

Abstract
Prion replication occurs via a template-assisted mechanism, which postulates that the folding pattern of a newly recruited polypeptide chain accurately reproduces that of a template. The concept of prion-like template-assisted propagation of an abnormal protein conformation has been expanded to amyloidogenic proteins associated with Alzheimer, Parkinson, Huntington diseases, amyotrophic lateral sclerosis and others. Recent studies demonstrated that authentic PrPSc and transmissible prion disease could be generated in wild type animals by inoculation of recombinant prion protein amyloid fibrils, which are structurally different from PrPSc and lack any detectable PrPSc particles. Here we discuss a new replication mechanism designated as “deformed templating,” according to which fibrils with one cross-β folding pattern can seed formation of fibrils or particles with a fundamentally different cross-β folding pattern. Transformation of cross-β folding pattern via deformed templating provides a mechanistic explanation behind genesis of transmissible protein states induced by amyloid fibrils that are considered to be non-infectious. We postulate that deformed templating is responsible for generating conformationally diverse amyloid populations, from which conformers that are fit to replicate in a particular cellular environment are selected. We propose that deformed templating represents an essential step in the evolution of transmissible protein states.
Keywords: amyloid fibrils, infectivity, neurodegenerative diseases, prion diseases, prion protein, template-assisted mechanism
Three general mechanisms have been put forward to explain the diversity in etiology of prion diseases.1 Spontaneous conversion of normal, cellular isoform of the prion protein, PrPC, into disease-related infectious isoform, PrPSc, is believed to underlie the initial events leading to sporadic forms of prion diseases (Fig. 1A). Inherited prion diseases have been linked to PRNP gene mutations, which appear to facilitate the conformational transition of PrPC into disease-related states (Fig. 1B). In prion diseases acquired through the transmission, PrPSc seeds initiate conformational conversion of PrPC into PrPSc (Fig. 1C). The current article proposes a previously unrecognized mechanism by which PrPSc might emerge.
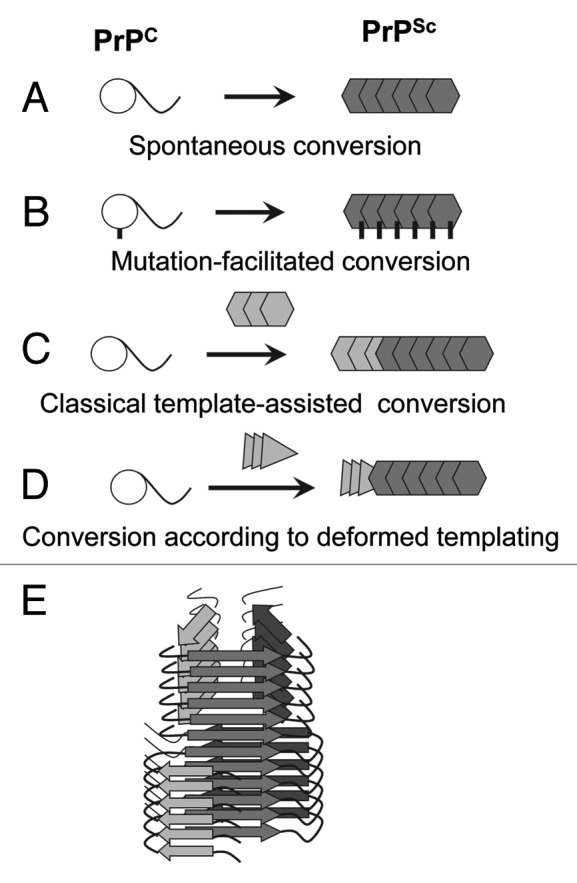
Figure 1. Four mechanisms for PrPSc formation. (A) Spontaneous conversion of PrPC into PrPSc is believed to underlie the sporadic forms of the prion diseases. (B) Disease-related mutations in the prion protein can facilitate the conversion of PrPC into PrPSc. (C) In prion diseases acquired via transmission, PrPSc replicates its pathogenic structure by recruiting and converting PrPC. According to the template-assisted model, the folding pattern of a newly recruited polypeptide chain accurately reproduces that of a PrPSc template. (D) A new mechanism referred to as “deformed templating” postulates that the formation of PrPSc de novo can be triggered by abnormal PrP structures substantially different from that of authentic PrPSc. Transformation from one cross-β folding pattern present in a template to a significantly different folding pattern, the one specific for PrPSc, occurs during deformed templating. (E) Schematic representation of a conformational switch in cross-β folding pattern within an individual fibril.
Regardless of etiology, PrPSc is believed to replicate its abnormal conformation via a template-assisted mechanism, which postulates that the folding pattern of a newly recruited PrP molecule accurately reproduces that of a PrPSc template.2 Existence of multiple prion strains within the same host suggests that multiple stable PrPSc conformations could be formed within the same amino acid sequence. Stability of any strain-specific disease phenotype and PrPSc properties during serial transmission of individual strains highlight the high fidelity of PrPSc replication and support the template-assisted mechanism. Substantial difficulties in generating authentic PrPSc in vitro experienced in the past point out that the PrPSc-specific folding pattern is not easily accessible.3 Nevertheless, authentic PrPSc can be produced in vitro in a highly efficient manner when a PrPSc template and appropriate co-factor environment are provided.4,5
Recent studies introduced a new concept that PrPSc and transmissible prion diseases can be induced by cross-β PrP structures substantially different from that of authentic PrPSc 6. Using recombinant full-length PrP (rPrP), amyloid fibrils were formed in vitro in the absence of cellular co-factors essential for producing authentic PrPSc and under solvent conditions, where PrPSc is largely denatured. As judged by FTIR spectroscopy and X-ray diffraction analysis, the PrP folding pattern within amyloid fibrils produced in the absence of co-factors was fundamentally different from that of brain-derived PrPSc 7,8. Moreover, using the sPMCA format that detects single PrPSc particles,9 the preparations of rPrP amyloid fibrils were found to be PrPSc free.6 Nevertheless, upon inoculation, rPrP fibrils induced a pathogenic process that led to transmissible prion diseases. These studies suggest that a new mechanism responsible for prion diseases different from the spontaneous conversion of PrPC into PrPSc or PrPSc-seeded conversion might exist (Fig. 1D).
When induced by rPrP amyloid fibrils formed in vitro, the disease pathogenesis displayed several peculiar features summarized below. First, development of the clinical disease was preceded by a long clinically silent stage that involved one or more serial passages, during which animals failed to show any detectible clinical symptoms.6,10 Second, while no clinical signs were detected during the clinically silent stage, significant amounts of partially proteinase K resistant PrP species referred to as atypical PrPres were observed during the silent stage.6 Atypical PrPres was structurally similar to rPrP amyloid seeds and, while clinically silent, appeared to represent one of the transmissible states of PrP. Slowly, atypical PrPres was replaced by authentic PrPSc, a process that coincided with development of the clinical stage of the disease. Third, significant transformation of neuropathological features including alteration in neurotropism was observed during progression from silent to clinical stages.6 These changes presumably reflect transformation of transmissible PrP states and evolution of authentic PrPSc.
Some of the above features (long clinically silent stage, transformation of neuropathological profile) are typically observed in cross-species prion transmission, a phenomenon referred to as a transmission barrier (reviewed in ref.11 and elsewhere). In cross-species transmission, the transmission barrier is attributed at least in part to species-specific differences in amino acid sequence of host PrPC and donor PrPSc. This is not the case for the pathogenic process induced by rPrP fibrils. A long clinically silent stage and transformation of neuropathological features were presumably due to a transition from a cross-β structure specific to rPrP fibrils to an alternative cross-β structure specific to PrPSc.
These studies on generating prion disease de novo using rPrP fibrils suggests that a new templating mechanism referred to as “deformed templating” different from the classical templating exists (Fig. 1D).6 According to deformed templating, daughter fibrils or particles can acquire a cross-β folding pattern different from that of seeds. If this mechanism is correct, rPrP fibrils with a structure different from that of PrPSc can trigger the formation of authentic PrPSc. The process of rPrP fibril-induced formation of PrPSc might involve a direct switching from one cross-β folding pattern present in rPrP fibrils to an alternative folding pattern that is specific for PrPSc (as depicted in Figure 1E) or occur in several steps. Nevertheless, recent studies on molecular imaging of individual fibrils provided a proof of principle that the conformational switching between two significantly different PrP folding patterns can occur within an individual fibril or particle.12 As a result of conformational switching, hybrid fibrils can be produced, where the cross-β spine changes its structure. The molecular details underlying this conformational switching remain to be elucidated. According to one model presented in Figure 1E, two global folding patterns might share a common structural motif which provides structural integrity to a hybrid structure. Because of only partial overlap between folding patterns in the two structures, seeding according to the deformed templating mechanism is expected to be substantially less effective than classical seeding where the folding pattern repeats itself. Poor seeding efficiency is consistent with a low attack rate and a long silent stage to clinical disease observed upon inoculating rPrP fibrils into wild type animals.6,10 Consistent with the deformed templating model, a number of studies illustrated that short amino acid stretches can serve as nucleation sites or facilitate transition of soluble proteins to self-replicating states.13,14 Moreover, the studies on yeast prion protein Sup35 revealed that short peptides derived from the Sup 35 primary sequence were sufficient to act as nucleation sites for triggering the Sup35 transition into a prion state and for specifying strain-specific features.15 Furthermore, another study on Sup 35 revealed that while large portions of the prion domain can be incorporated into cross-β sheets, very small segments appear to provide key links in bridging the species barrier.16 The results of these studies support the idea that short amino acid stretches could link β-sheets belonging to different folding patterns within hybrid fibrils.
Replication of abnormal protein states via a template-assisted mechanism is not unique to the prion protein. Recent years have witnessed an increasing number of studies where tissue extracts containing amyloid or aggregated forms of proteins or peptides linked to other neurodegenerative diseases were shown to seed aggregation of the same protein or peptide in a prion-like manner in experimental animal models or cultured cells (reviewed in refs.17,18 and elsewhere). While tissue-derived fibrils of tau, α-synuclein or Aβ were capable of seeding aggregation in vivo, it is yet unclear why amyloid fibrils produced in vitro from synthetic or recombinant analogs of these proteins failed to do so. It is likely that the amyloid ‘strains’ generated from recombinant proteins are not well adapted to replicate in the cellular environment perhaps due to their fast clearance or slow replication rates. By analogy to a long silent stage that accompanies genesis of mammalian prions,6 one can speculate that conformationally distinct amyloid states could be produced as a result of deformed templating triggered by seeds of recombinant tau, α-synuclein or Aβ fibrils, if sufficient experimental time is provided. Such a possibility has to be explored in future studies.
It is important to highlight the differences between deformed and classical templating. We postulate that deformed templating events are much rarer than replication according to classical templating. In fact, deformed templating could be considered as relatively rare byproduct of classical templating. While rare, the deformed templating events are expected to boost conformational diversity of fibril populations over time and generate heterogeneous conformers from which the conformers that are a better fit for replication in cellular environment could be selected. We propose that deformed templating is responsible for the molecular origin underlying the diversity of amyloid populations and represents an essential part in the evolution of transmissible protein states.
What is the spectrum of amyloid structures that could give rise to a truly pathogenic state capable of self-replicating in a cellular environment? Studies that employed transgenic mice with high PrPC expression level revealed that the efficiency in triggering prion disease and the incubation time to disease are variable depending on the conformation of rPrP amyloids inoculated into the animals.19,20 We do not know whether amyloids produced from non-prion proteins or peptides could trigger authentic PrPSc in vivo. In our experience, not every amyloid state produced in vitro from rPrP can trigger PrPSc and transmissible prion disease in wild type animals. This suggests that the relationship between a template conformation and its ability to give rise to authentic PrPSc is rather complex. On the other side of a spectrum is serum amyloid A (AA) derived amyloidosis, a process that appears to be one of the most promiscuous. Amyloidosis of AA protein can be triggered in animals by injecting not only AA fibrils but also amyloids produced from a broad range of proteins or peptides not related to the AA protein.21,22 Cross-seeding between non-homologous yeast prion proteins represents another example of promiscuous interactions between a fibrillar seed and a substrate in a cellular environment.23,24 The factors that control high fidelity vs. promiscuity in replication of abnormal protein states have yet to be explored. Nevertheless, the deformed templating model can provide a useful framework for future studies on this topic.
The studies on synthetic prions revealed that genesis of PrPSc could involve accumulation of transmissible yet clinically silent protein states such as atypical PrPres that appear to represent an intermediate step in the evolution of authentic PrPSc 6. While atypical PrPres did not cause the disease by itself, the role of atypical transmissible protein states in the etiology of neurodegenerative diseases should not be underestimated. Because much of the public health risk originates from asymptomatic stages, silent proteinopathies poses a potential risk for spreading neurodegenerative diseases via transmissible protein states that are clinically silent.
The implications of the new mechanism of deformed templating and clinically silent transmissible protein states are broad. How common are clinically silent transmissible states of the prion protein and other amyloidogenic proteins? Do clinically silent transmissible states play any role in the etiology of neurodegenerative diseases that are considered to be sporadic in origin? Do clinically silent transmissible states involved in different neurodegenerative diseases cross-communicate with each other? What are the implications of transmissible silent protein states for development of proper diagnostic approaches? Such questions will only be answered with more studies. Author: Please cite references 7 and 8 in the text.
Acknowledgments
This work was supported by NIH grant NS045585 to (I.V.B.)
Glossary
Abbreviations:
- PrPC
normal cellular isoform of the prion protein
- PrPSc
abnormal, disease-associated isoform of the prion protein
- rPrP
recombinant PrP
Footnotes
Previously published online: www.landesbioscience.com/journals/prion/article/19930
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen FE, Prusiner SB. Pathologic conformations of prion proteins. Annu Rev Biochem. 1998;67:793–819. doi: 10.1146/annurev.biochem.67.1.793. [DOI] [PubMed] [Google Scholar]
- 3.Legname G, Baskakov IV, Nguyen H-OB, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science. 2004;305:673–6. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 4.Castilla J, Saá P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature. 2003;425:717–20. doi: 10.1038/nature01979. [DOI] [PubMed] [Google Scholar]
- 6.Makarava N, Kovacs GG, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, et al. Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog. 2011;7:e1002419. doi: 10.1371/journal.ppat.1002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wille H, Bian W, McDonald M, Kendall A, Colby DW, Bloch L, et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proc Natl Acad Sci U S A. 2009;106:16990–5. doi: 10.1073/pnas.0909006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostapchenko VG, Sawaya MR, Makarava N, Savtchenko R, Nilsson KP, Eisenberg D, et al. Two amyloid States of the prion protein display significantly different folding patterns. J Mol Biol. 2010;400:908–21. doi: 10.1016/j.jmb.2010.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez-Montalban N, Makarava N, Ostapchenko VG, Savtchenk R, Alexeeva I, Rohwer RG, et al. Highly efficient protein misfolding cyclic amplification. PLoS Pathog. 2011;7:e1001277. doi: 10.1371/journal.ppat.1001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makarava N, Kovacs GG, Bocharova OV, Savtchenko R, Alexeeva I, Budka H, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119:177–87. doi: 10.1007/s00401-009-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hill AF, Collinge J. Subclinical prion infection. Trends Microbiol. 2003;11:578–84. doi: 10.1016/j.tim.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Makarava N, Ostapchenko VG, Savtchenko R, Baskakov IV. Conformational switching within individual amyloid fibrils. J Biol Chem. 2009;284:14386–95. doi: 10.1074/jbc.M900533200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ventura S, Zurdo J, Narayanan S, Parreño M, Mangues R, Reif B, et al. Short amino acid stretches can mediate amyloid formation in globular proteins: the Src homology 3 (SH3) case. Proc Natl Acad Sci U S A. 2004;101:7258–63. doi: 10.1073/pnas.0308249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ivanova MI, Sawaya MR, Gingery M, Attinger A, Eisenberg D. An amyloid-forming segment of beta2-microglobulin suggests a molecular model for the fibril. Proc Natl Acad Sci U S A. 2004;101:10584–9. doi: 10.1073/pnas.0403756101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tessier PM, Lindquist S. Prion recognition elements govern nucleation, strain specificity and species barriers. Nature. 2007;447:556–61. doi: 10.1038/nature05848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen B, Bruce KL, Newnam GP, Gyoneva S, Romanyuk AV, Chernoff YO. Genetic and epigenetic control of the efficiency and fidelity of cross-species prion transmission. Mol Microbiol. 2010;76:1483–99. doi: 10.1111/j.1365-2958.2010.07177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–40. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci. 2010;123:1191–201. doi: 10.1242/jcs.051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colby DW, Giles K, Legname G, Wille H, Baskakov IV, DeArmond SJ, et al. Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci U S A. 2009;106:20417–22. doi: 10.1073/pnas.0910350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colby DW, Wain R, Baskakov IV, Legname G, PAlmer CG, Nguyen H-O, et al. Protease-sensitive synthetic prions. PLoS Pathogen 2010; 6:e1000736. [DOI] [PMC free article] [PubMed]
- 21.Lundmark K, Westermark GT, Olsén A, Westermark P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc Natl Acad Sci U S A. 2005;102:6098–102. doi: 10.1073/pnas.0501814102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westermark P, Lundmark K, Westermark GT. Fibrils from designed non-amyloid-related synthetic peptides induce AA-amyloidosis during inflammation in an animal model. PLoS One. 2009;4:e6041. doi: 10.1371/journal.pone.0006041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN(+)] Cell. 2001;106:171–82. doi: 10.1016/S0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 24.Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci U S A. 2004;101:12934–9. doi: 10.1073/pnas.0404968101. [DOI] [PMC free article] [PubMed] [Google Scholar]