

SUMMARY
During progression of atherosclerosis, myeloid cells destabilize lipid-rich plaque in the arterial wall and cause its rupture, thus triggering myocardial infarction and stroke. Survivors of acute coronary syndromes have a high risk of recurrent events for unknown reasons. Here we show that the systemic response to ischemic injury aggravates chronic atherosclerosis. After myocardial infarction or stroke, apoE−/− mice developed larger atherosclerotic lesions with a more advanced morphology. This disease acceleration persisted over many weeks and was associated with markedly increased monocyte recruitment. When seeking the source of surplus monocytes in plaque, we found that myocardial infarction liberated hematopoietic stem and progenitor cells from bone marrow niches via sympathetic nervous system signaling. The progenitors then seeded the spleen yielding a sustained boost in monocyte production. These observations provide new mechanistic insight into atherogenesis and provide a novel therapeutic opportunity to mitigate disease progression.
INTRODUCTION
Today, survival following a first myocardial infarct (MI) approaches 90%. However, re-infarction occurs commonly and carries a high mortality. In a representative trial, new myocardial ischemia occurred in 54% of patients within the first year after MI1. The largest population study to date showed a 17.4% 1-year risk of re-infarction2. Conventional wisdom infers that these very high rates of secondary events reflect later stages of linear disease progression. This study tested the alternative hypothesis that a first infarct — triggering a burst of acute systemic inflammation aimed at repair of the injured heart —could accelerate atherosclerosis.
Monocytes infiltrate lesions and, together with their lineage descendant macrophages, instigate inflammation and deliver proteolytic enzymes that digest extracellular matrix and render atherosclerotic plaques unstable3–7. Elevated levels of circulating monocytes provide an expanded pool of inflammatory cells available for recruitment to growing arterial lesions, potentially promoting plaque rupture. Leukocytosis post MI predicts an increased risk of re-infarction and death8,9. During acute MI, blood monocyte levels spike, and these cells accumulate in the evolving myocardial wound10,11. Thus, the organism experiences an acute inflammatory event (e.g. MI) superimposed on a pre-existing chronic inflammatory disease (atherosclerosis), both of which involve the same myeloid cell type. Given the frequency of re-infarction, we investigated whether acute myocardial injury accelerates pre-existing chronic atherosclerosis.
We found that in apoE−/− mice with atherosclerosis, MI increased plaque size and induced a 'vulnerable' lesion morphology with higher inflammatory cell content and protease activity, fueled by persistently increased myeloid cell flux to atherosclerotic sites. Earlier clinical studies described an increase of hematopoietic stem and progenitor cells (HSPCs) in the circulation of patients shortly after MI12. We thus hypothesized that release of these progenitors may increase the availability of monocytes. We found that in response to heightened sympathetic nervous system (SNS) activity — provoked by pain, anxiety, and heart failure in patients with MI HSPCs departed bone marrow niches and produced prolonged amplified extramedullary monocytopoiesis in mice after coronary ligation.
MI accelerates atherosclerosis
Proteases, including metalloproteinases and cysteinyl cathepsins, can catabolize the extracellular matrix of the plaque’s fibrous cap and render it prone to rupture13,14. Therefore, protease activity may serve as a marker in mice of processes associated with lesion vulnerability in humans15. To test the hypothesis that MI changes the course of atherosclerotic disease, we serially imaged protease activity in aortic plaques of apoE−/− mice, before and 3 weeks after coronary ligation, using fluorescence molecular tomography fused to X-ray computed tomography (FMT/CT)16. Imaging showed a sharp increase of plaque protease activity within 3 weeks after MI (Fig. 1a,b). In parallel, expression of the inflammatory cytokine IL-6, MMP-9, myeloperoxidase, and Ly-6C increased in atherosclerotic plaques (Supplementary Fig. 1). The number of monocytes/macrophages per aorta expanded, particularly the inflammatory Ly-6Chigh monocyte subset (Fig. 1c). Plaque monocyte content also increased in apoE−/− mice without MI, reflecting the natural course of disease in these animals17,18. Yet innate immune cell accumulation accelerated distinctively after MI, as indicated by the significantly greater slope obtained when fitting the number of Ly-6Chigh monocytes in the aorta over time (Supplementary Fig. 2). Neutrophil presence in atheromata also increased (Supplementary Fig. 3) while mast cells did not (Supplementary Fig. 4). Histologic analysis affirmed increased accumulation of CD11b+ myeloid cells and larger lesion size after MI (Fig. 1d). The thickness of the fibrous cap decreased, covering larger necrotic cores (Supplementary Fig. 5). Ly-6Chigh monocytes isolated from atherosclerotic lesions exhibited higher levels of mRNAs encoding inflammatory genes. IL-1β and cathepsin B were expressed at higher levels 3 weeks after MI, whereas arginase-1 and TGFβ, markers associated with alternatively activated macrophages, were expressed at lower levels (Supplementary Fig. 6). Monocyte numbers in the blood and spleen increased consistently for up to 3 months after coronary ligation (Supplementary Fig. 7) but were unaltered in the bone marrow (Supplementary Fig. 8).
Figure 1. Increased inflammation in atherosclerotic plaques after MI.

a, Protease activity by FMT-CT before and 3 weeks after MI. Circles indicate aortic root (n = 10 per group). b, Protease activity in excised aortae by fluorescence reflectance imaging (FRI), expressed as target to background ratio (TBR, n = 10 per group). c, Flow cytometric quantification of myeloid cells and Ly-6Chigh monocytes in aorta (n = 5–9 per group). Dot plots 3 weeks after MI are shown. d, CD11b staining and lesion size (n = 9–10 per group). Scale bar represents 150 μm. Mean ± s.e.m., * P < 0.05, ** P < 0.01.
Extramedullary monocytopoiesis post MI
Because the spleen has the ability to host extramedullary hematopoiesis19–21, we measured splenic monocyte progenitor content following MI. Hematopoietic progenitor cell numbers in the spleen increased post-MI (Fig. 2, Supplementary Fig. 9) but not in the bone marrow (Supplementary Fig. 10). Proliferation of progenitors doubled in the spleen (Supplementary Fig. 11). In patients that died after an acute MI, we found increased numbers of c-kit+ cells in the spleen, some of which colocalized with the proliferation marker Ki-67 (Supplementary Fig. 12).
Figure 2. Elevated levels of progenitor cells in the spleen of apoE−/− mice after MI.

a, Quantification for HSPCs, MDPs, and GMPs at different time points after MI (n = 3–15 per group). The gating strategy is shown in Supplementary Fig. 10. b, Number of colony-forming units. Mean ± s.e.m., * P < 0.05, ** P < 0.01.
When we splenectomized mice at the time of MI, atherosclerosis did not accelerate(Supplementary Fig. 13). The number of progenitor cells in liver tissue after MI was much lower than in the spleen, however; splenectomy increased progenitor cell presence in the liver 4 days after MI (Supplementary Fig. 14). We concluded that the infarct-induced monocytosis resulted primarily from augmented production in the spleen, but other extramedullary sites may contribute22. This observation raised the question whether monocytes of splenic and bone marrow origin differ qualitatively. Surprisingly, Ly-6Chigh monocytes isolated from the spleen or bone marrow on day 4 after MI had significantly different mRNA levels in 11 of the 32 genes assessed (Supplementary Fig. 15). For instance, IL-1β and cathepsin B mRNA levels were 60- and 6-fold higher in inflammatory monocytes isolated from the spleen, matching the increased expression of these genes in Ly-6Chigh monocytes isolated from atherosclerotic plaque after MI (Supplementary Fig. 6). Therefore, post-MI extramedullary myelopoiesis may not only increase the availability of inflammatory cells but also change their functional program. To test whether another form of acute tissue injury prevalent in atherosclerotic patients would accelerate splenic myelopoiesis, we analyzed apoE−/−mice 6 weeks after ischemic stroke. The number of myeloid cells and Ly-6Chigh monocytes in atherosclerotic plaque increased after stroke, in parallel with expanded splenic monocytopoiesis (Supplementary Fig. 16).
Bone marrow HSPC release post MI
Since granulocyte macrophage progenitors (GMP) and macrophage dendritic cell progenitors (MDP) have a limited self-renewal capacity23,24, we hypothesized that upstream progenitors released from their bone marrow niches sustain the splenic proliferative activity after MI. Indeed, blood levels of HSPCs increased 2, 7 and 24-fold at 6, 48 and 96 hours after MI, respectively (Fig. 3a, ). The number of splenic FLK2− HSPCs increased dramatically after MI (Supplementary Fig. 17). This mobilization of upstream HSPCs with high capacity for self-renewal likely explains the long-term boost in splenic monocyte production in apoE−/− mice after MI.
Figure 3. β3 adrenergic receptor-mediated progenitor release after MI.

a, Flow cytometric analyses of HSPC in blood of C57BL/6 mice (n = 6–11 per group). b, Immunostaining for tyrosine hydroxylase (TH). Scale bar represents 10μm. Insets depict low magnification overview. Bar graph shows quantitation of TH+ area (n = 5 per group). c, Expression of HSPC retention factors (relative to Gapdh) in the bone marrow of C57BL/6 mice on day 4 after MI (n = 8 per group). Mean ± s.e.m., * P < 0.05, ** P < 0.01.
Anxiety, pain and impaired left ventricular function during MI can all activate the SNS. Accordingly, levels of tyrosine hydroxylase, the rate-limiting enzyme for production of noradrenaline in sympathetic fibers25, increased in the bone marrow of mice after MI and hence indicated a higher sympathetic tone (Fig. 3b). SNS activity may liberate hematopoietic stem cells from their niches by signaling through the β3 adrenergic receptor26. Nestin+ mesenchymal stem cells express this receptor which regulates the production of stem cell retention factors27. Because acute MI raises blood progenitor levels in patients12, we investigated whether SNS activity causes the release of HSPCs from the bone marrow after MI. Blood HSPCs decreased by 100, 75, and 50% at 6, 48, and 96 hours after MI in mice treated with a β3 receptor antagonist (Fig. 3a). The stem cell retention factor CXCL12, angiopoietin and stem cell factor (SCF)28 underwent similar regulation (Fig. 3c). Levels of the adhesion molecule VCAM-1, which also retains HSPCs in the bone marrow, decreased after MI but did not change after β3 receptor blocker administration (Fig. 3c). These data indicate that increased sympathetic tone after MI causes withdrawal of stem cell retention factors by β3 adrenoreceptor-expressing niche cells.
Treatment with a β3 adrenergic blocker reduced splenic accumulation of progenitors in wild type mice shortly after MI (Supplementary Fig. 18) and consequently diminished their output of myeloid cells (Supplementary Fig. 19). In apoE−/− mice 3 weeks after MI, β3 blocker treatment reduced the number of GMPs and their progeny in the spleen and blood (Supplementary Fig. 20). Retrospective analysis of a clinical trial29 revealed that prior β-blocker therapy was associated with a reduction in monocytes after an acute coronary syndrome (Supplementary Table 1). The mechanism that led to this decrease is unclear, also because some clinically used β-blockers have a lower affinity for the β3 receptor subtype30; however, these associative data show an interesting parallel to our findings in mice.
In apoE−/− mice after MI, β3 blocker treatment lowered protease activity, myeloid cell content, and mRNA levels of inflammatory cytokines in the plaque (Supplementary Fig. 21). When we adoptively transferred GFP+ GMPs to wild-type mice with MI, β3 blocker treatment did not alter their splenic differentiation (Supplementary Fig. 22). Sympathetic denervation with 6-hydroxydopamine (6-OHDA)26,31 increased bone marrow mRNA levels of the stem cell retention factor CXCL12, reduced levels of HSPCs in blood, decreased circulating monocyte levels, and attenuated the accumulation of myeloid cells in atherosclerotic lesions (Supplementary Fig. 23). Combination of β3 blockade and splenectomy showed no additive effects (Supplementary Fig. 24). Neither MI nor β3 blockade changed blood cholesterol and HDL levels (Supplementary Fig. 25).
IVM of HSPC departure from bone marrow
We adoptively transferred lineage− c-kit+ Sca-1+ Flk2− HSPCs labeled with a fluorescent membrane dye (DiD) to examine their release with serial intravital microscopy32. DiD+ cells were quantified after they had settled into the bone marrow, and then again 4 days after MI. Concomitant with the post-MI increase of progenitors in circulation, 52% of cells that were present during the first imaging session departed from the bone marrow, which was inhibited by the β3 receptor antagonist (Fig. 4). Post-imaging flow cytometry corroborated the trafficking of DiD+ cells (Supplementary Fig. 26). We next investigated the relocation of bone marrow cells to the spleen directly. Lineage− c-kit+ Sca-1+ Flk2 − HSPCs were harvested from CD45.2+ donors and labeled with a photoconvertible dye before transfer into CD45.1+ recipients. These cells engrafted into the skull bone marrow, where we photoconverted them with laser illumination. Only if mice underwent coronary ligation, photoconverted CD45.2+ DAPI+ cells were detected in splenic cell suspensions 4 days later (Supplementary Fig. 27).
Figure 4. Serial intravital imaging of progenitor release from the bone marrow.
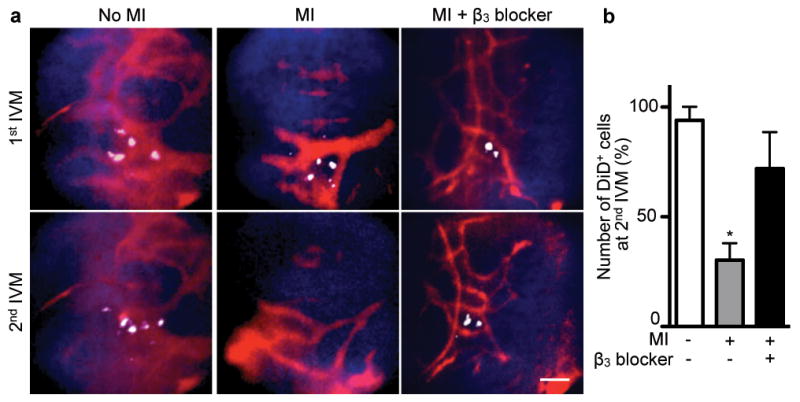
a, DiD labelled-HSPC Flk2− cells were imaged in the skull bone marrow by intravital microscopy (IVM) before and then again 4 days after MI. DiD labelled-HSPC are white, blood pool red, and bone is blue. The scale bar represents 50 μm. b, Change of HSPC presence between 1st and 2nd IVM session (n = 3 per group). Mean ± s.e.m., * P < 0.05.
Splenic HSPC engraftment after MI
Finally, we investigated the mechanisms of splenic progenitor seeding. The mRNA levels of stem cell factor (SCF) increased in splenic tissue after MI in parallel with the number of SCF+ cells in splenic sections (Fig. 5a,b). Antibody neutralization of SCF decreased retention and proliferation of adoptively transferred HSPCs in the spleen (Fig. 5c,d). Colocalisation studies identified CD31+ and occasionally nestin+ cells (Supplementary Fig. 28a,b) as a source of SCF, in agreement with a recent report on SCF's role in the splenic niche during the steady state33. We found adoptively transferred DiD+ Flk-2− HSPCs cells in close vicinity to CD31+ cells (Supplementary Fig. 28c). Neutralization of VLA-4, an integrin involved in stem cell retention34,35, reduced the number of adoptively transferred HSPCs in the spleen after MI, but not in the steady state (Supplementary Fig. 29).
Figure 5. Splenic progenitor engraftment after MI.

a, qPCR of SCF in spleen (n = 5–6 per group). b, Number of SCF+ cells in spleen of C57BL/6 mice 4 days after MI by immunofluorescence. c, Enumeration of adoptively transferred GFP+ HSPCs on day 4 after MI (n = 8 per group). d, Proliferation of endogenous HSPCs determined by BrdU incorporation (n = 8 per group). e, Paradigm. Mean ± s.e.m., * P < 0.05, ** P < 0.01.
DISCUSSION
This study reports that acute MI or stroke augments inflammation in atherosclerotic plaques at a distance. After an ischemic event, atherosclerotic plaque grew faster and displayed higher protease activity. We identified an increased supply of innate immune cells as a driving force for this phenomenon. On a systems level, pre-existing chronic inflammation flared when the organism experienced an additional acute inflammatory stimulus. Increased sympathetic nervous system activity after MI released upstream progenitors from bone marrow niches. On the receiving end, the spleen hosted these cells by increasing SCF production, leading to amplified extramedullary myelopoiesis (Paradigm, Fig. 5e). The pro-inflammatory changes in atherosclerotic plaques persisted for several months.
The evolutionary benefit of outsourcing myelopoiesis from the bone marrow may involve the protection of steady state "housekeeping" in this confined compartment. Unlike the bone marrow, the spleen is an organ that can rapidly expand in size. In the event of increased leukocyte need after acute injury, the myelopoietic system may proliferate in extramedullary sites to protect quiescent stem cells and to ensure unimpeded production of red cells, platelets and lymphocytes in the bone marrow.
Despite growing understanding of the chronic inflammatory nature of atherosclerosis3,6,7, specific anti-inflammatory therapy has yet to materialize. Given myeloid cells’ central role in disease promotion and their rapid turn-over in inflamed tissue, interrupting the monocyte supply chain may attenuate atherosclerosis. In our case, SNS inhibition abrogated stem cell release from the bone marrow. Since the regulation of progenitor cell migration is multifactorial35, there are other targets along this pathway that await exploration, including chemokine receptors and cytokines involved in stem cell activation. In addition, the innate immune response unleashed by acute ischemic injury may also change the “fluid phase” of blood by augmenting circulating acute phase reactants such as fibrinogen and plasminogen activator inhibitor-1, factors that promote thrombosis and counter endogenous fibrinolysis36. Our study suggests that patients with an ischemic complication of atherosclerosis experience a particularly vulnerable disease phase, and that interventions aiming at progenitors of innate immune cells could profoundly impact long-term outcomes.
METHODS SUMMARY
Wild-type C57BL/6J, C57BL/6.SJL, C57BL/6-Tg(UBC-GFP)30Scha/J and B6.129P2-Apoetm1Unc/J mice were used in these studies, which were approved by the Subcommittee on Animal Research Care at Massachusetts General Hospital (13th Street, Charlestown, MA). The patient studies were conducted in accordance with the Declaration of Helsinki. The studies were approved by the Research Committee of the Department of Pathology of the VUmc, Amsterdam and by the Ethikkommission Heidelberg University (Voss Str. 9, Heidelberg, Germany). Detailed procedures are available online at www.nature.com/nature.
Supplementary Material
Acknowledgments
We thank the CSB Mouse Imaging Program (Jessica Truelove, Derrick Jeon, Jessica Donahoe, Brett Marinelli), and Kamila Naxerova for helpful discussions. This work was funded by grants from the National Institute of Health R01-HL096576, R01-HL095629 (M.N.); R01-EB006432, T32-CA79443 (R.W.). F.L. was funded in part by Deutsche Forschungsgemeinschaft SFB 938/Z2. Figure 5e was produced using Servier Medical Art (www.servier.com).
Footnotes
AUTHOR CONTRIBUTIONS
P.D. and G.C. performed experiments, collected and analyzed the data, and contributed to writing the manuscript, R.G. did surgeries and performed experiments, Y.W., F.L., R.G., C.R., Y.I., B.T., A.L.C., T.H., M.D.M., F.La., M.E., P.W., M.T.W., A.T.C., A.M.L., H.W.M.N., J.J.P., B.B.R., J.B., J.S., H.A.K., C.V., S.A.M., D.A.M., and M.S.S. performed experiments, collected, analyzed and discussed data, M.A.M., M.J.P., P.L., C.P.L., F.K.S. and R.W. conceived experiments and discussed strategy and results; M.N. designed and managed the study and wrote the manuscript which was edited and approved by all co-authors.
COMPETING FINANCIAL INTERESTS
Marc S. Sabatine, David A. Morrow and Sabina A. Murphy received grant support from AstraZeneca and GSK. The remaining authors declare no competing financial interests.
References
- 1.Goldstein JA, et al. Multiple complex coronary plaques in patients with acute myocardial infarction. N Engl J Med. 2000;343:915–922. doi: 10.1056/NEJM200009283431303. [DOI] [PubMed] [Google Scholar]
- 2.Milonas C, et al. Effect of Angiotensin-converting enzyme inhibition on one-year mortality and frequency of repeat acute myocardial infarction in patients with acute myocardial infarction. Am J Cardiol. 2010;105:1229–1234. doi: 10.1016/j.amjcard.2009.12.032. [DOI] [PubMed] [Google Scholar]
- 3.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 4.Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 5.Randolph GJ. The fate of monocytes in atherosclerosis. J Thromb Haemost. 2009;7 (Suppl 1):28–30. doi: 10.1111/j.1538-7836.2009.03423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 7.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*) Annu Rev Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ernst E, Hammerschmidt DE, Bagge U, Matrai A, Dormandy JA. Leukocytes and the risk of ischemic diseases. JAMA. 1987;257:2318–2324. [PubMed] [Google Scholar]
- 9.Sabatine MS, et al. Relationship between baseline white blood cell count and degree of coronary artery disease and mortality in patients with acute coronary syndromes: a TACTICS-TIMI 18 (Treat Angina with Aggrastat and determine Cost of Therapy with an Invasive or Conservative Strategy- Thrombolysis in Myocardial Infarction 18 trial)substudy. J Am Coll Cardiol. 2002;40:1761–1768. doi: 10.1016/s0735-1097(02)02484-1. [DOI] [PubMed] [Google Scholar]
- 10.Nahrendorf M, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–2445. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massa M, et al. Increased circulating hematopoietic and endothelial progenitor cells in the early phase of acute myocardial infarction. Blood. 2005;105:199–206. doi: 10.1182/blood-2004-05-1831. [DOI] [PubMed] [Google Scholar]
- 13.Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, et al. In vivo imaging of proteolytic activity in atherosclerosis. Circulation. 2002;105:2766–2771. doi: 10.1161/01.cir.0000017860.20619.23. [DOI] [PubMed] [Google Scholar]
- 16.Nahrendorf M, et al. Hybrid in vivo FMT-CT imaging of protease activity in atherosclerosis with customized nanosensors. Arterioscler Thromb Vasc Biol. 2009;29:1444–1451. doi: 10.1161/ATVBAHA.109.193086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tacke F, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swirski FK, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robbins CS, et al. Extramedullary hematopoiesis generates Ly–6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364–374. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leuschner F, et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. 2012;209:123–137. doi: 10.1084/jem.20111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swirski FK, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Psaltis PJ, et al. Identification of a monocyte-predisposed hierarchy of hematopoietic progenitor cells in the adventitia of postnatal murine aorta. Circulation. 2012;125:592–603. doi: 10.1161/CIRCULATIONAHA.111.059360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kondo M, et al. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. 2003;21:759–806. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- 24.Geissmann F, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zigmond RE, Ben-Ari Y. Electrical stimulation of preganglionic nerve increases tyrosine hydroxylase activity in sympathetic ganglia. Proc Natl Acad Sci U S A. 1977;74:3078–3080. doi: 10.1073/pnas.74.7.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katayama Y, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–421. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 27.Mendez-Ferrer S, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- 29.Cannon CP, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–1504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 30.Hoffmann C, Leitz MR, Oberdorf-Maass S, Lohse MJ, Klotz KN. Comparative pharmacology of human beta-adrenergic receptor subtypes--characterization of stably transfected receptors in CHO cells. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:151–159. doi: 10.1007/s00210-003-0860-y. [DOI] [PubMed] [Google Scholar]
- 31.Kruszewska B, Felten SY, Moynihan JAB. Alterations in cytokine and antibody production following chemical sympathectomy in two strains of mice. J Immunol. 1995;155:4613–4620. [PubMed] [Google Scholar]
- 32.Lo Celso C, et al. Live–animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457:92–96. doi: 10.1038/nature07434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–462. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams DA, Rios M, Stephens C, Patel VP. Fibronectin and VLA-4 in haematopoietic stem cell-microenvironment interactions. Nature. 1991;352:438–441. doi: 10.1038/352438a0. [DOI] [PubMed] [Google Scholar]
- 35.Lo Celso C, Scadden DT. The haematopoietic stem cell niche at a glance. J Cell Sci. 2011;124:3529–3535. doi: 10.1242/jcs.074112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.