistent with this role for the Ca2+ ion is the known S state dependence of the affinity of Ca2+ to this site (93). It drops significantly in the S3 state, suggesting that in this state Ca2+ is less tightly bound, having a more flexible ligand sphere that potentially allows greater solvent access to the MnD1 ion.
FIGURE 8.

Scheme of the native Mn4O5Ca cluster in the S2 state and the Ca2+-depleted S2′ state represented by a hypothesized Mn4O5 cluster. In the putative S2′ state, the fast exchanging substrate water is already bound to MnD1III, filling the space of the Ca2+ ion. Ws and Wf denote the slowly and fast exchanging substrate waters, respectively (96, 99).
Besides μ-oxo bridge protonation, the loss of two positive charges is likely to be compensated by protonation of amino acid residues ligating the Ca2+ ion in the intact cluster. Other possibilities are the replacement of Ca2+ by monovalent Na+ in the samples or the absence of complete charge compensation, leaving the Mn4O5 cluster with an additional negative charge. It is evident that any of these modifications could have a critical effect on the catalytic capabilities of the cluster, especially with regard to proton-coupled electron transfer to YZ. In light of the proposed deprotonation sequence 1,0,1,2 for the individual oxidation steps starting from S0 (94), this would explain the Mn4O5 cluster being able to advance to S2′ but not from S2′YZ• to S3′.
Conclusions
This study demonstrates that Ca2+ is not required for conferring the critical electronic properties to the Mn4O5Ca cluster. This also confirms that Ca2+ is not essential for structural maintenance of the OEC. Its presence or absence does not affect the position of the only MnIII ion of the cluster in the S2/S2′ state (MnD1), and the contribution of the four manganese ions to the electronic states S2 and S2′ does not differ considerably. Thus, the necessity for Ca2+ in water splitting catalysis must be due to another functional role of the Ca2+ ion.
Although the exact mechanism of inhibition upon Ca2+ removal is still unclear, two models can be considered in terms of the two basic catalytic mechanisms proposed in the literature. (i) For mechanisms that involve O-O bond formation between a Ca2+-bound and a manganese-bound substrate water (be it a terminal ligand MnV = O or a μ-oxo bridge) (11, 95–97), inhibition due to Ca2+ depletion is readily explained. The enzyme is inactive, as it has lost a substrate binding site. It should be noted though that this model provides no rationale for the fact that the catalytic cycle is blocked at the stage of S2′YZ•. (ii) Instead, O-O bond formation has been proposed to follow a mechanism that results in the coupling of substrates bound to two manganese sites (be it between two terminal bound Mn-O ligands or involving a μ-oxo bridge via oxyl radical coupling) (10, 14, 16, 17, 98). Then, inhibition due to Ca2+ removal probably represents a secondary effect where the Ca2+ ion is critical for maintaining the H-bond network between YZ and the manganese cluster (6, 11, 30) as opposed (or in addition) to perturbation of substrate binding. Thus, Ca2+ removal would disable proton-coupled electron transfer during the S2′YZ•-to-S3′ transition, preventing substrate deprotonation and concomitant oxidation of MnD1III. Therefore, the elucidation of the mechanistic role of the Ca2+ ion in the OEC is tightly linked to understanding the mechanism of photosynthetic water splitting.
Supplementary Material
This work was supported by Max Planck Gesellschaft and the EU/Energy Network project SOLAR-H2 (FP7 Contract 212508).

This article contains supplemental Equation S1, Table S1, and Figs. S1–S4.
- OEC
- oxygen-evolving complex
- PS II
- photosystem II
- EXAFS
- extended X-ray absorption fine structure
- ENDOR
- electron-nuclear double resonance
- CW
- continuous wave
- RC
- reaction center
- ESE
- electron spin echo
- RF
- radio frequency
- HFI
- hyperfine interaction
- ZFS
- zero-field splitting
- DFT
- density functional theory
- mT
- millitesla
- MW
- microwave
- a.u.
- arbirtary units.
REFERENCES
- 1. Zouni A., Witt H. T., Kern J., Fromme P., Krauss N., Saenger W., Orth P. (2001) Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature 409, 739–743 [DOI] [PubMed] [Google Scholar]
- 2. Kamiya N., Shen J. R. (2003) Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 100, 98–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ferreira K. N., Iverson T. M., Maghlaoui K., Barber J., Iwata S. (2004) Architecture of the photosynthetic oxygen-evolving center. Science 303, 1831–1838 [DOI] [PubMed] [Google Scholar]
- 4. Loll B., Kern J., Saenger W., Zouni A., Biesiadka J. (2005) Toward complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature 438, 1040–1044 [DOI] [PubMed] [Google Scholar]
- 5. Guskov A., Kern J., Gabdulkhakov A., Broser M., Zouni A., Saenger W. (2009) Cyanobacterial photosystem II at 2.9 Å resolution and the role of quinones, lipids, channels, and chloride. Nat. Struct. Mol. Biol. 16, 334–342 [DOI] [PubMed] [Google Scholar]
- 6. Umena Y., Kawakami K., Shen J. R., Kamiya N. (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473, 55–60 [DOI] [PubMed] [Google Scholar]
- 7. Lubitz W., Reijerse E. J., Messinger J. (2008) Solar water-splitting into H2 and O2. Design principles of photosystem II and hydrogenases. Energy Environ. Sci. 1, 15 [Google Scholar]
- 8. Dau H., Zaharieva I. (2009) Principles, efficiency, and blueprint character of solar-energy conversion in photosynthetic water oxidation. Acc. Chem. Res. 42, 1861–1870 [DOI] [PubMed] [Google Scholar]
- 9. Messinger J., Renger G. (2008) in Primary Processes of Photosynthesis, Part 2: Principles and Apparatus (Renger G., ed.) pp. 291–349, Royal Society of Chemistry, Cambridge [Google Scholar]
- 10. Renger G., Renger T. (2008) Photosystem II. The machinery of photosynthetic water splitting. Photosynth. Res. 98, 53–80 [DOI] [PubMed] [Google Scholar]
- 11. McEvoy J. P., Brudvig G. W. (2006) Water-splitting chemistry of photosystem II. Chem. Rev. 106, 4455–4483 [DOI] [PubMed] [Google Scholar]
- 12. Nelson N., Yocum C. F. (2006) Structure and function of photosystems I and II. Annu. Rev. Plant Biol. 57, 521–565 [DOI] [PubMed] [Google Scholar]
- 13. Hillier W., Messinger J. (2005) in Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase (Wydrzynski T. J., Satho K., eds.) pp. 567–608, Springer, Dordrecht [Google Scholar]
- 14. Messinger J., Noguchi T., Yano J. (2012) in Molecular Solar Fuels, RSC Energy and Environment Series (Wydrzynski T. J., Hillier W., eds.) pp. 163–207, Royal Society of Chemistry, Cambridge [Google Scholar]
- 15. Yano J., Kern J., Irrgang K. D., Latimer M. J., Bergmann U., Glatzel P., Pushkar Y., Biesiadka J., Loll B., Sauer K., Messinger J., Zouni A., Yachandra V. K. (2005) X-ray damage to the Mn4Ca complex in single crystals of photosystem II. A case study for metalloprotein crystallography. Proc. Natl. Acad. Sci. U.S.A. 102, 12047–12052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kusunoki M. (2007) Mono-manganese mechanism of the photosystem II water splitting reaction by a unique Mn4Ca cluster. Biochim. Biophys. Acta 1767, 484–492 [DOI] [PubMed] [Google Scholar]
- 17. Siegbahn P. E. (2009) Structures and energetics for O2 formation in photosystem II. Acc. Chem. Res. 42, 1871–1880 [DOI] [PubMed] [Google Scholar]
- 18. Ames W., Pantazis D. A., Krewald V., Cox N., Messinger J., Lubitz W., Neese F. (2011) Theoretical evaluation of structural models of the S2 state in the oxygen evolving complex of photosystem II. Protonation states and magnetic interactions. J. Am. Chem. Soc. 133, 19743–19757 [DOI] [PubMed] [Google Scholar]
- 19. Ghanotakis D. F., Babcock G. T., Yocum C. F. (1984) Calcium reconstitutes high rates of oxygen evolution in polypeptide depleted photosystem II preparations. FEBS Letts. 167, 127–130 [Google Scholar]
- 20. Ghanotakis D. F., Topper J. N., Babcock G. T., Yocum C. F. (1984) Water-soluble 17- and 23-kDa polypeptides restore oxygen evolution activity by creating a high affinity binding site for Ca2+ on the oxidizing side of photosystem II. FEBS Lett. 170, 169–173 [Google Scholar]
- 21. Ono T., Inoue Y. (1988) Discrete extraction of the Ca2+ atom functional for O2 evolution in higher plant photosystem II by a simple low pH treatment. FEBS Lett. 227, 147–152 [Google Scholar]
- 22. Boussac A., Maison-Peteri B., Etienne A. L., Vernotte C. (1985) Reactivation of oxygen evolution of NaCl-washed photosystem II particles by Ca2+ and/or the 24-kDa protein. Biochim. Biophys. Acta Bioenerg. 808, 231–234 [Google Scholar]
- 23. Boussac A., Rutherford A. W. (1988) Nature of the inhibition of the oxygen-evolving enzyme of photosystem II induced by NaCl washing and reversed by the addition of Ca2+ or Sr2+. Biochemistry 27, 3476–3483 [Google Scholar]
- 24. Boussac A., Zimmermann J. L., Rutherford A. W. (1989) EPR signals from modified charge accumulation states of the oxygen evolving enzyme in Ca2+-deficient photosystem II. Biochemistry 28, 8984–8989 [DOI] [PubMed] [Google Scholar]
- 25. Sivaraja M., Tso J., Dismukes G. C. (1989) A calcium-specific site influences the structure and activity of the manganese cluster responsible for photosynthetic water oxidation. Biochemistry 28, 9459–9464 [DOI] [PubMed] [Google Scholar]
- 26. Miqyass M., van Gorkom H. J., Yocum C. F. (2007) The PSII calcium site revisited. Photosynth. Res. 92, 275–287 [DOI] [PubMed] [Google Scholar]
- 27. Yocum C. F. (2008) The calcium and chloride requirements of the O2 evolving complex. Coord. Chem. Rev. 252, 296–305 [Google Scholar]
- 28. Yachandra V. K., Yano J. (2011) Calcium in the oxygen-evolving complex. Structural and mechanistic role determined by X-ray spectroscopy. J. Photochem. Photobiol. B 104, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Latimer M. J., DeRose V. J., Yachandra V. K., Sauer K., Klein M. P. (1998) Structural effects of calcium depletion on the manganese cluster of photosystem II. Determination by x-ray absorption spectroscopy. J. Phys. Chem. B 102, 8257–8265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Diner B. A., Britt R. D. (2005) in Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase (Wydrzynski T. J., Satoh K., eds.) pp. 207–233, Springer, Dordrecht [Google Scholar]
- 31. Vrettos J. S., Stone D. A., Brudvig G. W. (2001) Quantifying the ion selectivity of the Ca2+ site in photosystem II. Evidence for direct involvement of Ca2+ in O2 formation. Biochemistry 40, 7937–7945 [DOI] [PubMed] [Google Scholar]
- 32. Boussac A., Rappaport F., Carrier P., Verbavatz J. M., Gobin R., Kirilovsky D., Rutherford A. W., Sugiura M. (2004) Biosynthetic Ca2+/Sr2+ exchange in the photosystem II oxygen-evolving enzyme of Thermosynechococcus elongatus. J. Biol. Chem. 279, 22809–22819 [DOI] [PubMed] [Google Scholar]
- 33. Dismukes G. C., Siderer Y. (1981) Intermediates of a polynuclear manganese center involved in photosynthetic oxidation of water. Proc. Natl. Acad. Sci. U.S.A. 78, 274–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Casey J. L., Sauer K. (1984) EPR detection of a cryogenically photogenerated intermediate in photosynthetic oxygen evolution. Biochim. Biophys. Acta Bioenerg. 767, 21–28 [Google Scholar]
- 35. Zimmermann J. L., Rutherford A. W. (1984) EPR studies of the oxygen-evolving enzyme of photosystem II. Biochim. Biophys. Acta Bioenerg. 767, 160–167 [Google Scholar]
- 36. Horner O., Rivière E., Blondin G., Un S., Rutherford A. W., Girerd J. J., Boussac A. (1998) SQUID magnetization study of the infrared-induced spin transition in the infrared-induced spin transition in the S2 state of photosystem II. Spin value associated with the g = 4.1 EPR signal. J. Am. Chem. Soc. 120, 7924–7928 [Google Scholar]
- 37. Boussac A. (1997) Inhomogeneity of the EPR multiline signal from the S2 state of the photosystem II oxygen-evolving enzyme. J. Biol. Inorg. Chem. 2, 580–585 [Google Scholar]
- 38. Åhrling K. A., Evans M. C., Nugent J. H., Pace R. J. (2004) The two forms of the S2 state multiline signal in photosystem II. Effect of methanol and ethanol. Biochim. Biophys. Acta 1656, 66–77 [DOI] [PubMed] [Google Scholar]
- 39. Force D. A., Randall D. W., Lorigan G. A., Clemens K. L., Britt R. D. (1998) ESEEM studies of alcohol binding to the manganese cluster of the oxygen-evolving complex of photosystem II. J. Am. Chem. Soc. 120, 13321–13333 [Google Scholar]
- 40. Åhrling K. A., Evans M. C., Nugent J. H., Ball R. J., Pace R. J. (2006) ESEEM studies of substrate water and small alcohol binding to the oxygen-evolving complex of photosystem II during functional turnover. Biochemistry 45, 7069–7082 [DOI] [PubMed] [Google Scholar]
- 41. Su J. H., Cox N., Ames W., Pantazis D. A., Rapatskiy L., Lohmiller T., Kulik L. V., Dorlet P., Rutherford A. W., Neese F., Boussac A., Lubitz W., Messinger J. (2011) The electronic structures of the S2 states of the oxygen-evolving complexes of photosystem II in plants and cyanobacteria in the presence and absence of methanol. Biochim. Biophys. Acta 1807, 829–840 [DOI] [PubMed] [Google Scholar]
- 42. Peloquin J. M., Campbell K. A., Randall D. W., Evanchik M. A., Pecoraro V. L., Armstrong W. H., Britt R. D. (2000) 55Mn ENDOR of the S2-state multiline EPR signal of photosystem II. Implications on the structure of the tetranuclear Mn cluster. J. Am. Chem. Soc. 122, 10926–10942 [Google Scholar]
- 43. Lorigan G. A., Britt R. D. (1994) Temperature-dependent pulsed electron paramagnetic resonance studies of the S2 state multiline signal of the photosynthetic oxygen-evolving complex. Biochemistry 33, 12072–12076 [DOI] [PubMed] [Google Scholar]
- 44. Kulik L., Epel B., Messinger J., Lubitz W. (2005) Pulse EPR, 55Mn-ENDOR, and ELDOR-detected NMR of the S2-state of the oxygen evolving complex in photosystem II. Photosynth. Res. 84, 347–353 [DOI] [PubMed] [Google Scholar]
- 45. Kulik L. V., Epel B., Lubitz W., Messinger J. (2005) 55Mn pulse ENDOR at 34 GHz of the S0 and S2 states of the oxygen-evolving complex in photosystem II. J. Am. Chem. Soc. 127, 2392–2393 [DOI] [PubMed] [Google Scholar]
- 46. Kulik L. V., Epel B., Lubitz W., Messinger J. (2007) Electronic structure of the Mn4OxCa cluster in the S0 and S2 states of the oxygen-evolving complex of photosystem II based on pulse 55Mn-ENDOR and EPR spectroscopy. J. Am. Chem. Soc. 129, 13421–13435 [DOI] [PubMed] [Google Scholar]
- 47. Pantazis D. A., Orio M., Petrenko T., Zein S., Lubitz W., Messinger J., Neese F. (2009) Structure of the oxygen-evolving complex of photosystem II. Information on the S2 state through quantum chemical calculation of its magnetic properties. Phys. Chem. Chem. Phys. 11, 6788–6798 [DOI] [PubMed] [Google Scholar]
- 48. Teutloff C., Pudollek S., Kessen S., Broser M., Zouni A., Bittl R. (2009) Electronic structure of the tyrosine D radical and the water-splitting complex from pulsed ENDOR spectroscopy on photosystem II single crystals. Phys. Chem. Chem. Phys. 11, 6715–6726 [DOI] [PubMed] [Google Scholar]
- 49. Cox N., Rapatskiy L., Su J. H., Pantazis D. A., Sugiura M., Kulik L., Dorlet P., Rutherford A. W., Neese F., Boussac A., Lubitz W., Messinger J. (2011) Effect of Ca2+/Sr2+ substitution on the electronic structure of the oxygen-evolving complex of photosystem II. A combined multifrequency EPR, 55Mn-ENDOR, and DFT study of the S2 state. J. Am. Chem. Soc. 133, 3635–3648 [DOI] [PubMed] [Google Scholar]
- 50. Stich T. A., Yeagle G. J., Service R. J., Debus R. J., Britt R. D. (2011) Ligation of D1-His-332 and D1-Asp-170 to the manganese cluster of photosystem II from Synechocystis assessed by multifrequency pulse EPR spectroscopy. Biochemistry 50, 7390–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Booth P. J., Rutherford A. W., Boussac A. (1996) Location of the calcium binding site in photosystem II. A Mn2+ substitution study. Biochim. Biophys. Acta Bioenerg. 1277, 127–134 [PubMed] [Google Scholar]
- 52. Berthold D. A., Babcock G. T., Yocum C. F. (1981) A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. EPR and electron-transport properties. FEBS Lett. 134, 231–234 [Google Scholar]
- 53. Arnon D. I. (1949) Copper enzymes in isolated chloroplasts. Polyphenoloxidase in β-vulgaris. Plant Physiol. 24, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Porra R. J., Thompson W. A., Kriedemann P. E. (1989) Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents. Verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. Biochim. Biophys. Acta Bioenerg. 975, 384–394 [Google Scholar]
- 55. Cinco R. M., Robblee J. H., Rompel A., Fernandez C., Yachandra V. K., Sauer K., Klein M. P. (1998) Strontium EXAFS reveals the proximity of calcium to the manganese cluster of oxygen-evolving photosystem II. J. Phys. Chem. B 102, 8248–8256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ono T., Inoue Y. (1989) Roles of Ca2+ in O2 evolution in higher plant photosystem II. Effects of replacement of Ca2+ site by other cations. Arch. Biochem. Biophys. 275, 440–448 [DOI] [PubMed] [Google Scholar]
- 57. Roberts A. G., Gregor W., Britt R. D., Kramer D. M. (2003) Acceptor and donor-side interactions of phenolic inhibitors in photosystem II. Biochim. Biophys. Acta 1604, 23–32 [DOI] [PubMed] [Google Scholar]
- 58. Reijerse E., Lendzian F., Isaacson R., Lubitz W. (2012) A tunable general purpose Q-band resonator for CW and pulse EPR/ENDOR experiments with large sample access and optical excitation. J. Magn. Reson. 214, 237–243 [DOI] [PubMed] [Google Scholar]
- 59. Epel B., Gromov I., Stoll S., Schweiger A., Goldfarb D. (2005) Spectrometer manager. A versatile control software for pulse EPR spectrometers Concepts Magn. Reson. Part B (Magn. Reson. Engineering) 26B, 36–45 [Google Scholar]
- 60. Stoll S., Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55 [DOI] [PubMed] [Google Scholar]
- 61. Sturgeon B. E., Ball J. A., Randall D. W., Britt R. D. (1994) 55Mn electron spin echo ENDOR of Mn2+ Complexes. J. Phys. Chem. 98, 12871–12883 [Google Scholar]
- 62. Rutherford A. W., Zimmermann J. L. (1984) A new EPR signal attributed to the primary plastosemiquinone acceptor in photosystem II. Biochim. Biophys. Acta Bioenerg. 767, 168–175 [Google Scholar]
- 63. Zheng M., Dismukes G. C. (1996) Orbital configuration of the valence electrons, ligand field symmetry, and manganese oxidation states of the photosynthetic water oxidizing complex. Analysis of the S2 state multiline EPR signals. Inorg. Chem. 35, 3307–3319 [DOI] [PubMed] [Google Scholar]
- 64. Kusunoki M. (1992) A new paramagnetic hyperfine structure effect in manganese tetramers. The origin of “multiline” EPR signals from an S2 state of a photosynthetic water-splitting enzyme. Chem. Phys. Lett. 197, 108–116 [Google Scholar]
- 65. Charlot M. F., Boussac A., Blondin G. (2005) Toward a spin coupling model for the Mn4 cluster in photosystem II. Biochim. Biophys. Acta 1708, 120–132 [DOI] [PubMed] [Google Scholar]
- 66. Teutloff C., Kessen S., Kern J., Zouni A., Bittl R. (2006) High-field (94 GHz) EPR spectroscopy on the S2 multiline signal of photosystem II. FEBS Lett. 580, 3605–3609 [DOI] [PubMed] [Google Scholar]
- 67. Lohmiller T., Cox N., Su J. H., Messinger J., Lubitz W. (2010) Proceedings of 15th International Congress on Photosynthesis Beijing, China, August 22–27, 2010, Zhejiang University Press/Springer, Dordrecht [Google Scholar]
- 68. Murray J. W., Barber J. (2006) Identification of a calcium-binding site in the PsbO protein of photosystem II. Biochemistry 45, 4128–4130 [DOI] [PubMed] [Google Scholar]
- 69. Zhang L. X., Liang H. G., Wang J., Li W. R., Yu T. Z. (1996) Fluorescence and Fourier-transform infrared spectroscopic studies on the role of disulfide bond in the calcium binding in the 33-kDa protein of photosystem II. Photosynth. Res. 48, 379–384 [DOI] [PubMed] [Google Scholar]
- 70. Heredia P., De Las Rivas J. (2003) Calcium-dependent conformational change and thermal stability of the isolated PsbO protein detected by FTIR spectroscopy. Biochemistry 42, 11831–11838 [DOI] [PubMed] [Google Scholar]
- 71. Kruk J., Burda K., Jemioła-Rzemińska M., Strzałka K. (2003) The 33-kDa protein of photosystem II is a low affinity calcium- and lanthanide-binding protein. Biochemistry 42, 14862–14867 [DOI] [PubMed] [Google Scholar]
- 72. Abramowicz D. A., Dismukes G. C. (1984) Manganese proteins isolated from spinach thylakoid membranes and their role in O2 evolution. II. A binuclear manganese-containing 34-kilodalton protein, a probable component of the water dehydrogenase enzyme. Biochim. Biophys. Acta 765, 318–328 [DOI] [PubMed] [Google Scholar]
- 73. Yamamoto Y., Shinkai H., Isogai Y., Matsuura K., Nishimura M. (1984) Isolation of an Mn-carrying 33-kDa protein from an oxygen-evolving photosystem-II preparation by phase partitioning with butanol. FEBS Lett. 175, 429–432 [Google Scholar]
- 74. Lu Y. K., Theg S. M., Stemler A. J. (2005) Carbonic anhydrase activity of the photosystem II OEC33 protein from pea. Plant Cell Physiol. 46, 1944–1953 [DOI] [PubMed] [Google Scholar]
- 75. Bondarava N., Beyer P., Krieger-Liszkay A. (2005) Function of the 23-kDa extrinsic protein of photosystem II as a manganese-binding protein and its role in photoactivation. Biochim. Biophys. Acta 1708, 63–70 [DOI] [PubMed] [Google Scholar]
- 76. Bondarava N., Un S., Krieger-Liszkay A. (2007) Manganese binding to the 23-kDa extrinsic protein of photosystem II. Biochim. Biophys. Acta 1767, 583–588 [DOI] [PubMed] [Google Scholar]
- 77. Bricker T. M., Frankel L. K. (2011) Auxiliary functions of the PsbO, PsbP, and PsbQ proteins of higher plant photosystem II. A critical analysis. J. Photochem. Photobiol. B 104, 165–178 [DOI] [PubMed] [Google Scholar]
- 78. Michoux F., Takasaka K., Boehm M., Nixon P. J., Murray J. W. (2010) Structure of CyanoP at 2.8 Å. Implications for the evolution and function of the PsbP subunit of photosystem II. Biochemistry 49, 7411–7413 [DOI] [PubMed] [Google Scholar]
- 79. Dasgupta J., Ananyev G. M., Dismukes G. C. (2008) Photoassembly of the water-oxidizing complex in photosystem II. Coord. Chem. Rev. 252, 347–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mulo P., Sirpiö S., Suorsa M., Aro E. M. (2008) Auxiliary proteins involved in the assembly and sustenance of photosystem II. Photosynth. Res. 98, 489–501 [DOI] [PubMed] [Google Scholar]
- 81. Grasse N., Mamedov F., Becker K., Styring S., Rögner M., Nowaczyk M. M. (2011) Role of novel dimeric photosystem II (PSII)-Psb27 protein complex in PSII repair. J. Biol. Chem. 286, 29548–29555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Komenda J., Michoux F., Nixon P. (2011) in Self-Healing at the Nanoscale, pp. 3–22, CRC Press [Google Scholar]
- 83. Pace R. J., Smith P., Bramley R., Stehlik D. (1991) EPR saturation and temperature dependence studies on signals from the oxygen-evolving centre of photosystem II. Biochim. Biophys. Acta Bioenerg. 1058, 161–170 [Google Scholar]
- 84. Åhrling K. A., Peterson S., Styring S. (1998) The S0 state EPR signal from the Mn cluster in photosystem II arises from an isolated S = 1/2 ground state. Biochemistry 37, 8115–8120 [DOI] [PubMed] [Google Scholar]
- 85. Siegbahn P. E. (2009) Water oxidation in photosystem II. Oxygen release, proton release, and the effect of chloride. Dalton Trans. 45, 10063–10068 [DOI] [PubMed] [Google Scholar]
- 86. Griffith J. S. (2009) The Theory of Transition-Metal Ions, Cambridge University Press, Cambridge, UK [Google Scholar]
- 87. Gerritsen H. J., Sabisky E. S. (1963) Paramagnetic resonance of trivalent manganese in rutile (TiO2). Phys. Rev. 132, 1507–1512 [Google Scholar]
- 88. Campbell K. A., Force D. A., Nixon P. J., Dole F., Diner B. A., Britt R. D. (2000) Dual-mode EPR detects the initial intermediate in photoassembly of the photosystem II Mn cluster. The influence of amino acid residue 170 of the D1 polypeptide on Mn coordination. J. Am. Chem. Soc. 122, 3754–3761 [Google Scholar]
- 89. Baldwin M. J., Stemmler T. L., Riggs-Gelasco P. J., Kirk M. L., Penner-Hahn J. E., Pecoraro V. L. (1994) Structural and magnetic effects of successive protonations of oxo bridges in high-valent manganese dimers. J. Am. Chem. Soc. 116, 11349–11356 [Google Scholar]
- 90. Yano J., Kern J., Sauer K., Latimer M. J., Pushkar Y., Biesiadka J., Loll B., Saenger W., Messinger J., Zouni A., Yachandra V. K. (2006) Where water is oxidized to dioxygen: structure of the photosynthetic Mn4Ca cluster. Science 314, 821–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pushkar Y., Yano J., Glatzel P., Messinger J., Lewis A., Sauer K., Bergmann U., Yachandra V. (2007) Structure and orientation of the Mn4Ca cluster in plant photosystem II membranes studied by polarized range-extended x-ray absorption spectroscopy. J. Biol. Chem. 282, 7198–7208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tso J., Sivaraja M., Dismukes G. C. (1991) Calcium limits substrate accessibility or reactivity at the manganese cluster in photosynthetic water oxidation. Biochemistry 30, 4734–4739 [DOI] [PubMed] [Google Scholar]
- 93. Boussac A., Rutherford A. W. (1988) Ca2+ binding to the oxygen evolving enzyme varies with the redox state of the Mn cluster. FEBS Lett. 236, 432–436 [Google Scholar]
- 94. Lavergne J., Junge W. (1993) Proton release during the redox cycle of the water oxidase. Photosynth. Res. 38, 279–296 [DOI] [PubMed] [Google Scholar]
- 95. Pecoraro V. L., Baldwin M. J., Caudle M. T., Hsieh W.-Y., Law N. A. (1998) A proposal for water oxidation in photosystem II. Pure Appl. Chem. 70, 925–929 [Google Scholar]
- 96. Messinger J., Badger M., Wydrzynski T. (1995) Detection of one slowly exchanging substrate water molecule in the S3 state of photosystem II. Proc. Natl. Acad. Sci. U.S.A. 92, 3209–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Dau H., Haumann M. (2008) The manganese complex of photosystem II in its reaction cycle. Basic framework and possible realization at the atomic level. Coord. Chem. Rev. 252, 273–295 [Google Scholar]
- 98. Yachandra V. K. (2005) in Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase (Wydrzynski T. J., Satoh K., eds.) pp. 235–260, Springer, Dordrecht [Google Scholar]
- 99. Hillier W., Wydrzynski T. (2008) 18O-water exchange in photosystem II. Substrate binding and intermediates of the water splitting cycle. Coord. Chem. Rev. 252, 306–317 [Google Scholar]
- 100. Messinger J., Robblee J. H., Bergmann U., Fernandez C., Glatzel P., Visser H., Cinco R. M., McFarlane K. L., Bellacchio E., Pizarro S. A., Cramer S. P., Sauer K., Klein M. P., Yachandra V. K. (2001) Absence of Mn-centered oxidation in the S2 → S3 transition. Implications for the mechanism of photosynthetic water oxidation. J. Am. Chem. Soc. 123, 7804–7820 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.