

Abstract
We have used recent structural advances in our understanding of the N-methyl-d-aspartate (NMDA) receptor amino terminal domain to explore the binding mode of multiple diaryl GluN2B-selective negative allosteric modulators at the interface between the GluN1 and GluN2B amino-terminal domains. We found that interaction of the A ring within the binding pocket seems largely invariant for a variety of structurally distinct ligands. In addition, a range of structurally diverse linkers between the two aryl rings can be accommodated by the binding site, providing a potential opportunity to tune interactions with the ligand binding pocket via changes in hydrogen bond donors, acceptors, as well as stereochemistry. The most diversity in atomic interactions between protein and ligand occur in the B ring, with functional groups that contain electron donors and acceptors providing additional atomic contacts within the pocket. A cluster of residues distant to the binding site also control ligand potency, the degree of inhibition, and show ligand-induced increases in motion during molecular dynamics simulations. Mutations at some of these residues seem to distinguish between structurally distinct ligands and raise the possibility that GluN2B-selective ligands can be divided into multiple classes. These results should help facilitate the development of well tolerated GluN2B subunit-selective antagonists.
Introduction
N-Methyl-d-aspartate (NMDA) receptors are ionotropic glutamate receptors that mediate excitatory postsynaptic signaling in the mammalian central nervous system. These receptors are widely expressed, require binding of both glycine and glutamate for activation, and have been implicated in physiological processes such as neuronal development, synaptic plasticity, and learning and memory (Citri and Malenka, 2008; Traynelis et al., 2010). Numerous pathological conditions have also been suggested to involve NMDA receptors, including ischemic damage (Hardingham and Bading, 2010), chronic pain (Wu and Zhuo, 2009), psychosis (Coyle et al., 2003), depression (Preskorn et al., 2008), and major degenerative disorders such as Parkinson's and Alzheimer's diseases (Reisberg et al., 2003; Hallett and Standaert, 2004; see also Mony et al., 2009a; Traynelis et al., 2010).
NMDA receptors are tetrameric assemblies of two glycine-binding GluN1 subunits and two glutamate-binding subunits, of which there are four types (GluN2A, GluN2B, GluN2C, GluN2D). The GluN2 subunit controls a wide range of functional properties of NMDA receptors and is differentially expressed throughout the central nervous system (Akazawa et al., 1994; Monyer et al., 1994). Each NMDA receptor subunit is composed of four discrete semiautonomous domains, which include the extracellular amino-terminal domain (ATD), the extracellular ligand-binding domain (LBD), the transmembrane domain, and the intracellular carboxyl-terminal domain (Fig. 1A; Sobolevsky et al., 2009). The GluN2 ATD regulates the agonist potency, deactivation time course, open probability and mean open/shut duration of different GluN2 subunits (Gielen et al., 2009; Yuan et al., 2009).
Fig. 1.

A ligand-based pharmacophore model was derived by structure-activity relationship studies mapped to the GluN1/GluN2B structure. A, a homology model representing the full-length NMDA receptor is shown with ifenprodil bound. TMD, transmembrane domain; CTD, carboxyl-terminal domain. B, a detailed representation of ifenprodil within the binding cleft interface between the GluN1 and GluN2B ATD is shown. The features of the general pharmacophore model for GluN2B antagonists are highlighted; the cyan spheres represent the hydrophobic features of rings A and B. The hydrogen bond donors are shown in blue (amine of the linker region and hydroxyl of ring B) where as the hydrogen bond acceptor is shown as a red sphere. C, a representation of the ligand-based pharmacophore model derived for ifenprodil and related compounds with the same color scheme for pharmacophore features as in B.
Since the discovery that the cerebral vasodilator ifenprodil has anti-ischemic actions (Carter et al., 1988; Gotti et al., 1988) and inhibits GluN1/GluN2B receptors with a 400-fold selectivity over the other GluN2 subunits (Williams, 1993), considerable effort has been directed toward the development of clinically useful GluN2B-selective NMDA receptor antagonists (Chenard et al., 1991; Mony et al., 2009a; Hansen et al., 2010a; Koller and Urwyler, 2010). Indeed, GluN2B antagonists have shown promising results in a number of clinical trials (Mony et al., 2009a; Traynelis et al., 2010). Multiple lines of evidence indicate that ifenprodil acts as a negative allosteric modulator, which may be one reason it is better tolerated than conventional NMDA antagonists. Ifenprodil and related compounds inhibit receptors incompletely (e.g., 90%) at saturating concentrations. Inhibition shows no voltage dependence and cannot be surmounted by increasing the concentration of glutamate or glycine (Williams, 1993; Kew et al., 1996). Ifenprodil and analogs show use-dependence in that the binding of glutamate increases the potency (i.e., decreases the IC50) of ifenprodil and vice versa (Kew et al., 1996). Ifenprodil has been proposed to show a higher potency for agonist-bound desensitized states than for other states (Kew et al., 1996).
Gallagher et al. (1996) first showed that ifenprodil's subunit-selectivity was controlled by the GluN2B amino terminal domain, and several subsequent mutagenesis studies have identified specific structural determinants of ifenprodil sensitivity in both the GluN1 and GluN2B subunits (Masuko et al., 1999; Perin-Dureau et al., 2002; Mony et al., 2009b). Karakas et al. (2011) used crystallographic methods to show that the binding site for ifenprodil lies at the interface of GluN1 and GluN2B ATD heterodimer rather than within a cleft of the bilobed ATD. These structural data shed further light on differences between the NMDA receptor ATDs and those of kainate and AMPA receptors. Whereas the ATDs of all ionotropic glutamate receptors have a bilobed architecture and consist of two domains (R1 and R2), NMDA receptors are unique compared with kainate and AMPA receptors in that the R1 and R2 lobes are oriented differently with respect to each other (Karakas et al., 2009, 2011; Farina et al., 2011). This difference in orientation of the R1 and R2 domains results in distinct association between the GluN1/GluN2B ATDs that differs in molecular detail from non-NMDA receptor ATDs, which can associate as homodimers or heterodimers (Zhao et al., 2012; Kumar et al., 2009, 2011) and extensive strong interactions between the R1-R1 and R2-R2 domains of the dimer. By contrast, the heterodimeric arrangement of the GluN1/GluN2B receptors shape a unique phenylethanolamine binding cavity that allows ifenprodil to reside between the R1-R1 and R1-R2 protein interfaces (Karakas et al., 2011). In the present study, we used a combination of mutagenesis and computational modeling based on crystallographic data to investigate the molecular details of binding of several classes of GluN2B-selective inhibitors to the GluN1/GluN2B ATD heterodimers. These results help elucidate the structural determinants of potency for this therapeutically relevant class of compounds.
Materials and Methods
Homology Modeling.
Amino acids are numbered with the initiating methionine set to 1. A protein family alignment was generated for the NMDA (GluN1/GluN2A–D), AMPA (GluA1–4), and kainate (GluK1–5) receptors using the program Muscle (Edgar, 2004). The unresolved loops of the GluN1/GluN2B crystal structure (Protein Data bank entry 3qel; 2.6-Å resolution; cocrystallized with ifenprodil) were built using Modeler 9v7 (Sali and Overington, 1994). The models were subjected to quality analysis using the PDBsum generator (http://www.ebi.ac.uk/pdbsum; Laskowski, 2009). Both the crystal structure and the subsequent models were prepared for further analysis using the protein preparation wizard (Schrödinger, Inc., Portland, OR). Side-chain optimization and the assigning of protonation states were performed and subsequently followed by a short minimization run to relieve the energetically unfavorable constraints. The sequence analysis and MULTISEQ modules from the VMD package were used to compare the proposed binding sites of the different GluN2 ATDs (Humphrey et al., 1996; Roberts et al., 2006).
Molecular Dynamics.
The heterodimer of GluN1/GluN2B was prepared for molecular dynamics (MD) simulation using the program Desmond (Bowers et al., 2006). Ligands and proteins were solvated using an orthorhombic box shape with a buffer distance of 10 Å using the simple point-charge water model. The overall system was neutralized at pH 7.0 using an ion concentration of 0.15 NaCl. The protonation state of ifenprodil used was as described by Kobayashi et al. (2006). The system was first relaxed with the use of the Desmond relaxation model. The completed equilibration run was followed by a 10-ns production run performed under NPT conditions using the Nose-Hoover thermostat (300 K) and particle-mesh Ewald electrostatics (Essmann et al., 1995) with a cutoff of 9 Å. Time-step calculations were performed every 2 fs. All figures from MD simulations were produced using VMD (Humphrey et al., 1996). Average structures were used to assess domain movement and prepared from the final 2 ns of the simulation. Domain movements of the different simulations were determined using structural alignments (backbone) of the R1 domains from both GluN1 and GluN2B followed by calculation of the center of mass of the R2 domains. The movement of the center of masses allows representation of ligand-induced shifts in the position of the R2 domains for the GluN1 and GluN2B. The residues used for alignment of the R1 were GluN1 residues 25 to 52, 58 to 95, 103 to 140, 277 to 297, and 316 to 341 and GluN2 residues 33 to 43, 65 to 132, 142 to 147, 289 to 310, and 335 to 341. The Cα atoms used for calculation of the center of mass of the R2 domains for GluN1 were 145 to 159, 162 to 183, 192 to 240, 245 to 250, and 265 to 270; for GluN2B, the Cα atoms used were 150 to 163, 168 to 174, 178 to 193, 197 to 206, 214 to 222, 226 to 249, 254 to 263, and 276 to 284. Movement of the center of mass domain was calculated from the average structure (last 2 ns) of the simulations performed under the different conditions. The root-mean-square fluctuation (RMSF) of the GluN1 and GluN2B subunits was calculated for the final 2 ns of the MD run by first aligning them to the backbone of the respective starting structures.
Molecular Docking.
Eighteen compounds were selected and prepared for docking by predicting their protonation states at pH 7.4 by using the program epik (Schrödinger, Inc.) with a range between 7.9 ± 1.5 and 10.0 ± 0.7 for the basic nitrogen. The resulting structures were subjected to a conformational search using confgen (Schrödinger, Inc.), and the most frequently occurring conformation was selected for further study. Glide (Schrödinger, Inc.) was used for docking, and the docking grids were generated for GluN2B using Gln110 as the center. The grid size was set to 33 Å, and the ligand diameter midpoint box was set to 14 Å for all three axes. Grids were generated with and without the water molecule facilitating the binding of the hydroxyl moiety of ifenprodil for all GluN2s. The extra precision scoring algorithm from Glide was used to identify the best scoring compounds during docking. All ligands were treated as flexible during docking, allowing for sampling of nitrogen inversion and ring conformations. Docking poses were restricted to 10 poses per ligand followed by postdocking minimization (minimization was performed with the Optimized Potentials for Liquid Simulations-all atoms force field) with a rejection threshold of 0.5 kcal/mol. The structure of the best-scoring docked pose for each of the 18 compounds in Table 1 is reported in the Supplemental data.
TABLE 1.
The structures of ligands docked to the hydrated model of GluN1/GluN2B
GluN2B-selective negative allosteric modulators that were used in docking studies to better understand the mechanism of action and their interactions within the GluN1/GluN2B interface are shown. The compounds are aligned with the general pharmacophore model (top) to illustrate how they interact with the binding interface. Published IC50 values were obtained from the references in the footnotes; for compound 93-138, the IC50 (0.384 μM) was determined from 20 oocytes as described under Materials and Methods.
| Compound | Structure | IC50 | Compound | Structure | IC50 |
|---|---|---|---|---|---|
| μM | μM | ||||
| Ifenprodila | 0.040 | Compound 68b | 0.065 | ||
| Ro-25-6981c | 0.009 | Compound 49b | 0.051 | ||
| Ro-63-1908d | 0.003 | Compound 15b | 0.028 | ||
| Traxoprodile | 0.0039 | Bensonprodilf | 0.008 | ||
| Eliprodilg | 1.0 | Compound 3ah | 0.002 | ||
| Compound 37ai | 0.0097 | Compound 3dh | 0.003 | ||
| Compound 29j | 0.051 | Compound 46bk | 0.005 | ||
| Compound 52b | 0.054 | Radiprodill | 0.003 | ||
| Compound 93–138 | 0.501 | Compound 12am | 0.018 |
Molecular Biology.
Site-directed mutagenesis was performed using the QuikChange protocol (Stratagene, La Jolla, CA) as described previously (Low et al., 2003). cDNAs for GluN1–1a (NR1–1a; GenBank accession numbers U11418 and U08261; hereafter GluN1), GluN2A (NR2A, GenBank accession number D13211), GluN2B (NR2B, GenBank accession number U11419), and GluN2D (NR2D, GenBank accession number L31611) were provided by Drs. S. Heinemann (Salk Institute, La Jolla, CA), S. Nakanishi (Kyoto University, Kyoto, Japan), and P. Seeburg (University of Heidelberg, Heidelberg, Germany).
Two-Electrode Voltage-Clamp Recordings from Xenopus laevis Oocytes.
Preparation and injection of cRNA, as well as two-electrode voltage-clamp recordings from X. laevis oocytes, was performed as described previously (Hansen et al., 2010b). In brief, oocytes were injected with 5 to 10 ng of cRNAs synthesized in vitro from linearized template cDNA. The ratio of GluN1 to GluN2 cRNA injected was 1:2. After injection, the oocytes were stored at 15°C in Barth's solution containing 88 mM NaCl, 2.4 mM NaHCO3, 1 mM KCl, 0.33 mM Ca(NO3)2, 0.41 mM CaCl2, 0.82 mM MgSO4, 5 mM Tris/HCl (pH adjusted to 7.4 with NaOH). Two-electrode voltage-clamp recordings were performed 2 to 3 days after injection at room temperature (23°C). The recording solution contained 90 mM NaCl, 1 mM KCl, 10 mM HEPES, 0.5 mM BaCl2, 0.01 mM EDTA (pH adjusted to 7.4 with NaOH). Solution exchange was computer controlled through an eight-modular valve positioner (Digital MVP Valve, Hamilton, CT). Voltage and current electrodes were filled with 0.3 and 3.0 M KCl, respectively, and current responses were recorded at a holding potential of −40 mV. Data acquisition and voltage control were accomplished with a two-electrode voltage-clamp amplifier (OC725; Warner Instruments, Hamden, CT). Only currents greater than 50 nA were included in the analysis. In all oocyte experiments, 100 μM glutamate and 100 μM glycine were used. Concentration-effect curves were constructed from data averaged from multiple oocytes, and fitted with the Hill equation: response (%) = (100 − minimum)/(1 − ([concentration]/IC50)nH) + minimum, where IC50 is the concentration of compound that inhibits 50% of the response, nH is the Hill slope, and the minimum is the steady-state minimal response at a maximally effective concentration of modulator. For some analyses, the minimum was forced to be 0.
Materials.
Ifenprodil hemitartrate (erythro enantiomers; Tocris, Ellisville, MO or Ascent Scientific, Princeton NJ), α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propanol (Ro-25-6981; Ascent Scientific), eliprodil (Tocris), and traxoprodil (CP101,606; Axon MedChem, Groningen, Netherlands) were made as 20 to 100 mM stocks in dimethyl sulfoxide and diluted to the desired concentration; final dimethyl sulfoxide content was never greater than 0.1% for all solutions. Compound 29 was synthesized as described in Tahirovic et al. (2008). Compounds 15, 49, and 68 were synthesized as described by Mosley et al. (2009).
Results
Ifenprodil Binding to the GluN1/GluN2B Interface.
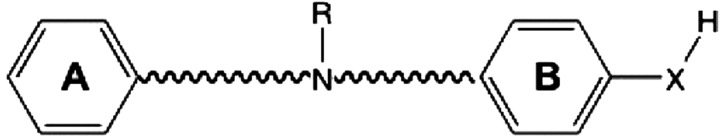
A large number of compounds have been synthesized in both industry and academia and tested for potential subunit-selective antagonistic effects on recombinant GluN1/GluN2B receptors (Chenard and Menniti, 1999; Hansen et al., 2010a; Koller and Urwyler, 2010). These synthetic efforts overwhelmingly suggest that active GluN2B-selective modulators consist of two nonpolar aromatic rings (an A ring and B ring) connected by a linker that typically contains a basic nitrogen (Fig. 1). In addition, the B ring often contains a hydrogen bond donor (Tamiz et al., 1998). Recent crystallographic data collected for an X. laevis GluN1 and rat GluN2B ATD heterodimer complex showed that the GluN2B-selective negative allosteric regulators occupy a binding site between the GluN1 and GluN2 protomers. Figure 1B shows the binding pocket for ifenprodil and highlights the hydrophobic (cyan) and hydrogen bond donor pharmacophore features (blue) buried within the dimer interface, which have a shared surface-accessible area of 1191 Å2 that accommodates ifenprodil (Karakas et al., 2011). To explore how the atomic contacts within the binding site vary for different ligands, we first built a hydrated model for the rat GluN1/GluN2B ATD dimer containing ifenprodil (1S,2S) for use in docking studies. This involved using the Rattus norvegicus sequence to generate the wild-type rat GluN1 and GluN2B structures, building loops for regions that were not resolved, optimizing hydrogen bond networks, and assigning protonation states of residues, followed by a short energy minimization. The models were used in docking studies and subsequent molecular dynamics simulations.
Eighteen compounds were selected for docking to the GluN1/GluN2B structures (Table 1). Evaluation of their atomic interactions and potency revealed several key features for ligand action at the GluN2B ATD heterodimer interface, a number of which were tested by mutagenesis (Table 2). In the next three sections, we consider the interactions of each of three sectors in the biaryl ifenprodil architecture (the A ring, biaryl linker, and B ring) with the ATD binding site.
TABLE 2.
The structural determinants of ifenprodil inhibition
Fitted IC50 values are shown to two significant figures and were determined from composite concentration-effect curves constructed from recordings in 8 to 41 oocytes (n) as described under Materials and Methods; the steady-state response was fixed at 0 for all conditions except GluN2B wild type. The following GluN2B mutations had less than 1.5-fold effect on the ifenprodil IC50 value: I50A, K51A, H127A, Q153A, Q180R, C232A, V258A, W285A, R328P (n = 4–20 oocytes per mutant). The ratio of IC50 values compared with control run during the same experiment.
| GluN2B mutation | IC50 | IC50 Mutant/IC50 Wild Type | n |
|---|---|---|---|
| μM | |||
| Wild type | 0.10 | 41 | |
| D101Aa | 47 | 246 | 8 |
| D102Aa | 0.34 | 1.8 | 8 |
| Q110A | 0.90 | 9.0 | 11 |
| Q110E | 0.039 | 0.39 | 25 |
| L205A | 0.28 | 2.8 | 14 |
| G212A,D213K | 2.1 | 21 | 20 |
| D213A | 0.79 | 7.9 | 14 |
| Y231A | 60 | 598 | 18 |
| T233Aa | 8.1 | 43 | 9 |
| T233S | 3.3 | 33 | 15 |
| S281A | 0.22 | 2.2 | 14 |
| Y282A | 12 | 121 | 23 |
| I299A | 0.28 | 2.8 | 14 |
Data are from mouse GluN2B coexpressed with rat GluN1, and IC50 values for ifenprodil were compared with IC50 from wild-type mouse GluN2B (0.19 μM). All other data are from rat GluN1/GluN2B receptors.
Molecular Interactions of the A Ring with the GluN1/GluN2B ATD Interface.
A number of studies suggest that the A-ring interactions with GluN2B (see Fig. 1B) are critical to activity (Chenard and Menniti, 1999, Tahirovic et al., 2008). Molecules lacking this ring are inactive (e.g., compound 6 in Chenard et al., 1991), and substitutions on this ring, such as dichloro in the case of propanolamines, strongly control potency (Tahirovic et al., 2008). The binding interface representing the hydrophobic A ring consists of residues from the R1 domains of both the GluN1 (Ala75, Tyr109, and Thr110) and GluN2B subunits (Pro78, Ile82, Ile111, and Phe114; Figs. 1B and 2). Among these, residues GluN1 Ala75, GluN2B Ile82, and GluN2B Phe114 are essential for ifenprodil sensitivity, presumably because they stabilize both the dimer interface and the pocket responsible for the hydrophobic character of the A-ring pharmacophore. This pocket is highly conserved with the exception of GluN2B Ile111 (Fig. 2), at which mutagenesis (I111S) had surprisingly little effect on ifenprodil potency (Karakas et al., 2011). Moreover, residues that facilitate the correct formation of the binding pocket, such as GluN1 His134, Ser108, Phe113, and Tyr114, are known to control ifenprodil potency (Masuko et al., 1999); these residues are not shown in Figs. 1 and 2 to improve clarity.
Fig. 2.

Residues that are homologous to known determinants of ifenprodil binding. A, color-coded model of the conservation found between the GluN2A-D subunits. Blue represents identical residues, whereas red shows similar residues. Note the high conservation in the LBD-TM (transmembrane) region compared with the lower conservation within the ATD region. B, two-dimensional illustration of the most important interactions formed between the GluN1/GluN2B interface and ifenprodil. A table gives the GluN2B residues that interact with ifenprodil and corresponding residues within the other GluN2 subtypes. C, summary of the sequence similarity between the different GluN2 subunits.
The A ring usually adopts a similar position among various ligands for all docked compounds, consistent with crystallographic data for ifenprodil and Ro 25-6981 (Fig. 3). The similarity of the ring contacts suggests that this set of interactions does not distinguish among ligands. Among the compounds docked, several series have demonstrated strongly enhanced potency with dichlorosubstituted A rings (Tahirovic et al., 2008; Mosley et al., 2010). For example, a 3,4-dichloro substitution of the benzene A ring increased the activity within two series of compounds by more than 100-fold (Tahirovic et al., 2008; Mosely et al., 2009). The apparent formation of a π-σ interaction between GluN2B Phe114 and the ligand A ring can in part explain the increased activity induced by the addition of chlorine atoms in the meta and para positions of the A ring (Imai et al., 2008; Matter et al., 2009). From the crystal structure of GluN1/GluN2B ATD in complex with ifenprodil and our docking studies, we speculate that a π-σ interaction takes place between the σ-hole of the para-positioned chloro and GluN2B Phe114 (e.g., Fig. 4B). The meta-positioned chloro is thought to form a halogen bond with the hydroxyl of GluN1 Thr110 (Fig. 2B) as well (Auffinger et al., 2004; Clark et al., 2007). These halogen substitutions should not only facilitate the binding within the dimer interface, but also decrease the solubility of the A ring, striking a good balance with respect to the desolvation energy, which can help to explain the observed 100-fold increase in activity after modification. The lower cost of desolvation by halogen substitution rather than strong hydrogen bond forming substituents such as a hydroxyl are further supported by the observation that the mono-substituted hydroxyl A rings are on average 90-fold less active than the 3,4-dichloro substituted A ring. Likewise, the 3,4-dihydroxy substituted A ring has an IC50 of greater than 100 μM (2000-fold lower potency than 2,3 dichloro; see Table 2 in Tahirovic et al., 2008).
Fig. 3.

An illustration of superimposed docking poses for ligands in Table 1 for the A and B rings of the GluN1/GluN2B diaryl negative allosteric modulators. The cyan spheres represent the pharmacophore features previously identified for the A and B rings.
Fig. 4.

Docking results of the various stereoisomers of ifenprodil. A, the docking poses of ifenprodil with stereochemistry of 1S,2S (gray) and 1S,2R (green) are shown. B, the docking poses of ifenprodil with stereochemistry of 1R,2R (gray) and 1R,2S (green) are shown.
Molecular Interactions of the Diaryl Linker with the GluN1/GluN2B ATD Interface.
The ATD interface that accommodates the linker region is conserved between GluN2A and GluN2B (Fig. 2) and is made up of residues from both the GluN1 (Tyr109, Gly112, Phe113, Ile133, and Leu135) and GluN2B R1 domains (Glu106, Ala107, and Gln110). This portion of the binding pocket apparently can accommodate a wide range of chemical moieties (Tamiz et al., 1998; Mony et al., 2009a; Hansen et al., 2010a). The linkers between the A and B rings of GluN2B-selective modulators are chemically diverse and include alkyl chains, aryl chains, amides, amines, ethers, piperidine rings, or piperazine rings. In general, the optimal linker length is often between 9 and 11 Å (Chenard et al., 1991; Marinelli et al., 2007; Tahirovic et al., 2008; Mosley et al., 2009). The piperidine ring of Ro 25-6981 and ifenprodil, with a pKa of 9.05 (Kobayashi et al., 2006), is protonated under physiological pH and forms a hydrogen bond with the oxygen of Gln110 (Karakas et al., 2011). Mutation of GluN2B Gln110 to Ala reduced ifenprodil potency by 10-fold (Table 2), consistent with the idea that this interactions helps to stabilize ligand orientation in the binding site. The hydroxyl group on the linkers of both (R,S)-ifenprodil and (S,S)-Ro 25-6981 depicted at the GluN1/GluN2B subunit interface (Karakas et al., 2011) makes a hydrogen bond with the carbonyl group of Ser132 (GluN1). However, if the common S stereoisomer at the hydroxylated center is inverted (e.g., R,R and S,R, respectively), the OH group can also make a hydrogen bond with Gln110 (GluN2B) (Fig. 4). The methyl groups of the two compounds project in opposite directions with that of ifenprodil protruding toward the GluN1 subunit interacting with Ile135, whereas that of Ro 25-6981 is directed toward the GluN2B subunit and interacts with Phe176 and Pro177 (Karakas et al., 2011). Figure 4 illustrates that the four stereoisomers of ifenprodil (R,S; S,R; S,S; R,R) accommodate all variations of inversion at the linker stereogenic centers without perturbation of the docking poses. This raises the possibility for engineering different potencies and activities within the linker region by designing functional groups to form different atomic interactions with either GluN1 or GluN2B residues in this region. Evaluation of the different stereoisomers of ifenprodil further emphasize this point. For example, (+)-erythro-ifenprodil is 4-fold more potent in neuroprotection assays than its (−) enantiomer, whereas the (−)-threo-ifenprodil is 4-fold more potent than its (+) enantiomer (Chenard et al., 1991; Hashimoto and London, 1995; Avenet et al., 1996). The 1S,2S-(+)-threo stereoisomer of the ifenprodil analog traxoprodil is less potent at α1 adrenergic receptor than the 1R,2R-(−)-threo compound [1-[(1R,2R)-1-hydroxy-1-(4-hydroxyphenyl)-2-propanyl]-4-phenyl-4-piperidinol (CP101,581)], which led to further development of compounds with this stereochemistry (Chenard et al., 1995).
Although the various stereoisomers of ifenprodil show differing potency (Chenard et al., 1991), the absolute stereochemistries of (±)-erythro- and threo-ifenprodil have not been determined. We investigated the ligand-protein interactions of (±)-threo- and (±)-erythro-ifenprodil by docking all possible stereoisomers. Each of the latter could be accommodated within the binding cavity with similar binding scores and with indistinguishable docking energies that fell within a −12.1 ± 1.0 kcal/mol window. However, a number of distinct molecular interactions differ among the stereoisomers (Fig. 4). For example, the ifenprodil hydroxyl group with (1R,2R) absolute stereochemistry prefers to form a hydrogen bond with GluN1 Ser132, whereas the hydroxyl group of (1R,2S)-ifenprodil is near both Ser132 (GluN1) and Gln110 (GluN2B), making hydrogen bonding possible to either of these acceptors. Furthermore, ifenprodil with (1S,2R) or (1S,2S) absolute stereochemistry prefers hydrogen bond formation with GluN2B Gln110 (NH2) and Glu106 (Fig. 4). Moreover, docking suggested that the mutation GluN2B (Q110E) should increase ifenprodil potency by allowing the formation of a hydrogen bond with the carboxylate of the glutamate residue. Evaluation of this mutation confirmed this prediction, as GluN2B (Q110E) increased ifenprodil (stereoisomers) potency by 2.5-fold compared with wild-type GluN2B (Table 2). Perin-Dureau et al. (2002) showed a significant reduction in ifenprodil potency when mutating GluN2B Glu106-to-Ala. Our docking studies suggest the possibility that Glu106 is able to form a hydrogen bond with the hydroxyl of ifenprodil, and the loss of this interaction may account for the reduction of ifenprodil potency. In addition, Glu106 makes interactions with Ala107 (NH) as well as it own (NH), which may be important for the structural integrity of the GluN2B subunit. Elimination of this intraprotein H-bond may also contribute to the reduction in potency for mutations at Glu106.
Docking studies identified a number of additional locations within the binding pocket that make atomic contacts with the linker region of modulators of GluN1/GluN2B. The most frequently observed hydrogen bonds between GluN1 and the di-aryl compounds were found to be with the carbonyl backbones of Tyr109, Ser132, Ile133, and Gly112 (e.g., traxoprodil, compound 12a, eliprodil, compound 68, trifluperidol). In addition, frequent atomic interactions were observed between the di-aryl compounds and Arg115 (GluN1), Glu106 (GluN2B), and Gln110 (GluN2B; e.g., ifenprodil, Ro-25-6981, compound 52). There did not seem to be a systematic pattern to experimentally determined IC50 values between these various interactions. The majority of compounds containing an ionizable piperidine ring [e.g., eliprodil, 1-[2-(4-hydroxy-phenoxy)-ethyl]-4-(4-methyl-benzyl)-piperidin-4-ol (Ro 63-1908), traxoprodil, besonprodil, compound 37a, and compound 46b] adjacent to the A ring form a hydrogen bond with GluN2B Glu110 and the ring nitrogen (if protonated), as observed for ifenprodil and Ro 25-6981. Two of these compounds (traxoprodil and compound 12a) were modeled to make a hydrogen bond between the carbonyl oxygen of GluN1 Tyr109 and the hydroxyl group at the C-4 position of the piperidine ring (Table 1). Traxoprodil is ∼2.5-fold more potent than ifenprodil and differs from ifenprodil by an additional hydroxyl group and shortening of the linker. The increased potency may reflect an increase in solubility as well as tighter binding at the GluN1/GluN2B interface. The presence of a piperidine ring and the shortened linker rigidifies this region, which seems to facilitate hydrogen bond formation with either GluN1 Ser132 or GluN2B Gln110 and GluN2B Glu106, depending on the stereochemistry of the compound (Table 1). Opening of the piperidine ring (acyclic analogs) apparently relaxes the strain imposed on binding to the interface, which may underlie the 25-fold reduced IC50 values (i.e., increased potency) of ifenprodil analogs (Tamiz et al., 1998; Marinelli et al., 2007). The lowest IC50 value was obtained by lengthening the carbon chain from four carbons to six (Tamiz et al., 1998; Marinelli et al., 2007).
Two different but related classes of compounds containing a 3,4-dichloro substituted A ring show that the amine is preferred at positions three and four of the linker chain (Tahirovic et al., 2008; Mosley et al., 2009; numbering from the A ring). If the nitrogen forms part of an amide bond, then position four is preferred (Mosley et al., 2009). Docking of these compounds (15, 49, and 52) shows that the amide makes hydrogen bonds with GluN2B Gln110 (NH) and GluN1 Arg115 (carbonyl). The propanolamines show a preference for the amine at position three, the amines being either secondary or tertiary (Tahirovic et al., 2008). Moreover, the stereogenic center in this series does not exhibit significant enantiomeric selectivity, with an R/S ratio ranging between 1 and 3 (Tahirovic et al., 2008). This phenomenon is similar to that observed for the hydroxyl stereogenic centers of ifenprodil and traxoprodil (Chenard et al., 1991), which are predicted to form hydrogen bonds with the carbonyl group of either GluN1 Ser132 or GluN2B Gln110 (see Fig. 4).
Molecular Interactions of the B Ring with the GluN1/GluN2B ATD Interface.
The B-ring binding pocket consists of residues Arg115, Ser132, and Ile135 from GluN1 and Thr174, Tyr175, Phe176, Pro177, Met207, Ser208, Thr233, and Glu236 of GluN2B. The hydrophobic region of this pocket is delineated by residues from the R1 domain of GluN1 (Ile135) and the R2 domain of the GluN2 (Pro177, Phe176, and Thr233) (see Figs. 1–5). This region is the most diverse among the different GluN2 subtypes and thus may play a role in the subunit selectivity of these compounds (Fig. 2). The B ring of more potent GluN2B-selective negative modulators often contains a phenol ring with the hydroxyl group in the para position with respect to the linker region, which makes a hydrogen bond with GluN2B Glu236 and a water molecule anchored by Tyr175 and Met207 in GluN2B located between β-strands β6 and β7 (Karakas et al., 2011; Figs. 1B and 5A).
Fig. 5.

Binding modes of two GluN2B-selective negative allosteric modulators (ifenprodil and compound 68) with the general pharmacophore model superimposed to highlight specific features of the binding pocket. A, the binding pose of ifenprodil within the GluN1/GluN2B interface is shown; cyan spheres, hydrophobic features; blue spheres, hydrogen bond donors; and red sphere, hydrogen bond acceptors. B, the binding pose (from docking) of a N-methylsulfonamide substituted B ring (compound 68) is shown. The simulations suggest that the N-methylsulfonamide can displace the water molecule that interacts with the phenol substituted B ring of ifenprodil. The water molecule that gets displaced is shown in A and is represented as a transparent water molecule in B (arrow).
Three residues (Glu236, Phe176, Thr233; see Fig. 2) involved in accommodating the B ring were first identified by Perin-Dureau et al. (2002) in an alanine screen of GluN2B. The mutation GluN2B(E236A) prevents the formation of a hydrogen bond with the hydroxyl group of the B ring of ifenprodil, which significantly reduces the potency. The mutation GluN2B(F176A) creates a void hydrophobic cavity that disrupts the B-ring binding pocket and breaks the π-σ interaction between Phe176 and the B ring, explaining the reduction in ifenprodil potency. We found a 42-fold reduction in ifenprodil activity in the mutant GluN2B(T233A) (Table 2; see also Perin-Dureau et al., 2002), which can be explained partially by the disruption of the hydrogen bond it forms with the backbone (NH) of GluN2B Glu236 at the start of α6-helix (R2-domain). Supporting this finding, mutations of GluN2B(E235A) and GluN2B(K234A) significantly reduce the ifenprodil sensitivity, which highlight the importance of this region for ifenprodil binding (Perin-Dureau et al., 2002). Crystallographic data also show that the methyl group of GluN2B Thr233 forms hydrophobic interactions with the B ring of ifenprodil (Fig. 2B). The residue corresponding to Thr233 in GluN2A is Ser232, raising the possibility that this contributes to the selectivity differences between GluN2A and GluN2B. Consistent with this prediction, we found that mutation of GluN2B (T233S) caused a 35-fold increase in IC50 (i.e., reduction in potency; Table 2).
The docking poses of the aromatic B ring were similar for all of the compounds docked (Fig. 3). Some of the most effective substitutions of the B ring among ifenprodil analogs have been 5-hydroxy-benzimidazole, para-N-phenylmethanesulfonamide, benzoxazolinone, and benzimidazolinone (Wright et al., 2000; Nagy et al., 2003; Barta-Szalai et al., 2004; McCauley et al., 2004; Tahirovic et al., 2008; Mony et al., 2009a; Mosley et al., 2009). Docking compounds with different B-ring moieties showed that the hydroxyl-containing groups exhibited a pose similar to ifenprodil and Ro 25-6981, forming hydrogen bonds with Glu236 and a water molecule (Figs. 1B and 5). However, the compounds with para-N-phenylmethanesulfonamide (e.g., propanolamines and compounds 15, 29, 49, 52, and 68), benzoxazolinone (besoprodil, radiprodil, and compound 3d) and benzimidazolinone (compounds 3a and 46b) moieties showed unfavorable binding poses when docked in the presence of the above-mentioned water molecule (Fig. 5). It is noteworthy that para-N-phenylmethanesulfonamide, benzoxazolinone, and benzimidazolinone moieties all possess a hydrogen bond donor similar to that of the phenolic B ring but additionally contain a hydrogen bond acceptor in the form of an oxygen atom that could replace the water molecule. Removal of the water molecule before docking yielded a hydrogen bond between the hydrogen bond donor groups (NH) and GluN2B Glu236, as well as hydrogen bonds between the hydrogen bond acceptors on the ligand and GluN2B Tyr175 and Met207 (Fig. 5). We therefore synthesized an analog identical to compound 29 in Tahirovic et al. (2008), except that the N-methylsulfonamide was replaced by a hydroxyl group (compound 93-138 in Table 1). Consistent with this idea, the presence of the hydroxyl group decreased potency 10-fold for this compound (Table 1).
We also investigated the determinants of subunit selectivity within the B-ring binding region by mutating residues that differ between the GluN2A and GluN2B. The loop region between the α-helix 5 and β-strand 7 could be important in GluN2B-selectivity by influencing the position of secondary structural elements even though it does not make direct atomic interactions with ifenprodil (Fig. 6). We mutated GluN1 Arg115 and Arg323, which interact with this loop region, to alanine (Table 3). Arg115 makes hydrogen bonds with GluN1 Gly112 and the backbone of GluN2B Met207 when bound with ifenprodil. The mutation GluN1 (R115A) caused a 3-fold increase in ifenprodil IC50 (i.e., reduction in potency). GluN1 Arg323 forms hydrogen bonds with GluN2B Asp213. The mutation GluN2B (D213A) and the double mutant GluN2B (G212A, D213K) showed 8- and a 24-fold reductions in ifenprodil potency, respectively (Table 2). These data suggest that the loop region between α-helix 5 and β-strand 7 in GluN2B facilitates the binding of ifenprodil and plays a role in stabilization of the GluN1/GluN2B complex interface.
Fig. 6.

GluN2B structural determinants of ifenprodil sensitivity are mapped onto the GluN1/GluN2B dimer. The GluN1 backbone is blue whereas the GluN2B backbone is orange with residues near ifenprodil (cyan) in gray. Residues in red are thought influence ifenprodil sensitivity as a result of structural alteration. Residues in green are thought to play a role in stabilizing the GluN1/GluN2B dimer interface.
TABLE 3.
Mutation studies of GluN1 and GluN2A
Fitted IC50 values were determined from composite concentration-effect curves constructed from recordings in 8 to 41 oocytes (n) as described under Materials and Methods at a holding potential of −40 mV; steady-state response was fixed at 0 for all conditions except GluN2B wild type. Data for wild-type GluN2B from Table 2 are included here for comparison.
| IC50 | Hill Slope | IC50mut/ IC50 WT | n | |
|---|---|---|---|---|
| μM | ||||
| GluN1/GluN2B | 0.10 | 1.0 | 41 | |
| GluN1(R115A)/GluN2B | 0.28 | 0.9 | 2.9 | 10 |
| GluN1(R323A)/GluN2B | 0.26 | 1.0 | 2.6 | 12 |
| GluN1/GluN2A | 47 | 1.2 | 9 | |
| GluN1/GluN2A(R181E) | 43 | 1.4 | 0.91 | 9 |
| GluN1/GluN2A(T208S) | 74 | 1.3 | 1.6 | 8 |
| GluN1/GluN2A(Q336R) | 58 | 1.1 | 1.2 | 14 |
| GluN1/GluN2A(S232T) | 44 | 1.0 | 0.93 | 10 |
Molecular Interactions Distant to the ATD Interface that Affect Ifenprodil Sensitivity.
A number of studies have identified residues distant from the binding site that have strong effects on the IC50 values for ifenprodil (e.g., Gallagher et al., 1996; Perin-Dureau et al., 2002; Fig. 6). Among these, we focused on closely spaced acidic residues (see Table 2) at which mutations can affect function (Perin-Dureau et al., 2002). To obtain functional data from full-length receptors on the role of Asp101, Asp102, and Asp104 in ifenprodil inhibition (Fig. 6), we evaluated a pair of GluN2B-selective ligands that either contain an ionizable amine (compound 68 in Mosley et al., 2009) or a nonionizable amide (compound 49 in Mosley et al., 2009) within the linker connecting two aromatic rings. Tables 2 and 4 summarize data showing that D101A shifts the ifenprodil analog IC50 values by more than 100-fold regardless of the ionization state of the chain nitrogen, consistent with the idea that these acidic residues do not sense distinct conformational changes induced by the ionized chain nitrogen compared with un-ionized form in GluN2B-selective modulators. A GluN2B-selective modulator with all chain nitrogens converted to nonionizable amides (e.g., compound 15 in Mosley et al., 2009) also remained sensitive to mutation of these aspartate residues (Table 4). These data suggest that disruption of a network of intraprotein interactions between GluN2B Asp101 and the hydroxyls of Thr103 and Ser130 as well as the main chain nitrogens of Gly129 and Ser130 reduces ifenprodil potency.
TABLE 4.
Role of Asp101, Asp102, and Asp104 in GluN1/GluN2B antagonism
Fitted IC50 values are shown to two significant figures and were determined from composite concentration-effect curves constructed from recordings in 6 to 36 oocytes as described under Materials and Methods; steady-state response was fixed at 0 for all conditions. IC50 data for D101A and D102A are from mouse GluN2B coexpressed with rat GluN1; all other data are from rat GluN1/GluN2B receptors.
| Compound | Structure | IC50 Wild Type | IC50 D101A/IC50 Wild Type | IC50 D102A/IC50 Wild Type | IC50 D104A/IC50 Wild Type |
|---|---|---|---|---|---|
| μM | |||||
| 68 | 0.017 | 177 | 2.3 | 9.8 | |
| 49 | 0.054 | 102 | 1.8 | 31 | |
| 15 | 0.069 | 51 | 1.9 |
To examine in detail the structural determinants of ifenprodil's actions, we compared the effect of mutations both in the binding pocket and distant to the pocket on the potency of ifenprodil and five analogs, which included Ro-25-6981, eliprodil, and traxoprodil (see Table 1), compound 68 (Mosley et al., 2009), and compound 15 (Mosley et al., 2009) (Table 5). We hypothesized that distant residues (e.g., Tyr282, Asp101, Asp102) might cause global changes in the conformation of the amino GluN2B terminal R1 and R2 domains that differentially alter the potency or nature of inhibition for different ligands if their binding poses produce different sets of long-range intraprotein rearrangements. GluN2B Tyr282, located at the hinge region of the bilobed ATD, has a strong effect on the degree of inhibition that can be achieved by saturating concentrations of ligands. It is noteworthy that mutation of Tyr282 showed different effects on various ligands, increasing the IC50 value for ifenprodil and CP101,606 by more than 15-fold compared with more modest effects on the IC50 values for other ligands (Table 5). By contrast, the potency of compound 68 was minimally affected by GluN2B(Y282A). It is noteworthy that the Hill slope was uniformly shallow for concentration-effect curves for GluN2B(Y282A), suggesting that the reduction in steady-state inhibition coupled with decreased potency at the interface may reveal a lower affinity inhibitory site, such as a channel-block site with an IC50 value that is similar to that for block of the mutant ATD dimer interface site (Perin-Dureau et al., 2002). Mutation of another residue distant to the interface binding site (Asp101) also shows variable effects on IC50 values for this set of ligands. GluN2B (D101A) exhibits an 800-fold reduction in potency for Ro-25-6981 but has a smaller effect on eliprodil (89-fold shift in potency; Table 5). These results suggest that this region of the protein may sense either different ligand conformations or the variable elements within the binding site that interact with substituents on the ligand. We also evaluated whether mutations at residues that contact the B ring of ifenprodil could distinguish among various ligands, because docking shows that differential atomic contacts exist for the diaryl linker and B ring for various ligands. Mutations of residues in contact with the B ring (Thr233, Tyr231) also reduced IC50 values in a manner that was ligand-dependent (Table 5). For example, T233A had a larger effect on eliprodil potency than Ro-25-2981. In addition, T233A is 6- to 8-fold more effective at disrupting eliprodil inhibition than ligands with a sulfonamide-substituted B ring (e.g., 15, 68), consistent with idea that additional interactions provided by the sulfonamide moiety further stabilize binding. Likewise, Y231A had a larger effect on B rings lacking the sulfonamide substituents. Table 5 summarizes the shift in potency and steady-state inhibition by all mutations tested for this set of ligands. We interpret these data to suggest that both contact residues as well as more distant determinants of actions can distinguish among different ligands. Mutations in this and other regions may be a useful way to categorize ligands in a manner that can be predictive of their properties
TABLE 5.
Distant residues of GluN2B differentially respond to binding of ifenprodil analogs
Recombinant GluN1/GluN2B expressed in X. laevis oocytes were recorded using two electrode voltage clamp. Fitted IC50 values for mutant receptors were determined from composite concentration-effect curves in 6–46 oocytes from 2 frogs as described under Materials and Methods; the steady-state response was allowed to float as a free parameter for all conditions. The fold change in IC50 values is shown for seven compounds at six different mutant receptors; the steady-state response in saturating ligand estimated from fitting is given in parentheses as percentage of control. IC50 values at rat receptors were 25 nM (compound 15, Mosley et al., 2009; n = 46), 9 nM (compound 68, Mosley et al., 2009; n = 42), 100 nM (ifenprodil, n = 12), 54 nM (Ro-25-6981; n = 37), 930 nM (eliprodil; n = 26), 23 nM (traxoprodil; n = 4). Similar values were obtained from mouse GluN2B (n = 4–21).
| IC50 Mutant/IC50 Wild Type (Fitted Steady-State Response) |
||||||
|---|---|---|---|---|---|---|
| Compound 15 | Compound 68 | Ifenprodil | Ro-25-6981 | Eliprodil | CP-101,606 | |
| fold (%) | ||||||
| Distant residues | ||||||
| D101Aa | 53 (0) | 372 (1) | 247 (0) | 815 (5) | 89 (0) | 200 (8) |
| D102Aa | 1.9 (5) | 2.0 (20) | 1.8 (20) | 1.2 (13) | 1.8 (14) | 1.6 (17) |
| Y282A | 3.2 (20) | 1.2 (64) | 17.2 (28) | 2.3 (38) | 4.2 (36) | 23 (53) |
| Contact residues | ||||||
| L205A | 2.1 (7) | 1.9 (10) | 3.0 (9) | 1.4 (14) | 2.4 (21) | 1.9 (10) |
| Y231A | 108 (8) | 433 (0) | 745 (0) | 648 (0) | 1361 (0) | N.D. |
| T233Aa | 16 (18) | 24 (9) | 43 (4) | 18 (11) | 120 (0) | 4.0 (22) |
N.D., not determined.
Values were determined from wild-type rat GluN1 plus mutant mouse GluN2B and compared with wild-type rat GluN1 plus wild-type mouse GluN2B.
Molecular Dynamics of the GluN1/GluN2B ATD Dimer and Domain Movement.
We compared the results of molecular dynamics simulations at 300 K on hydrated models of the ATD dimer of GluN1/GluN2B (see Materials and Methods) in the apo state to the ifenprodil-bound state (Fig. 7A). The goal of these simulations was to explore the conformational rearrangements within the amino terminal domains induced by negative allosteric modulators and thereby obtain insight into the mechanisms by which di-aryl compounds inhibit receptor function. Molecular dynamics simulations were performed for 10 ns from which the average position of Cα over the last 2 ns was calculated (see Materials and Methods). We initially focused on the changes in domain orientation between the R1 and R2 domains produced by ifenprodil binding for both GluN1 and GluN2 subunits. To do this, we aligned the dimer structure first on the R1 domain of GluN1 and determined the center of mass movement for the R2 domain of GluN1. The center of mass was analyzed because it captures overall domain movement better than evaluating motion of individual residues. We subsequently aligned the structures on the R1 domain of GluN2B, calculated the center of mass movement and vector displacement of the R2 domain of the GluN2B apo-state simulations and compared these quantities with those of the ifenprodil-bound GluN2B simulations. The R2 domain of the GluN1 ATD in the simulated ifenprodil-bound complex moved relative to the R1 domain in the apo-state simulation, raising the possibility that ifenprodil binding to the ATD heterodimer interface could trigger modest rearrangement in the relative orientation of the R1 to R2 domain. The net displacements of the R2 domain of GluN1 for the ifenprodil simulations compared with the apo state were 4.9 Å (Fig. 7). When this analysis was performed for the GluN2B R2 domain, the net displacement was found to be 2.8 Å compared with the apo state (Fig. 7). A comparison between the apo-state simulations and Ro-25-6981 simulations showed a net displacement similar to that of the ifenprodil-bound simulation for both subunits (Fig. 8 and 9). Moreover, these simulations suggest that both ligands have a similar impact on R1-R2 domain orientation within the wild-type GluN1/GluN2B ATD (Fig. 7B).
Fig. 7.

A schematic representation of the structural rearrangement observed for the R2 domain with respect to the R1 domain of the GluN1/GluN2B dimers simulated under different conditions. A, structural rearrangement of the R2 domain of GluN1/GluN2B simulations with ifenprodil shown in blue/orange and the apo-state shown in gray. An expanded view of the R2 domains superimposed for the apo- and ifenprodil-bound structures are shown below with GluN1 on the left and GluN2B on the right. B, a table containing the domain displacement (Ångstroms) (see Materials and Methods) of the structural rearrangement of the R2 domains for different GluN1/GluN2B simulations with ifenprodil and Ro-25-6981 compared with the apo state.
Fig. 8.

A schematic representation of structural rearrangement of the GluN1 R2 domains of the GluN1/GluN2B ATD dimer. A, an expanded view of GluN1 shows the net displacement of the R2 domain in simulations performed on the wild-type GluN1/GluN2B dimer in the presence (blue) and absence (green, apo) of ifenprodil. B, the relative 3-dimensional movement of the GluN1 R2 domain center of mass is plotted in the presence and absence of ifenprodil or Ro-25-6981; wild-type apo position indexed to coordinates 0,0,0 Å. The net domain displacement can be found in Table 6. C, the relative three-dimensional movement of the GluN1 R2 domain in GluN1/GluN2B(Y282A) mutant receptors is plotted in Ångstroms in the presence and absence of ifenprodil or Ro-25-6981; wild-type apo position indexed to coordinates 0,0,0 Å. The domain displacement can be found in Table 6.
Fig. 9.

A schematic representation of structural rearrangement of the GluN2 R2 domains of the GluN1/GluN2B ATD dimer. A, an expanded view of GluN2B shows the net displacement of the R2 domain in simulations performed on the wild-type GluN1/GluN2B dimer in the presence (blue) and absence (green, apo) of ifenprodil. B, the relative three-dimensional movement of the GluN2B R2 domain center of mass is plotted in the presence and absence of ifenprodil or Ro-25-6981; the wild-type apo position is indexed to coordinates 0,0,0 Å. The domain displacement can be found in Table 6. C, the relative three-dimensional movement of the GluN2B R2 domain center of mass in GluN1/GluN2B(Y282A) mutant receptors is plotted in the presence and absence of ifenprodil or Ro-25-6981; wild-type apo position indexed to coordinates 0,0,0 Å. The domain displacement can be found in Table 6.
We also investigated the effects on ligand-induced conformational rearrangements within simulations run on GluN1/GluN2B ATD heterodimers in which a GluN2B mutation distant to the ligand binding site were present (Y282A). These mutations differentially affect ifenprodil and Ro-25−6981 sensitivity; the GluN2B(Y282A) mutant produced a 17-fold reduction in potency of ifenprodil and a 2-fold reduction in potency of Ro-25−6981 (Table 5). For these simulations, we assumed similar overall folding of the ifenprodil-bound mutant and the wild-type ATD, which allows us to investigate the conformational rearrangement predicted from these simulations for ligand binding. A comparison of the molecular dynamics simulations of the apo-GluN1/GluN2B(Y282A) heterodimer and the apo-wild-type GluN1/GluN2B showed that the Y282A mutation altered the position of the R2 domains of GluN1 subunit relative to the R1 domain (Fig. 8C, green symbols). By contrast, the position for R2 domain in GluN2B of apo-GluN1/GluN2B(Y282A) ATD was similar to that for the apo-wild-type GluN1/GluN2B ATD (Fig. 9C, green symbols; Table 6). It is noteworthy that the GluN1 R2 domain of the ifenprodil-bound GluN1/GluN2B(Y282A) mutant simulation showed a pronounced domain movement of similar magnitude as ifenprodil-bound wild-type GluN1/GluN2B simulation (Table 6). However, the trajectory of this motion for mutant GluN2B(Y282A) ATD was distinctly different from wild-type ATD (Fig. 8, B and C, blue symbols), suggesting that this mutation could alter the manner by which ifenprodil influences domain orientation. As observed with apo configuration, there was little change (0.4 Å) in the final center of mass position of the GluN2B R2 domain displacement between the wild-type GluN1/GluN2B ATD and mutant GluN1/GluN2B(Y282A) ATD simulations in the presence of ifenprodil (Fig. 9, B and C, blue symbols; Table 6). That is, ifenprodil seems to stabilize the GluN2B(Y282A) R2 domain in a similar conformation to that of the wild-type (Table 6). These data suggest that residues such as GluN2B Tyr282 that are distant to the dimer interface can affect ifenprodil-induced changes in the orientation of R2 domain of GluN1 but have little effect on R2 domain of GluN2B in either the absence or presence of ifenprodil.
TABLE 6.
The net displacement for the R2 domains of the ATD GluN1/GluN2B heterodimer determined after 10 ns of molecular dynamics simulations
The simulations were performed after introducing site-directed mutations as well as in the presence of different ATD modulators.
| Simulation Comparison | Net R2 Displacement |
|
|---|---|---|
| GluN1 | GluN2B | |
| Å | ||
| GluN2B-wild type | ||
| Ifenprodil(wt)–apo(wt) | 4.9 | 2.8 |
| Ro-25-6981(wt)–apo(wt) | 4.4 | 3.1 |
| GluN2B Y282A mutants | ||
| Apo(wt)–Apo(Y282A) | 3.7 | 0.5 |
| Ifenprodil(Y282A)–apo(Y282A) | 3.8 | 3.1 |
| Ifenprodil(wt)–ifenprodil(Y282A) | 7.4 | 0.4 |
| Ro-25-6981(Y282A)–apo(Y282A) | 2.7 | 3.2 |
| Ro-25-6981(wt)–Ro-25-6981(Y282A) | 0.3 | 3.2 |
| Ro-25-6981(Y282A)–ifenprodil(Y282A) | 6.2 | 3.0 |
| GluN1 D130A mutants | ||
| Apo(wt)–Apo(D130) | 2.9 | 1.1 |
| Ifenprodil(D130A)–Apo(D130A) | 4.0 | 2.0 |
| Ifenprodil(wt)–ifenprodil(D130A) | 3.2 | 1.7 |
| GluN2B D101A mutants | ||
| Apo(wt)–apo(D101A) | 2.0 | 2.5 |
| Ifenprodil(D101A)–apo(D101A) | 1.5 | 5.5 |
| Ifenprodil(wt)–ifenprodil(D101A) | 3.0 | 1.6 |
wt, wild type.
We also performed these analyses for mutant and wild-type simulations of Ro-25-6981-bound ATD. The GluN1 R2 domain for the Ro-25-6981-bound GluN1/GluN2B(Y282A) mutant ATD simulation showed somewhat similar movement compared with wild-type Ro-25-6981 simulation (Fig. 8, B and C, red symbols; Table 6). The R2 GluN2B domain for both the wild-type and mutant GluN1/GluN2B(Y282A) ATD simulations showed displacement with a similar magnitude but different trajectory (Fig. 9, B and C, red symbols; Table 6). The different direction in which the R2 domains moves for the ifenprodil compared with Ro-25-6981 in GluN2B(Y282A) simulation is consistent with the general idea that these ligands may induce unique conformational changes and are therefore differentially sensitive to mutations that are made distant to their binding site. The fact that Ro-25-6981 possesses one additional easily rotated bond near the center of the molecule relative to ifenprodil implies a considerably more complex conformational energy surface compatible with an attenuated structural impact.
Molecular Dynamics of the GluN1/GluN2B ATD Dimer and Main Chain Flexibility.
We analyzed ATD Cα chain flexibility during the molecular dynamics simulations to identify regions of the protein through which intraprotein rearrangements might occur after ligand binding. To do this, we measured the RMSF for the Cα backbone atoms within the protein over the last 2 ns by aligning the GluN1 and GluN2B subunit individually to quantify the degree to which individual residues undergo movement within the simulated time frame (Fig. 10). It is noteworthy that two regions showed enhanced RMSF values compared with the average RMS over the entire ligand-bound ATD compared with the apo state, suggesting that ligand binding to the dimer interface can alter the manner in which these regions interact within the protein. As a control, we compared the B factor values of these regions (Cα atoms) to the average values of the crystal structures and found them to range between 41 and 61 (GluN1 residues 126–135) and 50 and 68 (GluN2 residues 261–271), just below the average values for the entire GluN1 (48–79) and GluN2 (64–80) ATD domains (Protein Data bank entries 3qel and 3qem), suggesting that these regions are not disordered loops and may play a role in domain movement. Both of these regions are distant from the residues lining the binding pocket, yet close to residues at which mutations are known to affect ifenprodil sensitivity. Specifically, residues 126 to 135 of GluN1, which include the η2 310-helix, showed strongly elevated RMSF values for the wild-type GluN1/GluN2B ATD bound to either ifenprodil or Ro-25-6981 compared with the apo-state simulation (Fig. 10C). These simulated data suggest that this region may play a role in conformational rearrangement of the GluN1/GluN2B interface. Consistent with the idea that this region is a critical determinant of ifenprodil's action, mutations at three residues within this sector (GluN1 Y128A, D130A, H134S) are known to reduce ifenprodil potency by 10- to 500-fold (Masuko et al., 1999). A second region (GluN2B residues 261–275) also showed strong ligand-induced movement within the simulation compared with the apo state (Fig. 10AB). Again, mutagenesis suggests multiple residues within this region (e.g., GluN2B Leu261 and Gly264) can control ifenprodil potency (Perin-Dureau et al., 2002). Furthermore, mutagenesis has also implicated Lys234, a residue that forms hydrogen bonds with Asp265 and Thr268 within this region, as a key determinant of ifenprodil sensitivity (Perin-Dureau et al., 2002). Whereas the molecular mechanism by which these two regions affect ifenprodil sensitivity remains unclear, we speculate that this zone stabilizes the GluN2B ATD clamshell allowing for conformational changes of the R2 domain to influence other regions of the receptor. Similar results were obtained for simulated binding of ifenprodil and Ro-25-6981 to wild-type ATD (Fig. 10C).
Fig. 10.

The RMSF calculated from molecular dynamics simulations performed on wild-type ATD heterodimers. The RMSF observed for the wild-type GluN1/GluN2B ATD heterodimers simulated in the (A) apo state or (B) ifenprodil-bound state are graphically illustrated. A thicker ribbon with warmer colors (yellow and red) represents regions with more fluctuation compared with overall average. The two boxes highlight regions of interest that showed increase fluctuation for ligand-bound ATD. In GluN1 this box encompasses residues 126 to 135 and in GluN2B the box indicates residues 261 to 275. C, the average RMSF over all ATD Cα carbons was 1.7 to 1.8 Å for GluN1 and 1.7 to 1.9 Å for GluN2B ATD residues in wild-type apo and ligand-bound simulations. C, the table summarizes the average and range of RMSF for residues within the two highlighted regions in A and B.
We also investigated simulated motion of the Cα chain in mutant ATD heterodimers to determine whether function-blocking mutations could also block the ligand-induced movement of the two regions of the polypeptide chains described above. Evaluation of residues GluN1 126 to 135 showed little or no change in RMSF for simulations with heterodimer ATD for GluN1(D130A), GluN2B(D101A), or GluN2B(Y282A) (Fig. 11) compared with wild type (Fig. 10). That is, none of these mutations markedly altered the ifenprodil-induced changes in GluN1 main-chain motility for residues 126 to 135 (Fig. 11C). By contrast, each of these three mutations substantially reduced ifenprodil-induced changes in mobility of GluN2B residues 261 to 275 (Fig. 11C). A similar result was found for Ro-25-6981 (Fig. 11C; Supplemental Fig. S1).
Fig. 11.

The RMSF reported for molecular dynamics simulations performed for mutant ATD heterodimers. A, the RMSF is illustrated for the GluN1/GluN2B(Y282A) ATD simulated in the apo state. B, the RMSF is similarly shown for the GluN1/GluN2B(Y282A) ATD simulated with ifenprodil bound. A thicker ribbon with warmer colors (yellow and red) represents regions with enhanced Cα fluctuations compared with average of all residues. The two boxes highlight regions of interest for which there were increased fluctuation in wild-type ATD. The boxes represent GluN1 residues 126 to 135 and GluN2B residues 261 to 275. C, the average RMSF over all GluN1 ATD Cα carbons was 1.6 to 2.7 Å and 1.6 to 2.1 for all GluN2B ATD residues across all simulations with mutant ATD. The table summarizes the average and range of RMSF for residues within the two highlighted regions in (A) and (B).
Given the caveats and methodological limitations of molecular dynamics, these simulations of the NMDA heterodimer amino terminal domain in the presence of different negative modulators and various mutants showed conformational rearrangements consistent with in vitro experimental results. Moreover, these simulated data independently highlight two regions distant to the binding site that have previously been identified as determinants of di-aryl compound sensitivity. These data are suggestive that protein rearrangement in these regions plays an important role in the negative allosteric modulation exerted by the di-aryl compounds investigated.
Structural Determinants of Subunit Selectivity.
We also evaluated potential structural determinants of subunit selectivity of regions identified as important for ifenprodil potency. We first assessed the loop region between α-helix 5 and β-strand 7 (residues 207–218 of GluN2B and residues 208–217 of GluN2A), which facilitates the binding of ifenprodil and stabilizes the GluN1/GluN2B interface. Chimeric GluN2A receptors containing this loop from GluN2B did not display enhanced ifenprodil sensitivity (Table 7). This GluN2A chimera was also subjected to three point mutations (S232T, I176Y, and L238Y) designed to eliminate additional differences between the GluN2A and GluN2B B-ring binding pocket. None of these mutations had detectable effects on ifenprodil potency of the mutant chimeric receptor, suggesting that this loop within the B-ring binding pocket is not sufficient to endow GluN2A with ifenprodil sensitivity (Table 7).
TABLE 7.
Structural determinants of subunit selectivity
Fitted IC50 values were determined from composite concentration-effect curves constructed from recordings in 4 to 41 oocytes (n) as described under Materials and Methods at a holding potential of −40 mV; the steady-state response was fixed at 0 for all conditions except GluN2B wild type. Data for wild-type GluN2B from Table 2 are included here for comparison.
| IC50 | IC50 Mutant/IC50 Wild Type | n | |
|---|---|---|---|
| μM | |||
| GluN1/GluN2A(2B-loop) | 48 | 1.03 | 20 |
| GluN1/GluN2A(2B-loop,S232T) | 53 | 1.14 | 8 |
| GluN1/GluN2A(2B-loop,I176Y) | 36 | 0.77 | 9 |
| GluN1/GluN2A(2B-loop,L238Y) | 50 | 1.06 | 8 |
| GluN1/GluN2A | 47 | 9 | |
| GluN1/GluN2A(2B-ATD-LBD linker) | 49 | 1.1 | 11 |
| GluN1/GluN2B | 0.10 | 41 | |
| GluN1/GluN2B(2A-ATD-LBD linker) | 0.14 | 1.4 | 4 |
| GluN1/GluN2D | 205 | 9 | |
| GluN1/GluN2D(2B-ATD-LBD linker) | 155 | 0.75 | 14 |
Previous studies have shown that the 15- to 16-residue linker region between the ATD and the ligand binding domain is highly divergent and can affect a wide range of NMDA receptor properties (Gielen et al., 2009; Yuan et al., 2009). The potential role of this divergent region in controlling ifenprodil sensitivity has not been previously evaluated. To examine whether the ATD-LBD linker can affect ifenprodil inhibition, we first transferred the 15-residue ATD-LBD linker from GluN2B (390-VWPRMCPETEEQEDD-404) into GluN2A. This mutation did not enhance the sensitivity of GluN2A to ifenprodil (Table 7). Likewise, transferring the ATD-LBD linker from GluN2B to GluN2D yielded chimeric receptors that remain largely insensitive to ifenprodil. We also transferred the ATD-LBD linker from GluN2A to GluN2B, which yielded chimeric receptors that showed sensitivity to ifenprodil similar to that of wild-type receptors, suggesting that this linker region has minimal impact on ifenprodil binding to the GluN2B ATD (Table 7). Together, these data suggest that the linker is not a critical determinant of ifenprodil subunit selectivity.
Discussion
We have evaluated the determinants of binding for multiple structurally diverse GluN2B-selective diaryl ligands to the subunit interface within the GluN1/GluN2B ATD heterodimer. Without exception, these ligands seem capable of binding within the pocket formed by the heterodimer interface between the GluN1 and GluN2B amino terminal domains, each of the two aromatic rings adopting similar positions regardless of their structure. However, docking studies supported by mutagenesis suggest multiple atomic contacts that can vary depending on ligand structure, and these provide opportunities to alter binding and perhaps allosteric function of GluN2B ligands. These variations suggest that ligand structure can be optimized by rational drug design to either enhance potency or circumvent off-target binding. In addition, these studies show that there is more variation in terms of atomic contacts for one aromatic ring (referred to as the B ring) than the other ring (A ring).
From the perspective of protein structure, our results provide insight into mutagenesis work previously published by multiple groups (e.g., Masuko et al., 1999; Perin-Dureau et al., 2002; Mony et al., 2009b). By providing explanations for the effects of function-changing mutations at the atomic level, our data suggest atomic interpretations of what were previously complex data, particularly for residues distant from the binding site. Furthermore, covarying ligand structure and amino acid identity at critical regions has suggested potential heterogeneity in the way ligand binding to the ATD dimer interface induces downstream functional effects. That is, some ligands that adopt similar binding poses are differentially sensitive to function-blocking mutations distant from the ligand-binding site. The differential sensitivity to these mutations of structurally distinct ligands raises the possibility that the GluN2B-selective negative allosteric modulators could be subcategorized according to the manner in which they trigger downstream intraprotein conformational changes distant from the ligand binding pocket. Furthermore, the differential sensitivity of these ligands provides a rationale for more detailed study of how these compounds interact within the binding cleft. Understanding in detail how different ligands interact with the GluN2B receptor could allow the design of compounds with the best set of therapeutically relevant properties.
We have also explored protein motion using molecular dynamics simulations run for the hydrated ATD heterodimers both in the presence and absence of ligand. Multiple caveats need to be associated with the relatively short (10 ns) simulations performed with only a portion of the protein, which are unlikely to fully sample all available conformations. Nevertheless, the data reinforce several important concepts suggested from mutagenesis and docking studies. First, there seem to be consistent ligand-induced changes in the orientation of the individual domains of the bilobed clamshell ATDs for both GluN1 and GluN2B. We interpret the changes in the center of mass displacement to suggest that ifenprodil binding at the interface can alter the orientation of the R2 subdomains within the ATD. This may be important for the overall mechanism of inhibition. Second, evaluation of motion within the molecular dynamics simulations (after steady-state equilibrium) shows increased mobility of two regions of the heterodimers. We found increased motion for GluN1 residues 126 to 135 and GluN2B residues 261 to 275. Separate experimental lines of evidence previously implicated these regions as important determinants of ifenprodil-induced inhibition (Masuko et al., 1999; Perin-Dureau et al., 2002; Mony et al., 2009b). Thus, our independent finding that ligand binding induces increased motion of the Cα chain in these specific regions suggests that our simulations have correctly captured some features of ifenprodil-induced effects. Furthermore, our simulations with function-blocking mutations show an uncoupling of motion in this sector from ifenprodil binding, consistent with this protein subsection being critical for ifenprodil's action. From these simulations, we speculate that ligand binding can induce sufficient intraprotein rearrangement to alter the associated atomic contacts in these regions, which leads to increased mobility as side chains and main chain hydrogen bonds undergo increased moment-to-moment breakage and restoration. Although we cannot extract the nature of the long-range conformational changes with this form of analysis, these data nonetheless cast a spotlight on this protein environment as a critical downstream element for ifenprodil's actions.
Supplementary Material
Acknowledgments
We thank Kimberly Vellano, Phuong Le, and Jing Zhang for excellent technical assistance. We thank Pierre Paoletti for sharing mutant cDNA for D101A, D102A, and T233A in mouse GluN2B. We thank Shashank Dravid, Chris Makinson, and Yesim Tahirovic for sharing unpublished data. We are likewise appreciative of OpenEye Software's policy of providing academic institutions with free use of their creative software. We thank Dr. Bert Chenard for critical comments on the manuscript.
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant NS36654]. D.C.L., J.P.S., and S.F.T. are coinventors on Emory-owned intellectual property, and hold an equity position in NeurOp Inc., which has licensed this intellectual property. J.P.S. and S.F.T. serve as consultants to NeurOp Inc., and D.C.L. serves on the Board of Directors for NeurOp Inc.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
- NMDA
- N-methyl-d-aspartate
- ATD
- amino-terminal domain
- LBD
- ligand-binding domain
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- MD
- molecular dynamics
- RMSF
- root-mean-square fluctuation
- Ro-25-6981
- α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propanol
- CP101,606
- traxoprodil
- CP101,581
- 1-[(1R,2R)-1-hydroxy-1-(4-hydroxyphenyl)-2-propanyl]-4-phenyl-4-piperidinol
- Ro 63-1908
- 1-[2-(4-hydroxy-phenoxy)-ethyl]-4-(4-methyl-benzyl)-piperidin-4-ol.
Authorship Contributions
Participated in research design: Burger, Yuan, Geballe, Snyder, and Traynelis.
Conducted experiments: Burger, Yuan, Karakas, and Geballe.
Performed data analysis: Burger, Yuan, Karakas, Geballe, Furukawa, Snyder, and Traynelis.
Wrote or contributed to the writing of the manuscript: Burger, Yuan, Karakas, Furukawa, Geballe, Liotta, Snyder, and Traynelis.
References
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. (1994) Differential expression of five N-methyl-d-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol 347:150–160 [DOI] [PubMed] [Google Scholar]
- Auffinger P, Hays FA, Westhof E, Ho PS. (2004) Halogen bonds in biological molecules. Proc Natl Acad Sci USA 101:16789–16794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avenet P, Léonardon J, Besnard F, Graham D, Frost J, Depoortere H, Langer SZ, Scatton B. (1996) Antagonist properties of the stereoisomers of ifenprodil at NR1A/NR2A and NR1A/NR2B subtypes of the NMDA receptor expressed in Xenopus oocytes. Eur J Pharmacol 296:209–213 [DOI] [PubMed] [Google Scholar]
- Barta-Szalai G, Borza I, Bozó E, Kiss C, Agai B, Proszenyák A, Keseru GM, Gere A, Kolok S, Galgóczy K, et al. (2004) Oxamides as novel NR2B selective NMDA receptor antagonists. Bioorg Med Chem Lett 14:3953–3956 [DOI] [PubMed] [Google Scholar]
- Bowers KJ, Dror RO, Shaw DE. (2006) The midpoint method for parallelization of particle simulations. J Chem Phys 124:184109. [DOI] [PubMed] [Google Scholar]
- Butler TW, Blake JF, Bordner J, Butler P, Chenard BL, Collins MA, DeCosta D, Ducat MJ, Eisenhard ME, Menniti FS, et al. (1998) (3R,4S)-3-[4-(4-fluorophenyl)-4-hydroxypiperidin-1-yl]chroman-4,7-diol: a conformationally restricted analogue of the NR2B subtype-selective NMDA antagonist (1S,2S)-1-(4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)- 1-propanol. J Med Chem 41:1172–1184 [DOI] [PubMed] [Google Scholar]
- Carter C, Benavides J, Legendre P, Vincent JD, Noel F, Thuret F, Lloyd KG, Arbilla S, Zivkovic B, MacKenzie ET. (1988) Ifenprodil and SL 82.0715 as cerebral anti-ischemic agents. II. Evidence for N-methyl-d-aspartate receptor antagonist properties. J Pharmacol Exp Ther 247:1222–1232 [PubMed] [Google Scholar]
- Clark T, Hennemann M, Murray JS, Politzer P. (2007) Halogen bonding: the sigma-hole. Proceedings of “Modeling interactions in biomolecules II”, Prague, September 5th-9th, 2005. J Mol Model 13:291–296 [DOI] [PubMed] [Google Scholar]
- Chenard BL, Bordner J, Butler TW, Chambers LK, Collins MA, De Costa DL, Ducat MF, Dumont ML, Fox CB, Mena EE. (1995) (1S,2S)-1-(4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol: a potent new neuroprotectant which blocks N-methyl-d-aspartate responses. J Med Chem 38:3138–3145 [DOI] [PubMed] [Google Scholar]
- Chenard BL, Menniti FS. (1999) Antagonists selective for NMDA receptors containing the NR2B subunit. Curr Pharm Des 5:381–404 [PubMed] [Google Scholar]
- Chenard BL, Shalaby IA, Koe BK, Ronau RT, Butler TW, Prochniak MA, Schmidt AW, Fox CB. (1991) Separation of alpha 1 adrenergic and N-methyl-d-aspartate antagonist activity in a series of ifenprodil compounds. J Med Chem 34:3085–3090 [DOI] [PubMed] [Google Scholar]
- Citri A, Malenka RC. (2008) Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33:18–41 [DOI] [PubMed] [Google Scholar]
- Coyle JT, Tsai G, Goff D. (2003) Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann NY Acad Sci 1003:318–327 [DOI] [PubMed] [Google Scholar]
- Edgar RC. (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577 [Google Scholar]
- Farina AN, Blain KY, Maruo T, Kwiatkowski W, Choe S, Nakagawa T. (2011) Separation of domain contacts is required or heterotetrameric assembly of functional NMDA receptors. J Neurosci 31:3565–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. (1997) Ro 25–6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vitro. J Pharmacol Exp Ther 283:1285–1292 [PubMed] [Google Scholar]
- Gallagher MJ, Huang H, Pritchett DB, Lynch DR. (1996) Interactions between ifenprodil and the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem 271:9603–9611 [DOI] [PubMed] [Google Scholar]
- Gielen M, Siegler Retchless B, Mony L, Johnson JW, Paoletti P. (2009) Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 459:703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill R, Alanine A, Bourson A, Buttelmann B, Fischer G, Heitz MP, Kew JN, Levet-Trafit B, Lorez HP, Malherbe P, et al. (2002) Pharmacological characterization of Ro 63-1908 (1-[2-(4-hydroxy-phenoxy)-ethyl]-4-(4-methyl-benzyl)-piperidin-4-ol), a novel subtype-selective N-methyl-d-aspartate antagonist. J Pharmacol Exp Ther 302:940–948 [DOI] [PubMed] [Google Scholar]
- Gotti B, Duverger D, Bertin J, Carter C, Dupont R, Frost J, Gaudilliere B, MacKenzie ET, Rousseau J, Scatton B. (1988) Ifenprodil and SL 82.0715 as cerebral anti-ischemic agents. I. Evidence for efficacy in models of focal cerebral ischemia. J Pharmacol Exp Ther 247:1211–1221 [PubMed] [Google Scholar]
- Hallett PJ, Standaert DG. (2004) Rationale for and use of NMDA receptor antagonists in Parkinson's disease. Pharmacol Ther 102:155–174 [DOI] [PubMed] [Google Scholar]
- Hansen KB, Furukawa H, Traynelis SF. (2010a) Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol Pharmacol 78:535–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Mullasseril P, Dawit S, Kurtkaya NL, Yuan H, Vance KM, Orr AG, Kvist T, Ogden KK, Le P, et al. (2010b) Implementation of a fluorescence-based screening assay identifies histamine H3 receptor antagonists clobenpropit and iodophenpropit as subunit-selective N-methyl-d-aspartate receptor antagonists. J Pharmacol Exp Ther 333:650–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11:682–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, London ED. (1995) Interactions of erythro-ifenprodil, threo-ifenprodil, erythro-iodoifenprodil, and eliprodil with subtypes of sigma receptors. Eur J Pharmacol 273:307–310 [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38 [DOI] [PubMed] [Google Scholar]
- Imai YN, Inoue Y, Nakanishi I, Kitaura K. (2008) Cl-pi interactions in protein-ligand complexes. Protein Sci 17:1129–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, Furukawa H. (2009) Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J 28:3910–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, Furukawa H. (2011) Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature 475:249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew JN, Trube G, Kemp JA. (1996) A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurones. J Physiol 497:761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Washiyama K, Ikeda K. (2006) Inhibition of G protein-activated inwardly rectifying K+ channels by ifenprodil. Neuropsychopharmacology 31:516–524 [DOI] [PubMed] [Google Scholar]
- Koller M, Urwyler S. (2010) Novel N-methyl-d-aspartate receptor antagonists: a review of compounds patented since 2006. Expert Opin Ther Pat 20:1683–1702 [DOI] [PubMed] [Google Scholar]
- Kumar J, Schuck P, Mayer ML. (2011) Structure and assembly mechanism for heteromeric kainate receptors. Neuron 71:319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Schuck P, Jin R, Mayer ML. (2009) The N-terminal domain of GluR6-subtype glutamate receptor ion channels. Nat Struct mol Biol 16:631–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA. (2009) PDBsum new things. Nucleic Acids Res 37:D355–D359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low CM, Lyuboslavsky P, French A, Le P, Wyatte K, Thiel WH, Marchan EM, Igarashi K, Kashiwagi K, Gernert K, et al. (2003) Molecular determinants of proton-sensitive N-methyl-d-aspartate receptor gating. Mol Pharmacol 63:1212–1222 [DOI] [PubMed] [Google Scholar]
- Marinelli L, Cosconati S, Steinbrecher T, Limongelli V, Bertamino A, Novellino E, Case DA. (2007) Homology modeling of NR2B modulatory domain of NMDA receptor and analysis of ifenprodil binding. ChemMedChem 2:1498–1510 [DOI] [PubMed] [Google Scholar]
- Masuko T, Kashiwagi K, Kuno T, Nguyen ND, Pahk AJ, Fukuchi J, Igarashi K, Williams K. (1999) A regulatory domain (R1–R2) in the amino terminus of the N-methyl-d-aspartate receptor: effects of spermine, protons, and ifenprodil, and structural similarity to bacterial leucine/isoleucine/valine binding protein. Mol Pharmacol 55:957–969 [DOI] [PubMed] [Google Scholar]
- Matter H, Nazaré M, Güssregen S, Will DW, Schreuder H, Bauer A, Urmann M, Ritter K, Wagner M, Wehner V. (2009) Evidence for C-Cl/C-Br… pi interactions as an important contribution to protein-ligand binding affinity. Angew Chem Int Ed Engl 48:2911–2916 [DOI] [PubMed] [Google Scholar]
- McCauley JA, Theberge CR, Romano JJ, Billings SB, Anderson KD, Claremon DA, Freidinger RM, Bednar RA, Mosser SD, Gaul SL, et al. (2004) NR2B-selective N-methyl-d-aspartate antagonists: synthesis and evaluation of 5-substituted benzimidazoles. J Med Chem 47:2089–2096 [DOI] [PubMed] [Google Scholar]
- Mony L, Kew JN, Gunthorpe MJ, Paoletti P. (2009a) Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br J Pharmacol 157:1301–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mony L, Krzaczkowski L, Leonetti M, Le Goff A, Alarcon K, Neyton J, Bertrand HO, Acher F, Paoletti P. (2009b) Structural basis of NR2B-selective antagonist recognition by N-methyl-d-aspartate receptors. Mol Pharmacol 75:60–74 [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. (1994) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12:529–540 [DOI] [PubMed] [Google Scholar]
- Mosley CA, Acker TM, Hansen KB, Mullasseril P, Andersen KT, Le P, Vellano KM, Bräuner-Osborne H, Liotta DC, Traynelis SF. (2010) Quinazolin-4-one derivatives: A novel class of noncompetitive NR2C/D subunit-selective N-methyl-d-aspartate receptor antagonists. J Med Chem 53:5476–5490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley CA, Myers SJ, Murray EE, Santangelo R, Tahirovic YA, Kurtkaya N, Mullasseril P, Yuan H, Lyuboslavsky P, Le P, et al. (2009) Synthesis, structural activity-relationships, and biological evaluation of novel amide-based allosteric binding site antagonists in NR1A/NR2B N-methyl-d-aspartate receptors. Bioorg Med Chem 17:6463–6480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott DD, Doherty JJ, Zhang S, Washburn MS, Fendley MJ, Lyuboslavsky P, Traynelis SF, Dingledine R. (1998) Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nature Neurosci 1:659–667 [DOI] [PubMed] [Google Scholar]
- Nagy J, Boros A, Dezso P, Kolok S, Fodor L. (2003) Inducible expression and pharmacology of recombinant NMDA receptors, composed of rat NR1a/NR2B subunits. Neurochem Int 43:19–29 [DOI] [PubMed] [Google Scholar]
- Perin-Dureau F, Rachline J, Neyton J, Paoletti P. (2002) Mapping the binding site of the neuroprotectant ifenprodil on NMDA receptors. J Neurosci 22:5955–5965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. (2008) An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-d-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol 28:631–637 [DOI] [PubMed] [Google Scholar]
- Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ, and Memantine Study Group (2003) Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med 348:1333–1341 [DOI] [PubMed] [Google Scholar]
- Roberts E, Eargle J, Wright D, Luthey-Schulten Z. (2006) MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics 7:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Overington JP. (1994) Derivation of rules for comparative protein modeling from a database of protein structure alignments. Protein Sci 3:1582–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, Gouaux E. (2009) X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462:745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahirovic YA, Geballe M, Gruszecka-Kowalik E, Myers SJ, Lyuboslavsky P, Le P, French A, Irier H, Choi WB, Easterling K, et al. (2008) Enantiomeric propanolamines as selective N-methyl-d-aspartate 2B receptor antagonists. J Med Chem 51:5506–5521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamiz AP, Whittemore ER, Schelkun RM, Yuen PW, Woodward RM, Cai SX, Weber E, Keana JF. (1998) N-(2-(4-hydroxyphenyl)ethyl)-4-chlorocinnamide: a novel antagonist at the 1A/2B NMDA receptor subtype. Bioorg Med Chem Lett 8:199–200 [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K. (1993) Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol 44:851–859 [PubMed] [Google Scholar]
- Wright JL, Gregory TF, Kesten SR, Boxer PA, Serpa KA, Meltzer LT, Wise LD, Espitia SA, Konkoy CS, Whittemore ER, et al. (2000) Espitia SA, Konkoy CS, Whittemore ER, Woodward RM. Subtype-selective N-methyl-d-aspartate receptor antagonists: synthesis and biological evaluation of 1-(heteroarylalkynyl)-4-benzylpiperidines. J Med Chem 43:3408–3419 [DOI] [PubMed] [Google Scholar]
- Wu LJ, Zhuo M. (2009) Targeting the NMDA receptor subunit NR2B for the treatment of neuropathic pain. Neurotherapeutics 6:693–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF. (2009) Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J Neurosci 29:12045–12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Berger AJ, Brown PH, Kumar J, Balbo A, May CA, Casillas E, Jr, Laue TM, Patterson GH, Mayer ML, et al. (2012) Analysis of high-affinity assembly for AMPA receptor amino-terminal domains. J Gen Physiol 139:371–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.