

Schizophrenia is a major therapeutic challenge of modern medicine, and one of the last frontiers of brain research. The illness is defined by delusions, hallucinations, disorganized behavior, and cognitive difficulties such as memory loss. It occurs in ≈1% of the world population and usually first appears in early adulthood. Although antipsychotic medications have dramatically improved the lives of patients with schizophrenia, the causes of the illness remain unknown.
Of the many contemporary theories of schizophrenia, the most enduring has been the dopamine hypothesis. As originally put by Van Rossum in 1967 (ref. 1, p. 321), “When the hypothesis of dopamine blockade by neuroleptic agents can be further substantiated, it may have fargoing consequences for the pathophysiology of schizophrenia. Overstimulation of dopamine receptors could be part of the aetiology … [emphasis added].” Indeed, this speculative sentence by Van Rossum foreshadows the title of the important work by Abi-Dargham et al. (2) in this issue of PNAS: “Increased baseline occupancy of D2 receptors by dopamine in schizophrenia.”
The discovery of the antipsychotic/dopamine receptor (3, 4), now commonly known as the dopamine D2 receptor, led to repeated confirmation that it is the primary site of action for all antipsychotics (3–5), including clozapine and quetiapine (6). All these drugs have different potencies at the receptor. The potency depends on the drug's dissociation constant at D2, which, in turn, relates to the rate of release of the drug from the D2 receptor. For example, the dopamine D2 receptor releases clozapine and quetiapine more rapidly than it does any of the other antipsychotic drugs (7, 8).
Given the tight correlation between the clinical potency and the D2-blocking action of the antipsychotic medications, dopamine overactivity could be the common denominator in the psychotic element of schizophrenia. This possibility has been actively investigated. Dopamine overactivity can be presynaptic (an excess of dopamine release from dopamine nerve terminals) or postsynaptic (an increase in the density of D2 receptors or an increase in postreceptor action). The innovative report by Abi-Dargham et al. (2) sheds light on both pre- and postsynaptic aspects by using an indirect method to measure the levels of endogenous dopamine in patients and controls.
Although numerous postmortem studies have consistently revealed D2 receptors to be elevated in the striata of patients with schizophrenia (9), the majority of the postmortem tissues examined have come from patients who have been treated with antipsychotics, raising the probability that the drugs themselves contributed to the elevation of D2 receptors. To measure the density of D2 receptors in never-medicated patients with schizophrenia, D2-selective ligands have been used with in vivo brain imaging methods (10–12). The results have not been consistent. Data with [11C]methylspiperone show elevated D2 receptors in schizophrenia (ref. 10, but see also ref. 12), whereas data with [11C]raclopride do not show such elevation (ref. 11 and discussed later in this paper). One major reason for this discrepancy is the quantitatively different effects of endogenous dopamine on [11C]methylspiperone and [11C]raclopride (see references in ref. 7).
Hence, one way to resolve this discrepancy is to measure D2 receptors after partial depletion of endogenous dopamine in patients. The work of Abi-Dargham et al. (2) provides this resolution. Fig. 1 summarizes the principle used by Abi-Dargham et al. Fig. 1 (Top) illustrates that the radiobenzamide (S)-(−)-3-[123I]iodo-2-hydroxy-6-methoxy-N-[(1-ethyl-2-pyrrolidinyl)methyl]benzamide ([123I]IBZM) binds to the same number of D2 receptors in control and schizophrenia individuals. That is, the “binding potential” was the same in both sets of subjects. However, after partial depletion of endogenous dopamine by oral ingestion of α-methylparatyrosine over 2 days, the binding of [123I]IBZM rose by 19% in schizophrenia but only by 9% in control subjects (Fig. 1, Bottom). In fact, when Abi-Dargham et al. examined the number of D2 receptors after partially removing the obscuring effect of endogenous dopamine, the D2 receptors were significantly elevated in schizophrenia patients as compared with control subjects. When the authors examined the data by subgroups, the results of increased receptors reached significance for previously medicated patients, but exhibited only a trend for patients who had never been medicated with antipsychotic drugs. Despite this lack of statistical significance in this latter group of patients, the empirical findings of Abi-Dargham et al. indicate that an increase in dopamine D2 receptors must occur, because it is not possible for patients to show a greater increase yet not have a higher number of D2 receptors. Thus, the paper by Abi-Dargham et al. provides support for both an increase in the level of dopamine as well as an increase in the number of D2 receptors in schizophrenia, compared to control subjects.
Figure 1.
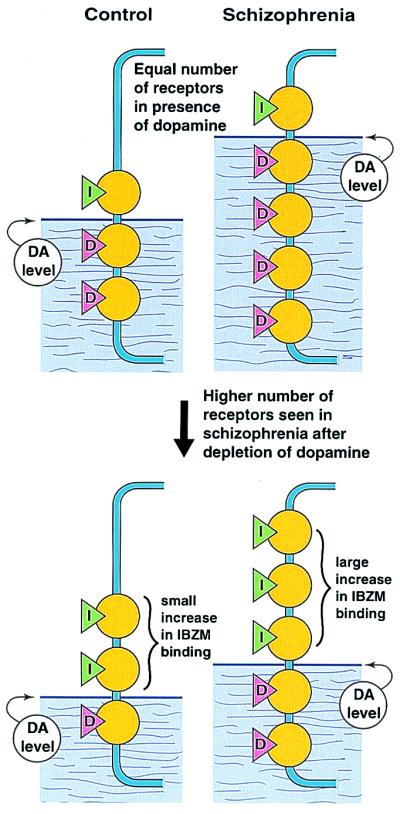
Method and findings of Abi-Dargham et al. (2) to reveal an increased occupancy of dopamine D2 receptors in schizophrenia. (Top) The number of dopamine D2 receptors, measured by the [123I]IBZM binding potential (green triangles with I), were the same in the brain striata of control and schizophrenia subjects. The levels of synaptic dopamine (pink triangles with D), which is higher in patients compared to control subjects, normally occupies most of the D2 receptors, masking the difference between control and schizophrenia individuals. (Bottom) After partial depletion of endogenous brain dopamine by oral ingestion of α-methylparatyrosine over 2 days, the binding of [123I]IBZM rose in both the control and schizophrenia subjects, but that for the patients rose significantly higher.
Schizophrenia, as compared with control subjects, also is associated with an increased releasability of dopamine (13, 14). A high release rate of dopamine reduces the binding of radiobenzamides to tissues (15, 16), but enhances the binding of radiospiperone (17, 18). Competition with endogenous dopamine, as well as dopamine-induced internalization of the D2 receptors, may account for the lessened binding of radiobenzamides to the tissue (13, 14), because the benzamides are generally water-soluble and have less ready access to vesicle-associated receptors. Radiospiperone compounds, by contrast, are highly lipid-soluble and readily permeate cell membranes to reach internalized receptors.
In addition to the two schizophrenia-associated factors of increased D2 receptors and increased dopamine release, there is a third factor. Dopamine D2 receptors exist in monomer, dimer, and oligomeric forms (19). The D2 monomer, but not the D2 dimer, is selectively labeled by a photolabel of radiospiperone (19). This finding is in contrast to a benzamide photolabel (for nemonapride), which readily binds to both monomers and dimers of D2 (19). This important distinction between benzamides and butyrophenones may explain why more D2 receptors are detected in schizophrenia (as compared to controls) by radiospiperone, even without depletion of endogenous dopamine. This finding is illustrated in Fig. 2, where the control individual has three D2 receptors, two in the dimer form and one in the monomer form. It is proposed that in schizophrenia, under the influence of increased release of endogenous dopamine, all three exist in the monomer form. Thus, radioraclopride binding would show no difference, but the binding of radiospiperone would be higher in the schizophrenia brain (as compared to controls) because of an increased number of monomers.
Figure 2.
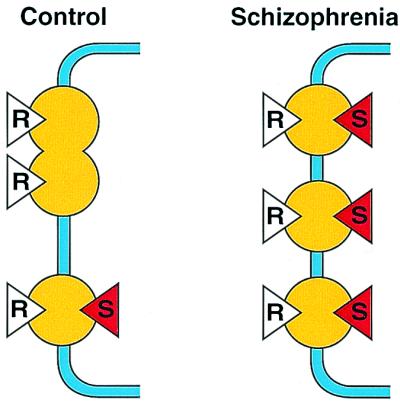
Possible model to account for the increased number of dopamine D2 receptors in schizophrenia seen with [11C]methylspiperone but not with [11C]raclopride. It is known that the photolabel of spiperone ([125I]azidophenethylspiperone) primarily or selectively labels monomers of D2 receptors, whereas the benzamide photolabel ([125I]azido-iodo-nemonapride) unselectively labels monomers, dimers, and oligomers of D2 receptors (see text). These findings suggest that even if there is no increase in the total population of D2 receptors in schizophrenia, an increase in the proportion of monomers caused by the increased level of dopamine in schizophrenia (see Fig. 1) would result in an increase in the binding of [11C]methylspiperone (red triangle with S) in schizophrenia but not with [11C]raclopride (white triangle with R).
The dopamine hypothesis has been much criticized. For instance, although therapeutic doses of most antipsychotics occupy 60% to 80% of the D2 receptors in patients, clozapine and quetiapine have been apparent exceptions, exhibiting clinical efficacy with only 10% to 45% occupation of D2 receptors (see references in ref. 7). It therefore has been suggested that the dopamine hypothesis of schizophrenia be extended into a serotonin-dopamine hypothesis. However, recent work on imaging both D2 and serotonin-2 receptors in patients taking antipsychotics fails to find evidence for a contribution from the occupation of serotonin receptors (20). For example, the threshold for clinical antipsychotic action remains at 65% occupation of D2 receptors in first-episode patients, whether one uses haloperidol, which has no serotonin-receptor blocking action, or risperidone or olanzapine, which block all serotonin-2 receptors but at doses far below those needed for clinical efficacy. Similarly, the threshold for extrapyramidal signs, which is ≈80% D2 occupancy, remains unaltered despite the presence of 100% block of serotonin-2 receptors for risperidone or olanzapine. It should also be noted that therapeutic doses of clozapine and quetiapine transiently occupy high levels of D2 receptors in patients, but the effect lasts for only the first few hours (6). Thus, the D2-occupying properties of clozapine and quetiapine are remarkable only for their short duration of action; they otherwise support the dopamine hypothesis of schizophrenia, as originally outlined by Van Rossum (1).
There is more to schizophrenia than psychosis. The psychological abnormalities and cognitive difficulties in schizophrenia precede and outlive the psychosis. The hypothesis of dopamine dysregulation is the best explanation for the psychotic episode in schizophrenia; the pathophysiology of other psychological and cognitive abnormalities in schizophrenia remains unclear. A combination of susceptibility genes (21) and other factors contributes to schizophrenia, and the net result dysregulates the dopamine neurotransmission system, leading to high release of dopamine, more D2 receptors, and an apparent predominance of monomer forms of D2. This dopamine dysregulation leads to the psychotic episode. Further research needs to uncover underlying mechanisms that predispose the brain to the dysregulation of the dopamine system (22). Until then, the dopamine hypothesis remains the main path to the origin and treatment of clinical signs and symptoms of psychosis in schizophrenia.
Footnotes
See companion article on page 8104.
References
- 1.Van Rossum J. In: Neuropsychopharmacology, Proceedings Fifth Collegium Internationale Neuropsychopharmacologicum. Brill H, Cole J, Deniker P, Hippius H, Bradley P B, editors. Amsterdam: Excerpta Medica; 1967. pp. 321–329. [Google Scholar]
- 2.Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles L S, Weiss R, Cooper T B, Mann J J, Van Heertum R L, et al. Proc Natl Acad Sci USA. 2000;97:8104–8109. doi: 10.1073/pnas.97.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seeman P, Chau-Wong M, Tedesco J, Wong K. Proc Natl Acad Sci USA. 1975;72:4376–4380. doi: 10.1073/pnas.72.11.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burt D R, Creese I, Snyder S H. Mol Pharmacol. 1976;12:800–812. [PubMed] [Google Scholar]
- 5.Seeman P, Lee T, Chau-Wong M, Wong K. Nature (London) 1976;261:717–719. doi: 10.1038/261717a0. [DOI] [PubMed] [Google Scholar]
- 6.Kapur, S., Zipursky, R., Jones, C., Shammi, C. S., Remington, G. & Seeman, P. (2000) Arch. Gen. Psychiatry, in press. [DOI] [PubMed]
- 7.Seeman P, Tallerico T. Am J Psychiatry. 1999;156:876–884. doi: 10.1176/ajp.156.6.876. [DOI] [PubMed] [Google Scholar]
- 8.Kapur S, Seeman P. J Psychiatr Neurosci. 2000;25:161–166. [PMC free article] [PubMed] [Google Scholar]
- 9.Seeman P. Neuropsychopharmacology. 1992;7:261–284. [PubMed] [Google Scholar]
- 10.Wong D F, Pearlson G D, Tune L E, Young L T, Meltzer C C, Dannals R F, Ravert H T, Reith J, Kuhar M J, Gjedde A. J Cereb Blood Flow Metab. 1997;17:331–342. doi: 10.1097/00004647-199703000-00010. [DOI] [PubMed] [Google Scholar]
- 11.Farde L, Wiesel F-A, Stone-Elander S, Halldin C, Nordström A-L, Hall H, Sedvall G. Arch Gen Psychiatry. 1990;47:213–219. doi: 10.1001/archpsyc.1990.01810150013003. [DOI] [PubMed] [Google Scholar]
- 12.Nordström A-L, Farde L, Eriksson L, Halldin C. Psychiatry Res Neuroimaging. 1995;61:67–83. doi: 10.1016/0925-4927(95)02732-d. [DOI] [PubMed] [Google Scholar]
- 13.Laruelle M, Abi-Dargham A, van Dyck C H, Gil R, De Souza C D, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi S S, et al. Proc Natl Acad Sci USA. 1996;93:9235–9240. doi: 10.1073/pnas.93.17.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Breier A, Su T P, Saunders R, Carson R E, Kolachana B S, de Bartolomeis A, Weinberger D R, Weisenfeld N, Malhotra A K, Eckelman W C, et al. Proc Natl Acad Sci USA. 1997;94:2569–2574. doi: 10.1073/pnas.94.6.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barton A C, Black L E, Sibley D R. Mol Pharmacol. 1991;39:650–658. [PubMed] [Google Scholar]
- 16.Itokawa M, Toru M, Ito K, Tsuga H, Kameyama K, Haga T, Arinami T, Hamaguchi H. Mol Pharmacol. 1996;49:560–566. [PubMed] [Google Scholar]
- 17.Chugani D C, Ackermann R F, Phelps M E. J Cereb Blood Flow Metab. 1988;8:291–303. doi: 10.1038/jcbfm.1988.64. [DOI] [PubMed] [Google Scholar]
- 18.Bischoff S, Gunst F. J Recept Signal Transduction Res. 1997;17:419–431. doi: 10.3109/10799899709036618. [DOI] [PubMed] [Google Scholar]
- 19.Zawarynski P, Tallerico T, Seeman P, Lee S P, O'Dowd B F, George S R. FEBS Lett. 1998;441:383–386. doi: 10.1016/s0014-5793(98)01588-9. [DOI] [PubMed] [Google Scholar]
- 20.Kapur S, Zipursky R B, Remington G. Am J Psychiatry. 1999;156:286–293. doi: 10.1176/ajp.156.2.286. [DOI] [PubMed] [Google Scholar]
- 21.Brzustowicz L M, Hodgkinson K A, Chow E W C, Honer W G, Bassett A S. Science. 2000;288:678–682. doi: 10.1126/science.288.5466.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertolino A, Breier A, Callicott J H, Adler C, Mattay V S, Shapiro M, Frank J A, Pickar D, Weinberger D R. Neuropsychopharmacology. 2000;22:125–132. doi: 10.1016/S0893-133X(99)00096-2. [DOI] [PubMed] [Google Scholar]