

Abstract
When mTOR inhibitor rapalogs prevent cap-dependent translation of cell cycle proteins like cmyc, continuing tumor cell growth depends on cap-independent translation, which is mediated by internal ribosome entry sites (IRESes) located in the 5′UTR of transcripts. To investigate if rapalog-induced activation of MNK kinases played a role in such IRES activity, we studied multiple myeloma (MM) cells. Rapamycin activated MNK1 kinase activity in MM cell lines and primary specimens by a MAPK-dependent mechanism. Pharmacological inhibition of MNK activity or genetic silencing of MNK1 prevented a rapalog-induced upregulation of c-myc IRES activity. Although rapamycin, used alone, had little effect on myc protein expression, when combined with a MNK inhibitor, myc protein expression was abrogated. In contrast, there was no inhibition of myc RNA, consistent with an effect on myc translation. In a rapamycin-resistant MM cell lines as well as a resistant primary MM specimen, co-exposure to a MNK inhibitor or MNK1 knockdown significantly sensitized cells for rapamycin-induced cytoreduction. Studies in MNK-null murine embryonic fibroblasts additionally supported a role for MNK kinases in rapamycin-induced myc IRES stimulation. These results indicate that MNK kinase activity plays a critical role in the fail safe mechanism of IRES-dependent translation when mTOR is inhibited. As kinase activity also regulated sensitivity to rapamycin, the data also provide a rationale for therapeutically targeting MNK kinases for combined treatment with mTOR inhibitors.
Keywords: Multiple myeloma, rapamycin, mTOR inhibitors, c-myc, IRES activity, MNK kinases
INTRODUCTION
Previous work in glioblastoma, prostate cancer and multiple myeloma (MM) models demonstrates that sensitivity of tumor cells to rapalog mTOR inhibitors is, at least in part, dependent upon AKT activity (1-4). When rapalogs inhibit cap-dependent translation, tumor cell growth depends on cap-independent translation of critical proteins, which is mediated by internal ribosome entry sites (IRESes) in the 5′UTR of mRNAs. Specifically for D-cyclin and cmyc translation occurring during mTOR inhibition, IRES activity of these transcripts is curtailed by AKT (5). Thus, hyperactive AKT prevents this fail-safe mechanism of translation and, during mTOR inhibition, cellular levels of cyclin and myc fall precipitously resulting in G1 arrest. In contrast, cells with quiescent AKT activity demonstrate upregulation of cyclin/myc IRES activity upon exposure to rapamycin ensuring maintained levels and continual cell cycle transit.
Our work (5, 6) also demonstrates that both ERK and p38 MAPK pathways are critical for this IRES activity observed during mTOR inhibition. The participation of these pathways in myc IRES function had also previously been shown to be operative during genotoxic and apoptotic responses (7, 8). The mechanism by which these pathways facilitate IRES activity is unknown. However, a possible central candidate are the MNK kinases. These signal proteins are downstream target substrates of both the ERK and p38 MAPK pathway (9). In addition, they are activated secondary to mTOR inhibition (10). Furthermore, a single report in Aplysia neurons demonstrated an elevation of the cap-independent/dependent translation ratio upon MNK over-expression (11). Finally, the MNK kinases have been reported to induce post-translational modification of hnRNP A1 (12), a bona fide myc/cyclin IRES-trans acting factor (ITAF), which binds to the IRES and enhances its activity. In fact, hnRNP A1 presence is required for myc and D-cyclin IRES activation during mTOR inhibition.
The two major MNK kinases, MNK 1 and MNK 2, both have MAPK-binding motifs. However, differences in their C-termini result in differential functional responses. MNK 1 has low basal activity but can be activated by either p38 or ERK signaling (9, 13, 14). Activation is associated with phosphorylation in threonine residues in the T activation loop of the kinase. In contrast, MNK 2 has high basal activity that does not increase upon activation of MAPKs and ERK/p38 inhibitors do not affect MNK 2 activity (13, 14). MNK kinases are thought to be the only kinases that phosphorylate eIF-4E on S209 (15). To phosphorylate eIF-4E, MNK kinases must first bind to eIF-4G (16, 17). Interestingly, MNK1/MNK2 knock-out mice appear normal (15), suggesting that the MNKs are critical for stress responses rather than normal basal physiology. In keeping with that theory, MNK-deficient cells are more sensitive to serum starvation (18). Thus, it seems possible that these kinases could protect the cell from the stress of mTOR inhibition by supporting the remaining mechanism of protein translation occurring through the IRES. We, thus, in the current study, addressed the role of MNK kinase activation in myeloma cells stressed by exposure to rapamycin.
RESULTS
Rapamycin activates the MNK-1 kinase in MM cells through MAPK-dependent pathways
The response of multiple myeloma (MM) cells to challenge with mTOR inhibitors is, in part, regulated by IRES activity. For example, D-cyclin translation in rapamycin-treated MM cells can be maintained by cyclin-D IRES activity which is promoted by the MAPK ERK pathway (6). This fail safe mechanism of cyclin translation affords resistance to rapamycin-induced G1 arrest. When ERK is inhibited, IRES activity is restricted and, along with reduced cap-dependent translation, cyclin levels rapidly fall and G1 arrest ensues. The c-myc IRES is also a well characterized IRES (19) and its function is specifically up-regulated in myeloma cells (20, 21). Of interest, myc IRES activity is also regulated by both the ERK and p38 MAPK pathways (5, 7, 8). To test whether myc IRES activity occurring in MM cells during mTOR inhibition was related to MNKs, we first tested the ability of rapamycin (RAP) to activate these kinases. ANBL-6, U266 or 8226 MM cell lines were incubated with rap for 3 hrs followed by immunoblot assays for expression of phosphorylated MNK. The antibody detects phosphorylated MNK1 as well as MNK2. As shown in fig 1A, rap successfully upregulated phosphorylation of MNK in all 3 cell lines while having no significant effect on total MNK levels. In time course experiments, rap induced MNK phosphorylation as early as 60 mins (fig 1B). In addition, the activation of MNK kinases was temporally correlated with enhanced phosphorylation of eIF-4E (fig 1B), a substrate of MNKs (15), suggesting that rap enhanced kinase activity of MNKs as well as their phosphorylation state.
Figure 1.
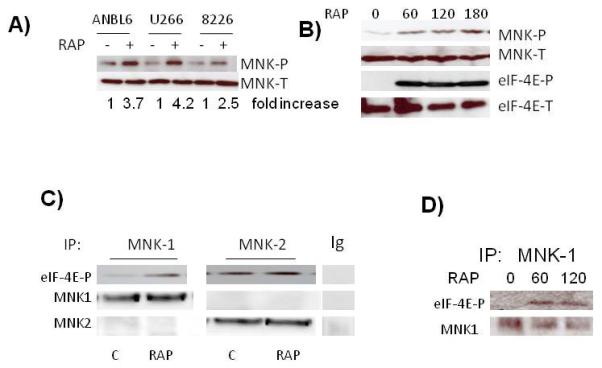
Activation of MNK kinases in MM cells. A) MM cell lines exposed to rapamycin (100 nM) for 3 hrs followed by immunoblot assay for phospho-MNK and total MNK expression. Fold increase is determined by densitometric ratio of MNK-P/MNK-total and represents the mean of 3 independent experiments. Rap-induced increase was significant (p<0.05) in all three cell l ines. B) ANBL-6 cells exposed to rapamycin for 0, 60, 120 or 180 mins, followed by immunoblot assay for phospho-MNK, total MNK, phospho-eIF-4E and total eIF-4E expression. C) MNK1 or MNK2 immunoprecipitated with anti-MNK1, anti-MNK2 antibodies or non-specific IgG, from control or rapamycin (100 nM for 60 mins)-treated ANBL-6 extracts and tested for ability to phosphorylate eIF-4E in vitro. eIF-4E phosphorylation determined by immunoblot. D) MNK1 immunoprecipitated from MM cells after 0, 60 or 120 mins of exposure to rapamycin (100 nM) and tested for phosphorylation of eIF-4E in vitro.
To further confirm enhanced kinase function and ascertain if MNK1 or MNK2 was being activated, we performed in vitro kinase assays (Figs 1C and 1D). MM cells were exposed to DMSO alone (control) or to rap at 100 nM. After 1 hr, either MNK1 of MNK2 was immunoprecipitated from protein lysates and tested for its ability to phosphorylate eIF-4E in vitro. As shown by immunoblot (fig 1C), the immunoprecipitating antibodies were specific for MNK1 or MNK2 without cross reactivity. The immunoprecipitated MNK1 from rap-treated MM cells demonstrate a significantly increased ability to phosphorylate eIF-4E relative to control cells. In contrast, MNK2 kinase activity was constitutively higher but there was no effect of RAP. A second limited time course experiment (fig 1D) further demonstrated the ability of rap to increase MNK1 kinase activity by 60 mins of exposure. These data indicate that rapamycin primarily stimulates MNK1 kinase activity.
Both ERK and p38 MAPK pathways have been described as potential activators of MNKs (5, 7, 8). In our MM cell model, Rap activated both pathways, demonstrated by the enhanced phosphorylation of ERK or p38 (fig 2A). The effect on ERK peaked at 30 mins while the effect on p38 lagged behind, occurring sometime between 30 and 60 mins. To identify if these pathways mediate MNK1 activation in MM cells, we used inhibitors of the ERK (U0126) or p38 (SB203580) pathways. Specificity of the inhibitors is presented in fig 2B. As shown, U0126, used at 1 or 10uM (U1 and U2 respectively), effectively inhibited ERK phosphorylation but had no effect on phosphorylation of hsp27, a p38 MAPK substrate. Conversely, the SB203580 p38 inhibitor used at 12.5 and 25 uM (SB1 and SB2 respectively) successfully inhibited p38-mediated phosphorylation of hsp27 but had no non-specific inhibitory effect on ERK phosphorylation. In Rap-stimulated cells, MNK phosphorylation was most inhibited by the p38 inhibitor. Thus, the p38 MAPK stress-activated pathway appears more important in MNK phosphorylation than the ERK pathway, at least in MM cells stressed by rapamycin exposure. Although these data are consistent with previous reports (5, 7, 8) indicating MNK kinases are downstream of the p38 pathway, it should be noted that, in lung cancer cells, rapamycin-induced eIF-4E phosphorylation is relatively unaffected by a p38 inhibitor (22).
Figure 2.
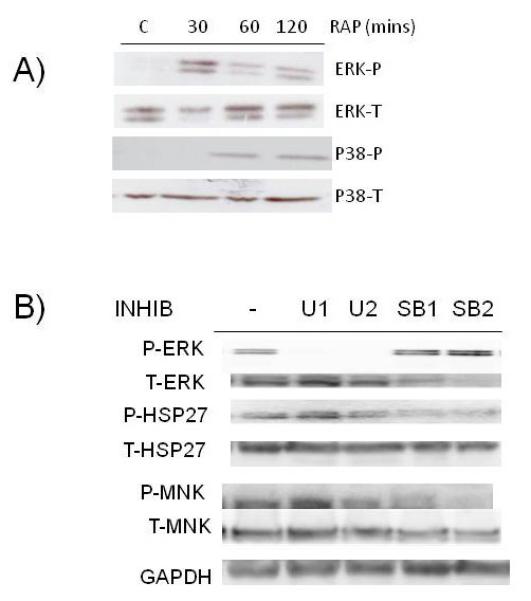
Role of MAPK pathways in MNK phosphorylation. A) ANBL-6 MM cells exposed to rapamycin (100 nM) for 30, 60 or 120 mins (C=control, no rap), followed by immunoblot assay for phospho-ERK, total-ERK, phospho-p38 or total p38 expression. B) MM cells pre-treated for 30 mins with U0126 at 1 or 10 uM (U1 and U2) or SB203580 at 12.5 or 25 uM (SB1 and SB2). Rapamycin then added at 100 nM for additional 3 hrs and immunoblot assay performed for expression of phospho-ERK, total ERK, phospho-HSP 27, total HSP-27, phospho-MNK, total MNK or GAPDH.
MNK inhibition curtails rapamycin-induced myc IRES activity
We initially used the MNK1/MNK2 inhibitor CGP57380 to test effects on IRES activity. Figure 3A demonstrates the ability of CGP to prevent rapamycin-induced MNK activity as shown by phosphorylation of the eIF-4E MNK substrate. At the lowest concentration (12.5 uM), eIF-4E phosphorylation was decreased by 80% (mean of 3 separate experiments) while it was completely ablated at 25 or 50 uM. At this early time point (3 hrs), there was no effect of the MNK inhibitor on cell survival. To test effects of the inhibitor on myc IRES activity, MM cells were transfected with either the pRF or pRmF dicistronic reporter constructs as shown in fig 3B and subsequently treated with rapamycin +/− CGP. The c-myc 5′UTR, containing its IRES, was subcloned into the intracistronic space between the Renilla and firefly luciferase open reading frames in the pRF vector to yield the pRmF vector. The pRmF reporter’s firefly luciferase translation is driven by the myc 5′UTR and is a reflection of IRES-dependent, cap-independent translation while Renilla expression is due to cap-dependent, IRES-independent translation. Results are normalized for transfection efficiency by co-transfection with a beta-galactosidase construct. A previous study (23) has shown that firefly luciferase expression in these MM cells transfected with pRmF is not due to presence of a cryptic promoter in the 5′UTR. In addition, the ANBL-6 maintains a relatively low level of activated AKT allowing significant myc IRES activity (23).
Figure 3.
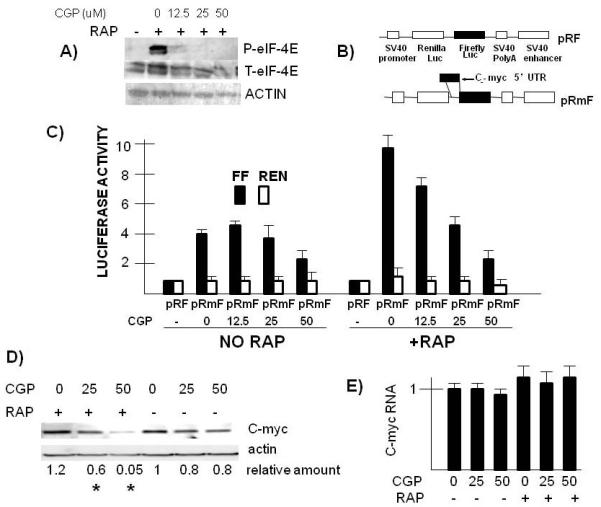
Effect of MNK inhibitor on myc IRES activity and myc expression. A) ANBL-6 MM cells pre-treated with the CGP MNK inhibitor for 30 mins at varying concentrations. Followed by addition of rapamycin (100 nM) for 3 hrs and then immunoblot assay for phospho-eIF-4E, total eIF-4E and actin. B) Reporter constructs used to assay for myc IRES activity. C) Firefly (FF, dark bars) or Renilla (Ren, open bars) luciferase expression in pRF versus pRmF-transfected ANBL-6 cells treated+/−rapamycin (100 nM) and +/− CGP (used at 0, 12.5, 25 or 50 uM). All lluciferase activity is normalized to the luciferase values (both Renilla and Firefly) obtained for pRF in the absence of added rapamycin and CGP (designated “1”). Results represent means+/−SD of quadruplicate samples. D) ANBL-6 MM cells pre-treated with CGP at 0, 25 or 50 uM followed by addition of rapamycin (or not) at 100 nM for 8 hrs. Immunoblot assay then performed for c-myc or actin expression. Relative amount of c-myc protein determined densitometrically (ratio of c-myc/actin) and represents means of three independent experiments. Asterix denotes a value significantly lower (p<0.05) than control (no CGP). E) Experiment performed as in “D” but assay is real time PCR for c-myc RNA expression (data are means+/−SDs of 3 experiments).
Figure 3C is a representative experiment of 4 separate experiments, each with identical results. In the absence of rapamycin, the presence of the myc 5′UTR in the intracistronic space in the pRmF vector increased firefly luciferase (FF, black bars) to 4.5 × fold versus that of the pRF control vector. In contrast, the presence of the 5′UTR had no effect on Renilla expression. Rapamycin exposure significantly stimulated IRES activity as the increase in firefly expression due to the myc 5′UTR was now 9.3 × fold versus that of the pRF vector in rapamycin treated cells. This approximate 2 × fold increase in IRES activity was consistent across all four experiments (mean increase of 2.3+/−0.5, mean+/−SD) and was statistically significant at the p<0.05 level. It is difficult to discern in figure 3C, but rapamycin had a modest inhibitory effect on Renilla luciferase activity with an approximately 25% reduction. In rapamycin-stressed MM cells, the addition of the CGP MNK inhibitor had a concentration-dependent inhibitory effect on IRES activity assayed by firefly luciferase expression. This was statistically significant (p<0.05) at all CGP concentrations. In contrast, the inhibitor had less of an effect on myc IRES activity (firefly luciferase expression) in the basal state in the absence of rapamycin with only 50 uM having a significant effect. There was no consistent effect of the CGP inhibitor on Renilla luciferase expression except at the highest concentration (50 uM) in rapamycin-treated cells where expression was reduced to 50% of control.
The ability of CGP to inhibit myc IRES activity in rapamycin-challenged cells is associated with curtailed myc protein expression. As shown in fig 3D, rapamycin, used alone, has little effect on c-myc expression presumably due to the stimulation of IRES-mediated cap-independent translation. In addition, the CGP inhibitor, used by itself, has only a minimal effect. However, inhibiting MNK activity with CGP during rapamycin exposure significantly prevents myc protein expression. As shown in fig 3E, the CGP-induced inhibition of protein expression is not associated with an inhibition of myc RNA. These results indicate that MNK activity in rapamycin-treated MM cells plays a role in the upregulated myc IRES activity observed and helps maintain myc expression through post-transcriptional activity.
Effects of MNK silencing on myc IRES activity
The above data with the CGP inhibitor suggested a role for MNK1 activation in the rapamycin-induced upregulation of myc IRES activity. However, as there may be non-specific effects of the inhibitor, we knocked down MNK1 by shRNA transfection of MM cells. In ANBL-6 MM cells, we targeted 2 separate sequences of MNK1 (shRNA 1-1 and 1-4). As shown in fig 4A, knockdown of MNK1 was much more successful with shRNA 1-4 than with 1-1. In addition, the knockdown was relatively specific for MNK1 as MNK2 RNA and protein levels were only minimally affected.
Figure 4.
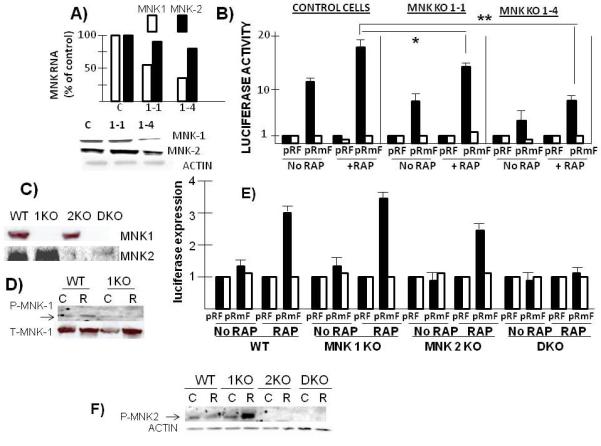
Effect of silencing MNKs on myc IRES activity- A) MNK1 knocked down in ANBL-6 MM cells by shRNAs targeting two separate sequences (1-1 and 1-4). Control (C) cells infected with shRNA targeting scrambled sequence. Shown is real time PCR for MNK1 or MNK2 RNA expression and immunoblot for protein expression. B) Stably knocked down cells transfected with pRF or pRmF reporter plasmids followed by treatment+/−rapamycin (100nM) for 3 hrs and luciferase expression assayed. Black bars are firefly and white bars are Renilla luciferase. Data are means+/−SD of 4 separate experiments. *=significantly different than control, p<0.05; **=significantly different at p=0.01. C) MNK wild type, MNK1 knocked out (1KO), MNK2 knocked out (2KO) or MNK1/MNK2 double knock out (DKO) mouse embryonic fibroblasts (MEFS) assayed for MNK1 or MNK2 expression. D) WT or MNK1 knocked out MEFs treated with rapamycin (R) (100 nM for 3 hrs) or without (control (C)) followed by immunoblot assay for phospho-MNK1 or total MNK1. E) Different MEF cell lines transiently transfected with pRF or pRmF reporter plasmids, treated with or without rapamycin (100 nM) and luciferase expression assayed as described above in figure 3C legend. Data represent means+/−SD of 3 independent experiments. F) Different MEF cell lines treated with rapamycin (R; 100nM for 3 hrs) or without (control (C)), followed by immunoblot assay for phospho-MNK2 and actin.
These cell lines were then tested in the myc IRES reporter assay by transiently transfecting either pRF or pRmF reporter plasmids, followed by treatment with or without rapamycin and assessment of luciferase expression. In control cells transfected with shRNA targeting scrambled sequence, exposure to rapamycin induced a 1.8 × fold increase in IRES activity (firefly luciferase expression, black bars in figure 4B). In both MNK-1 knockout cell lines, the basal IRES activity was modestly but significantly decreased and the rapamycin-induced upregulation was blunted. The 1-4 MNK knockout line was more inhibited in its IRES activity. The inhibition of IRES activity in the basal state by the MNK knockouts was comparable to inhibition of activity induced by rapamycin. However, in a second MM cell lines, U266, the rapamycin-induced IRES response was more inhibited than the basal response (suppl fig 1). In that cell line, we only targeted one sequence of MNK1with shRNA and obtained a successful knock down. In control U266 cells (transfected with shRNA against scrambled sequence), rapamycin increased IRES activity 4 × fold (firefly luciferase expression, black bars). In contrast, the rapamycin-induced increase in IRES activity was significantly curtailed in the MNK1 knocked-out U266 cell line (only 1.8x fold increase).
In a third MM cell line, OPM-2, a slightly different pattern of IRES responses occurred (suppl fig 2). Although rapamycin was capable of inducing ERK, MNK and eIF-4E phosphorylation (supp fig 2A), it did not significantly enhance IRES activity (supp fig 2C) when used at 20 nM. We have previously demonstrated (24) how heightened AKT activity prevents a rapamycin-induced IRES response because of phosphorylation and inactivation of the myc IRES ITAF, hnRNP A1. This is the likely explanation for the OPM-2 results as this cell line expressed heightened AKT activation due to its PTEN null state (4). Nevertheless, the MNK inhibitor CGP, used at 20 uM, significantly inhibited eIF-4E phosphorylation (suppl fig 2B) and myc IRES activity (suppl fig 2C) in OPM-2 cells in the presence or absence of rapamycin. These data in U266 as well as OPM-2 cells demonstrate MNK-dependent IRES activity is a generalized finding in MM cells and not singular to ANBL-6 cells.
Further support for a role for MNK kinases in myc IRES activity comes from experiments with MNK-null murine embryonic fibroblasts (MEFs). These cell lines have been previously used (10) to demonstrate a rapamycin-induced activation of MNK activity. Fig 4C confirms the absence of MNK1 or MNK2 in these cell lines. Immunoprecipitation of MNK1 from protein extracts demonstrated in vitro kinase activity against eIF-4E when extracts were obtained from wild type (WT) or MNK2 knock-out (2KO) lines but not when obtained from MNK1 knockout (1KO) or MNK1/MNK2 double knock out (double) cell lines (supplemental fig 3). Conversely, immunoprecipitated MNK2 has kinase activity when obtained from WT or MNK1 knockout (1KO) cell lines but not from 2KO or double knock out cells (suppl fig 3). Fig 4D also demonstrates the ability of rapamycin to increase MNK1 phosphorylation in WT MEFs, which was ablated in the MNK1 null cells. The cell lines were then transiently transfected with the pRF or pRmF reporter constructs, treated with or without rapamycin and luciferase expression evaluated (fig 4E). In wild type (WT) MEFs, there is very minimal myc IRES activity in the basal state but activity (ie., firefly luciferase expression, black bars) increases 3x fold following exposure to rapamycin. There was no increase in Renilla expression seen in rapamycin-treated cells. A significant rapamycin-induced increase in firefly expression was likewise seen in both MNK1 and MNK2 knock out MEFs. However, this response was ablated in the double knockout cell line (DKO). Thus, in genetically knocked out MEFs, the rapamycin IRES response can be supported by either MNK1 or MNK2 but the response is lost when both MNKs are absent.
To explain why the MNK1 KO MEFs were not inhibited in rapamycin-induction of myc IRES activity, we considered the possibility that, in these cells, rapamycin could activate MNK2 which might facilitate IRES activity. We, thus, treated each of the MEF cell lines with and without rapamycin and tested MNK2 phosphorylation. As shown in fig 4F, rapamycin was capable of robust MNK2 phosphorylation in MNK1 knocked out MEFs although no activation was seen in WT MEFs. These data suggest that, in genetically knocked out MEFs, MNK2 can become activated by rapamycin if MNK1 is absent.
To confirm the specificity of effects of MNK silencing in MEFs, we stably re-expressed either FLAG-tagged MNK1, MNK2 or both MNK1/MNK2 in the double knockout MEF cell line. In supplemental figure 4A, a Western blot demonstrates expression of the transgenes (top panel) with a corresponding rescuing of eIF-4E phosphorylation, confirming kinase activity of the re-expressed proteins. These cell lines were then transiently transfected with pRF and pRmF reporter plasmids, treated+/− rapamycin and reporter expression assayed. As shown in suppl fig 4B, re-expression of either MNK1, MNK2 or both MNK1/MNK2, rescued the ability of rapamycin to significantly enhance myc IRES activity (firefly luciferase expression), thus confirming that its is the loss of MNK1/MNK2 in the DKO MEFs that prevents the IRES response.
Effects of MNK inhibition on cellular responses to rapamycin
To test if paralysis of MNK1 affected cellular growth responses to rapamycin, the ANBL-6 MM cell line was treated for 48 or 72 hrs with the CGP MNK inhibitor (25 uM), rapamycin (100 nM) or the combination of both drugs. ANBL-6 cells were resistant to rapamycin used alone as shown in fig 5A. Furthermore, MNK inhibition with CGP used alone had no significant effect on MM cell recovery as well. However, a significant cytoreduction was present when CGP was added to rapamycin. Relative resistance to rapamycin, which was reversed by CGP, was also seen in U266 MM cells (fig 5B).
Figure 5.
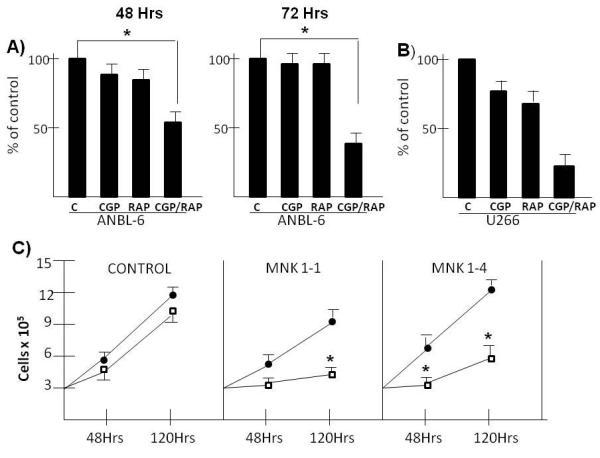
Effect of MNK paralysis on responses to rapamycin. A) ANBL-6 MM cells treated +/− CGP (25 uM) +/− rapamycin (100nM) for 48 or 72 hrs. Viable cell recovery then enumerated by trypan blue exclusion. Data represent means+/−SD of 4 separate experiments. The only significant difference (p<0.05) are in the combined CGP/rapamycin-treated groups at both time points (designated by an asterix). B) U266 cells similarly treated with RAP+/−CGP. Data are means+/−SD, n=3. Significant differences (p<0.05) designated by asterix. C) ANBL-6 MM cells stably transfected with shRNA targeting scrambled sequence (control) or MNK1 sequences (1-1 and 1-4) and treated with or without rapamycin (100 nM) for 48 or 120 hrs followed by enumeration of viable cells. Closed circles are control, non-treated cells and open squares are rapamycin-treated. Data represent means+/−SD of 3 separate experiments. *=significantly different from control (non-rapamycin-treated), p<0.05.
Further confirmation of a role for MNK1 in MM cell rapamycin responses was obtained by using the two ANBL-6 MNK1 knockout transfectants, 1-1 and 1-4. Their growth over 120 hrs in the absence of rapamycin (black circles, fig 5C) is comparable to control cells transfected with scrambled sequence. However, the MNK1 knocked out cell lines are considerably more sensitive to rapamycin (open squares) as shown in fig 5C. Rapamycin has no effect on cell growth in control cells but is effective in preventing growth in MNK1 knocked-out cell lines. These data support the notion that the activation of MNK1 in rapamycin-treated cells serves as a protective factor.
To test if MNKs played a role in rapamycin responses in primary MM cells, we first tested for induction of MNK phosphorylation. As shown in fig 6A, Western blot analysis demonstrated a rapamycin-induction of MNK phosphorylation in four primary MM specimens, although the degree of induction was variable. In two MM specimens, we were fortunate to harvest sufficient numbers of purified MM cells for further study. One sample was exposed to rapamycin (100 nM)+/− the CGP MNK inhibitor at 25 or 50 uM. As shown in fig 6B, both concentrations of CGP successfully prevented MNK activity, shown by abrogated eIF-4E phosphorylation. This specimen is relatively resistant to rapamycin used alone in terms of c-myc down regulation, similar to ANBL-6 MM cell line. However, CGP can inhibit myc expression in the presence or absence of rapamycin although myc downregulation is considerably more effective in the presence of rapamycin. The combination of CGP and rapamycin is also more effective at preventing survival of primary MM cells (fig 6C). After 48 hrs of culture, rapamycin, used at 100 nM, or CGP at 50 uM, have no significant effect. However, concurrent exposure to both agents significantly inhibited viable recovery of primary cells.
Figure 6.
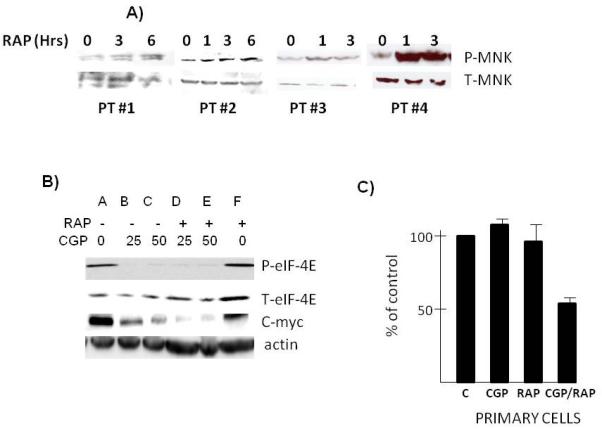
Effect of MNK inhibition in primary MM cells. A) Primary MM cells obtained from bone marrow biopsies of 4 patients and treated with or without rapamycin (100 nM) for 1,3 or 6 hrs followed by immunoblot assay for phospho-MNK and total MNK. B) Primary MM cells pre-treated with CGP at 0, 25 or 50 uM for 30 mins followed by addition of rapamycin (100 nM) for 6 hrs and then immunoblot assay performed for phospho-eIF-4E, total eIF-4E, c-myc and actin expression. C) Primary MM cells exposed to CGP (50 uM), rapamycin (100 nM) or the combination of the two drugs for 72 hrs followed by assessment of viable recovery by trypan blue assay. Data are means+/−SD of 4 wells/group. Control (C) cells not treated represent 100%.
DISCUSSION
Our previous work has documented the regulatory influence of IRES-dependent, cap-independent translation upon tumor responses to mTOR inhibitors (5). TORC1 inhibition, induced by rapalogs, primarily results in restrained cap-dependent translation of cell cycle proteins like c-myc and D-cyclins with attendant G1 arrest. As the only remaining mechanism for myc/cyclin translation in rapalog-treated cells, IRES activity can determine whether overall myc/cyclin levels are maintained or significantly depressed. The results of the current study demonstrate that MNK kinase activity is a key regulator of rapamycin-induced IRES activity. MNK activity was enhanced by exposure to rapamycin in conjunction with myc IRES function and downregulation of MNK activity with the CGP inhibitor or by MNK knockdown, prevented IRES stimulation and sensitized to rapamycin cytoreduction.
The MAPK-dependence of rapamycin-induced MNK phosphorylation mirrors the MAPK-dependence of myc IRES activity. Activation of the p38 MAPK cascade during apoptosis (8) or genotoxic stress (7) is necessary for the stimulation of IRES function. The ERK MAPK pathway also participates in upregulated IRES function during genotoxic stress (7). In some rapamycin-treated MEF cell lines, both MAPK pathways are involved in enhanced myc IRES activity (5). The identification of a role for MNK kinases in IRES activity could explain the involvement of the MAPK cascades. It is likely that which MAPK cascade mediates IRES stimulation is cell line- and stimulus-dependent. It is also likely that MNK kinases stimulate IRES activity directly or indirectly via phosphomodulation of one or more IRES-trans-acting factors (ITAFs) that are critical for the myc IRES. Alternatively, MNK activity may lead to changes in ITAF expression. Candidate ITAFs include hnRNP A1, which can be phosphorylated by MNK kinases (12) or PCBP1, whose expression is dependent on p38 activity, at least in neuronal cells (25). Both hnRNP A1 and PCBP1 are required for myc IRES activity (24, 26).
In myeloma cells, rapamycin primarily stimulated MNK1 activity and MNK1 knockdown curtailed upregulated IRES function. However, significant rapamycin-mediated stimulation still occurred (fig 4B). As loss of MNK1 function can be compensated for by MNK2, it is possible that, in MNK1 shRNA-silenced MM cells, rapamycin could stimulate MNK2 activity with resulting maintenance of some IRES activity. This notion is supported by the results of the IRES reporter assay in MNK-null MEFs. Although rapamycin stimulated MNK1 in these MEFs, loss of MNK1 did not affect rapamycin-stimulated IRES activity although double MNK1/MNK2 knock-out MEFS had abrogated activity. The ability of rapamycin to induce MNK2 phosphorylation specifically in MNK-1 knocked out MEFs is consistent with MNK2 compensation for the MNK1 null state.
Myc IRES activity is specifically enhanced in MM cells (20, 21). IRES activity could be particularly important in this tumor model because continuing ER stress, due to heightened Ig synthesis, restrains mTOR-mediated cap-dependent translation. A recent finding (27) of MM-specific over-expression of DEPTOR, an mTOR inhibitor, is consistent with this idea. Heightened MM IRES activity could also be one reason for ineffectiveness of rapalogs in MM patients (28). Although our ANBL-6 MM cell line and primary specimen were resistant to rapamycin-induced growth inhibition, concurrent paralysis of MNKs with CGP or shRNA knockdown allowed for significant cytoreduction. In contrast, there was minimal effect of MNK inhibition on MM cells not challenged with rapamycin. These results suggest that MNK activity may be less critical for IRES function in the basal state, at least for these myeloma cell types. However, in other MM clones, constitutive MAPK signaling, due to MM growth factor stimulation (29) or RAS mutation (30) could result in upregulated MNK activity and myc IRES function. Nevertheless, these results suggest MNK kinases could be potential therapeutic targets in MM patients.
MATERIALS & METHODS
Cell lines, reagents, plasmids, transfections
The MM cell lines were obtained from ATCC. The MEF cell lines have been previously described (10,15). The pRF construct was a kind gift of Dr. A. Willis (University of Leicester). The myc IRES was cloned into pRF as previously described (5) to obtain pRmF. The plko.1 lentiviral vectors targeting MNK1 were purchased from Sigma-Aldrich. shRNA 1-1 has the ID# TRCW0000006232 and shRNA 1-4 is TRC0000199013. Virus particles expressing these shRNAs were produced and titers determined by the UCLA viral vector core. Lentiviral infection of MM cells was performed as previously described (31). After infection, clones were selected in puromycin. Rapamycin and CGP57380 were purchased from Calbiochem. All antibodies were purchased from Cell Signaling Technology, including the anti-phospho-MNK antibody.
Primary myeloma specimens
Primary MM cells were purified from bone marrow of patients by negative selection as described (31) using the RosettesSep antibody cocktail method (Stem Cell technologies). The purity by microscopy and CD138 flow analysis was >99% plasma cells.
Evaluation of protein and RNA expression
Western blot was performed as described (31). Real time PCR for myc RNA and GAPDH RNA was performed as described (23). All real time PCR samples were run in triplicate.
MNK In vitro kinase assay
The MNK kinase assay was carried out as described (32) with modifications. Cells were lysed with ice-cold cell lysis buffer(20 mM Tris-HCl (pH 7.5) 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, 1MM PMSF). Direct covalent attachment of Mnk1 (Santa Cruz #sc-133107) and Mnk2 (Sigma #M0696) antibodies to the agarose beads were performed with Pierce Direct IP Kit according to the manufacture’s protocol (Thermo Fisher Scientific #26468). Labeled beads were washed three times with kinase buffer (20 mM HEPES, 10 mM MgCl2, 10 mM sodium b-glycerophosphate,, pH 7.4) before 1ug purified GST fusion eIF-4E protein(purchased from BPS Bioscience, #40530) was added. Reaction mixtures were incubated at 30°C for 30 minutes in the presence of 25 mM ATP. After SDS-PAGE, phosphorylated eIF-4E proteins were detected on Western blots to assay Mnks activity.
Myc IRES activity
The dicistronic pRF or pRmF reporter constructs were transfected into cell lines using Lipofectamine Plus (Invitrogen) and normalized for transfection efficiency by cotransfection with pSVβGal (promega). Transfection efficiency was generally 5-10%. A transfection efficiency of at least 5% was required for carrying out a dicistronic reporter assay. After 12-14 hrs, cells were treated and were then harvested, followed by detection of Renilla luciferase, firefly luciferase and β-galactosidase activities as previously described (5). All luciferase activity is normalized to the luciferase values (both Renilla and firefly) obtained for pRF in the absence of any treatment, which is designated as a value of “1”.
Cell survival assays and statistics
Quantitative increases in protein phosphorylation on Western blots were evaluated by densitometric analysis of ratio of phosphorylated-protein/total protein signal of treated MM cells. All Western blots were repeated 3 times and the mean fold increase (n=3) in drug-treated groups versus non-treated cells is shown under the gels in the figures. The t-test was used to determine significance of differences between groups. The viable recovery data shown in figs 5 & 6 are means. Percent viable recovery is determined by enumeration of trypan blue-negative viable cells with comparison to that of cells not exposed to any drugs.
Supplementary Material
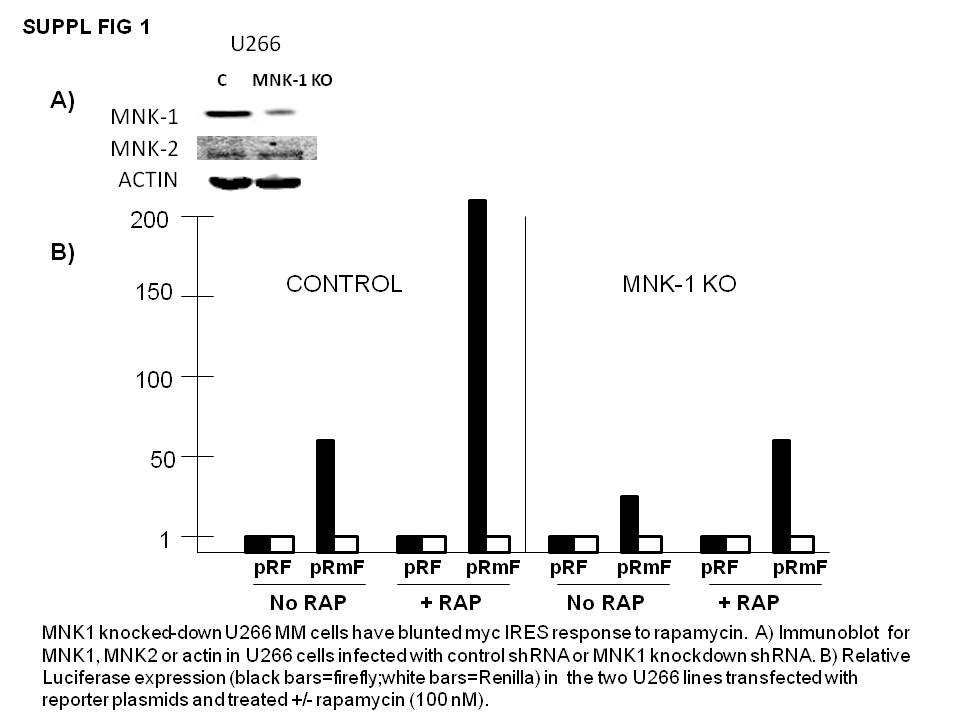{kind=link}
{kind=link}
{kind=link}
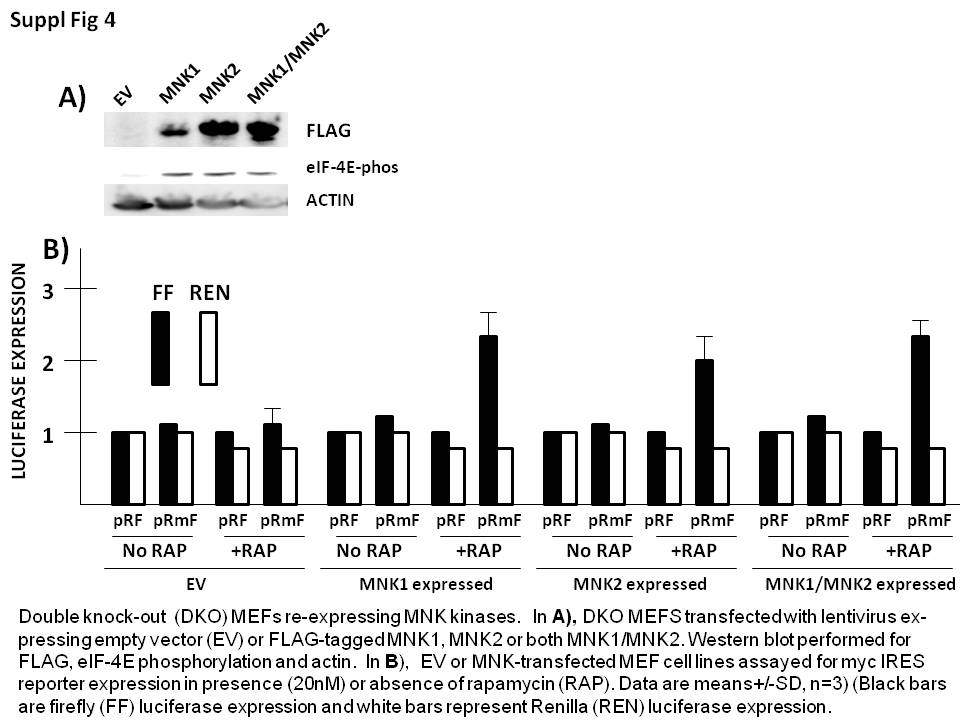{kind=link}
Acknowledgements
The authors thank the UCLA Jonsson Comprehensive Cancer Center vector core lab for assistance in generating the lentiviral shRNA vectors.
Supported by research funds of the Veteran’s Administration, the Multiple Myeloma Research Foundation and the Department of Defense and NIH grants RO1 CA109312 and RO1 CA111448
Footnotes
Conflict of interest: The authors declare no conflict of interest
References
- 1.Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Peterson R, et al. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc. Natl. Acad. Sci. 2001;98:10314–10319. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gera J, Mellinghoff I, Shi Y, Rettig M, Tran C, Hsu JH, et al. AKT activity determines sensitivity to mTOR inhibitors by regulating cyclin D1 and c-myc expression. J. Biol. Chem. 2004;279:2737–2746. doi: 10.1074/jbc.M309999200. [DOI] [PubMed] [Google Scholar]
- 3.Frost P, Moatomed F, Hoang B, Shi Y, Gera J, Yan H, et al. In vivo antitumor effects of the mTOR inhibitor CCI-779 against human multiple myeloma cells in a xenograft model. Blood. 2004;104:4181–4187. doi: 10.1182/blood-2004-03-1153. [DOI] [PubMed] [Google Scholar]
- 4.Shi Y, Gera J, Hu L, Hsu JH, Bookstein R, Li W, et al. Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779. Cancer Res. 2002;62:5027–5034. [PubMed] [Google Scholar]
- 5.Shi Y, Sharma A, Wu H, Lichtenstein A, Gera J. Cyclin D1 and c-myc IRES dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38 MAPK- and ERK-dependent pathway. J. Biol. Chem. 2005;280:10964–109973. doi: 10.1074/jbc.M407874200. [DOI] [PubMed] [Google Scholar]
- 6.Frost P, Shi Y, Hoang B, Gera J, Lichtenstein A. Regulation of D-cyclin translation inhibition in myeloma cells treated with mTOR inhibitors: rationale for combined treatment with ERK inhibitors and rapamycin. Mol Cancer Ther. 2009;8:83–93. doi: 10.1158/1535-7163.MCT-08-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subkhankulova T, Mitchell SA, Willis AE. IRES-mediated initiation of c-myc protein synthesis following genotoxic stress. Biochem. J. 2001;359:183–192. doi: 10.1042/0264-6021:3590183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoneley M, Chappell SA, Jopling CL, Dickens M, MacFarlane M, Willis AE. Cmyc protein synthesis is initiated from the IRES during apoptosis. Mol. & Cell. Biol. 2000;20:1162–1169. doi: 10.1128/mcb.20.4.1162-1169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parra JL, Buxade M, Proud CG. Features of the catalytic domains and C termini of the MAPK signal-integrating kinases MNK1 and MNK2 determine their differing activities and regulatory properties. J. Biol. Chem. 2005;280:37623–37633. doi: 10.1074/jbc.M508356200. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Yue P, Chan C-B, Ye K, Ueda T, Watanabe-Fukunaga R, et al. Inhibition of mTOR induces PI3-kinase-dependent and MNK-mediated eIF-4E phosphorylation. Mol. & Cell. Biol. 2007;27:7405–7413. doi: 10.1128/MCB.00760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross G, Dyer JR, Castellucci VF, Sossin WS. MNK is a negative regulator of cap-dependent translation in Aplysia neurons. J. of Neurochem. 2006;97:79–91. doi: 10.1111/j.1471-4159.2006.03704.x. [DOI] [PubMed] [Google Scholar]
- 12.Buxade M, Parra JL, Rousseau S, Shpiro N, Marquez R, Morrice N, et al. The MNKs are novel components in the control of TNF alpha biosynthesis andphosphorylate and regulate hnRNP A1. Immunity. 2005;23:177–189. doi: 10.1016/j.immuni.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Scheper GC, Morrice NA, Kleijn M, Proud CG. The mitogen-activated protein kinase signal-integrating kinase MNK2 is an eIF-4E kinase with high levels of basal activity in mammalian cells. Mol. Cell. Biol. 2001;21:743–754. doi: 10.1128/MCB.21.3.743-754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheper GC, Parra J-L, Wilson ML, van Kollenburg B, Vertegaal ACO, et al. The N and C termini of the splice variants of the human MNK2 determine activity and localization. Mol. Cell. Biol. 2003;23:5692–5705. doi: 10.1128/MCB.23.16.5692-5705.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R. MNK2 and MNK1 are essential for constitutive and inducible phosphorylation of eIF-4E but not for cell growth or development. Mol. Cell Biol. 2004;24:6539–6549. doi: 10.1128/MCB.24.15.6539-6549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parra-Palau JL, Scheper GC, Wilson ML, Proud CG. Features in the N and C termini of the MAPK-integrating kinase MNK1 mediate its nucleocytoplasmic shuttling. J. Biol. Chem. 2003;278:44197–44204. doi: 10.1074/jbc.M302398200. [DOI] [PubMed] [Google Scholar]
- 17.Pyronnet S, Imataka H, Gingras AC, Fukunaga R, Hunter T, Sonenberg N. Human eIF4G recruits MNK1 to phosphorylate eIF-4E. EMBO J. 1999;18:270–279. doi: 10.1093/emboj/18.1.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chrestensen CA, Shuman JK, Eschenroeder A, Worthington M, Gram H, Sturgill TW. MNK1 and MNK2 regulation in HER2-overexpressin breast cancer lines. J. Biol. Chem. 2007;282:4243–4252. doi: 10.1074/jbc.M607368200. [DOI] [PubMed] [Google Scholar]
- 19.Stoneley M, Willis AE. Cellular IRESes: structures, trans-acting factors and regulation of gene expression. Oncogene. 2004;23:3200–3207. doi: 10.1038/sj.onc.1207551. [DOI] [PubMed] [Google Scholar]
- 20.Paulin FEM, West MJ, Sullivan NF, Whitney R,L, Lyne L, Willis AE. Aberrant translational control of the c-myc gene in multiple myeloma. Oncogene. 1996;13:505–513. [PubMed] [Google Scholar]
- 21.Chappell SA, LeQuesne JPC, Paulin FEM, deSchoolmeester ML, Stoneley M, Soutar RL, et al. A mutation in the c-myc IRES leads to enhanced internal ribosome entry in multiple myeloma: a novel mechanism of oncogene de-regulation. Oncogene. 2000;19:4437–4440. doi: 10.1038/sj.onc.1203791. [DOI] [PubMed] [Google Scholar]
- 22.Sun S-Y, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al. Activation of AKT and eIF4E survival pathways by rapamycin-mediated mTOR inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 23.Shi Y, Frost P,J, Hoang B, Benavides A, Sharma S, Gera J, et al. IL-6-induced stimulation of c-myc translation in multiple myeloma cells is mediated by myc IRES function and the RNA-binding protein hnRNP A1. Cancer Res. 2008;68:10215–10222. doi: 10.1158/0008-5472.CAN-08-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jo OD, Martin J, Bernath A, Masri J, Lichtenstein A, Gera J. Heterogeneous nuclear ribonucleoprotein A’1 regulates cyclin D and c-myc IRES function through AKT signaling. J Biol Chem. 2008;283:23274–23287. doi: 10.1074/jbc.M801185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Y, Sun Y, Mao O, Jin KL, Greenberg D. Expression of poly(C) binding proteins is differentially regulated by hypoxia and ischemia in cortical neurons. Neuroscience. 2002;110:191–198. doi: 10.1016/s0306-4522(01)00522-x. [DOI] [PubMed] [Google Scholar]
- 26.Evans JR, Mitchell SA, Spriggs KA, Ostrowski J, Bomsztyk K, Ostarek D, et al. Members of the poly (rC) binding protein family stimulate the activity of the c-myc IRES in vitro and on vivo. Oncogene. 2003;22:8012–8020. doi: 10.1038/sj.onc.1206645. [DOI] [PubMed] [Google Scholar]
- 27.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:1–14. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farag SS, Zhang S, Jansak BS, Wang X, Kraut E, Chan K, et al. Phase II trial of temsirolimus in patients with relapsed or refractory multiple myeloma. Leuk Res. 2009;33:1475–1480. doi: 10.1016/j.leukres.2009.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogata A, Chauhan D, Teoh G, Treon S, Urashima M, Schlossman RL, et al. IL-6 triggers cell growth via the RAS-dependent mitogen-activated protein kinase cascade. J. of Immunol. 1997;159:2212–2221. [PubMed] [Google Scholar]
- 30.Paquette R,L, Berenson J, Lichtenstein A, McCormick F, Koeffler HP. RAS mutations in multiple myeloma. Oncogene. 1990;5:1569–1663. [PubMed] [Google Scholar]
- 31.Hoang B, Frost P, Shi Y, Belanger E, Benavides A, Pezeshkpour G, et al. Targeting TORC2 in multiple myeloma with a new mTOR kinase inhibitor. Blood. 2010;116:4560–4568. doi: 10.1182/blood-2010-05-285726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinses MNK1 and MNK2. EMBO J. 1997;16:1909–1920. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.