

Abstract
A library of complexes that included iron, cobalt, nickel, and copper chelates of cyclam, cyclen, DOTA, DTPA, EDTA, tripeptide GGH, tetrapeptide KGHK, NTA, and TACN was evaluated for DNA nuclease activity, ascorbate consumption, superoxide and hydroxyl radical generation, and reduction potential under physiologically relevant conditions. Plasmid DNA cleavage rates demonstrated by combinations of each complex and biological coreactants were quantified by gel electrophoresis, yielding second-order rate constants for DNAsupercoiled to DNAnicked conversion up to 2.5 ×106 M-1min-1, and for DNAnicked to DNAlinear up to 7 ×105 M-1min-1. Relative rates of radical generation and characterization of radical species were determined by reaction with the fluorescent radical probe TEMPO-9-AC and rhodamine B. Ascorbate turnover rate constants ranging from 9.1×10-3 to 8.2 min-1 were determined, although many complexes demonstrated no measureable activity. Inhibition and Freifelder-Trumbo analysis of DNA cleavage supported concerted cleavage of dsDNA by a metal associated ROS in the case of Cu2+(aq), Cu-KGHK, Co-KGHK, and Cu-NTA and stepwise cleavage for Fe2+(aq), Cu-cyclam, Cu-cyclen, Co-cyclen, Cu-EDTA, Ni-EDTA, Co-EDTA, Cu-GGH, and Co-NTA. Reduction potentials varied over the range from -362 mV to +1111 mV versus NHE, and complexes demonstrated optimal catalytic activity in the range of the physiological redox coreactants ascorbate and peroxide (-66 to +380 mV).
Keywords: Artificial nuclease, gene therapy, oxidation, reactive oxygen species, catalysis, multiple turnover, redox chemistry, metal, chelate, metallodrug
INTRODUCTION
There is significant interest in the development of artificial nucleases, especially those that cause sequence-specific inactivation of disease-associated genes. Because diseases such as cancer and HIV ultimately stem from genetic sources, they require a gene modification strategy at the DNA level, rather than downstream products, to effect a cure. Current anticancer strategies include the use of DNA-binding cisplatin derivatives,1,2 DNA intercalators3,4 and alkylating drugs5,6 as antineoplastic agents. Progress in gene therapy has been unsatisfactory, since even the most successful attempts tend to result in secondary cancers as a result of unintended insertional mutagenesis.7,8 Another important approach for anti-cancer and anti-HIV therapy is the use of compounds that incorporate both a DNA-damaging hydrolytic or redox-active metal complex and a stable gene-specific targeting sequence, such that the compound can both specifically hybridize to and damage the targeted disease-associated gene, leaving other genes unaffected.9-14 In addition to gene targeting and cell delivery, two major factors critical to the success of redox-dependent DNA-targeting compounds are their abilities to efficiently utilize biologically-available redox coreactants, as required, and to function with multiple turnover.15-19 Artificial nucleases must be very efficient in order to compete with the innate cellular DNA repair machinery.
With these factors in mind, the goal of this study was to determine and compare the DNA nuclease activities, oxidative pathways, and reduction potentials of various metal-chelate complexes that are likely candidates for incorporation into gene-specific artificial nucleases. These studies also served to elucidate the factors required for optimal nuclease activity, as well as identify several metal-chelates with many, if not all of the desired properties for use in gene-selective nuclease catalysts. A library consisting of combinations of the transition metals Fe2+, Co2+, Ni2+, and Cu2+ with the chelators cyclam, cyclen, DOTA, DTPA, EDTA, tripeptide GGH, tetrapeptide KGHK, NTA, and TACN was studied (Scheme 1). These ligands reflect the broad range of carboxylate and cyclic amine chelates that commonly form the basic framework for DNA cleavage agents, and also build on our prior studies of the natural ATCUN peptide sequence.16-18,20,21 DNA cleavage rates, coreactant consumption rates, radical generation rates, characterization of reactive intermediates, coreactant selectivity, and reduction potentials were determined. As expected, the chelators were found to significantly modulate the behavior of these transition metals, resulting in a wide variety of nuclease activity, observed mechanisms, and reduction potentials. Reactivity was also highly dependent on the available redox coreactants.
Scheme 1.
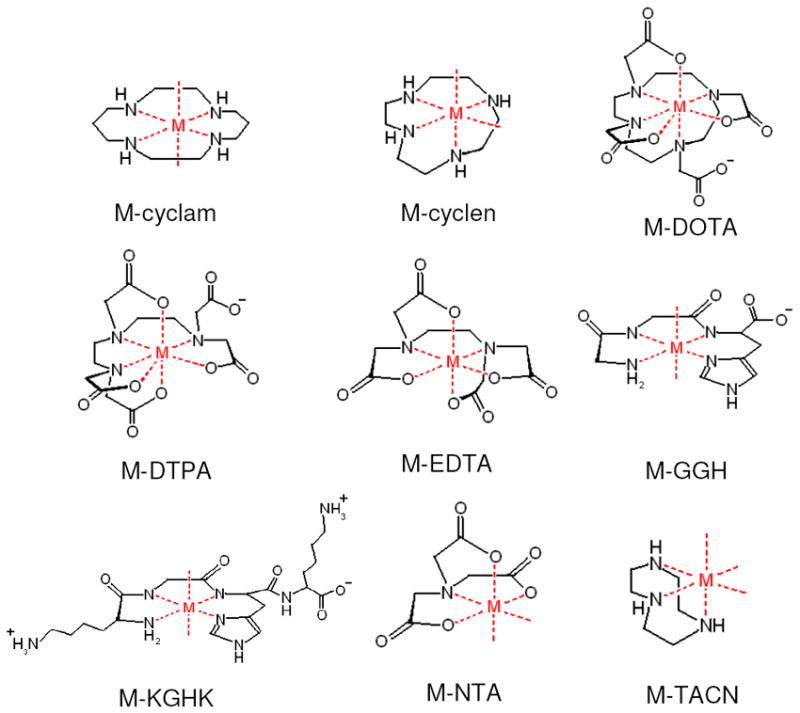
A summary of the metal-chelates used in this study, where M = Fe2+, Co2+, Ni2+, or Cu2+. These complexes are expected to accommodate up to a 6-coordinate metal coordination geometry, although Fe-DOTA and Fe-DTPA have typically been shown to adapt a 7-coordinate metal coordination geometry. The M-cyclam and ATCUN (GGH and KGHK) complexes tend to leave two trans open coordination sites, while M-cyclen complexes have two cis open coordination sites as a result of the smaller chelate size relative to the metal. Finally, M-TACN complexes have 3 open coordination sites.22,31-37
These results are useful for interpretation of the metal-mediated mechanisms responsible for the observed DNA nuclease activities. While many of the metal complexes studied herein have been the focus of prior reports,12,18,20,22-30 comparison of data is hindered by the range of experimental conditions used. Moreover, a variety of mechanisms and rates of DNA cleavage have been reported, reflecting variations of solution pH and buffers, and the use of solution coreactants of varying redox potentials, all of which precludes an accurate comparison of the reactivity of these metal complexes. The studies reported here were conducted under uniform conditions, allowing a high degree of confidence when making comparisons between metal-chelates or assays. The results obtained and conclusions drawn should provide a benchmark for future work in this area.
MATERIALS AND METHODS
Chemicals and Reagents
The chelators 1,4,7,10-tetraazacyclododecane (cyclen) and 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) were obtained from Macrocyclics. Tripeptide GGH-OH (GGH) and tetrapeptide KGHK-OH (KGHK) were obtained from Bachem. 1,4,8,11-tetraazacyclotetradecane (cyclam), diethylenetriaminepentaacetic acid (DTPA), nitrilotriacetic acid (NTA), and 1,4,7-triazacyclononane (TACN) were purchased from Sigma. Ethylenediaminetetraacetic acid (EDTA) was purchased from Aldrich. Fe(II) sulfate heptahydrate, Co(II) chloride hexahydrate, Ni(II) tetrahydrate, and Cu(II) chloride dihydrate were purchased from ACROS, J.T.Baker, Aldrich, and J. T. Baker, respectively. Salts, NaCl and NaOH, were purchased from Fisher, and HEPES was purchased from Sigma. Ascorbic acid and rhodamine B were purchased from Fluka. Stabilized 30% hydrogen peroxide solution and dibasic sodium phosphate were purchased from Sigma-Aldrich. TEMPO-9-AC was purchased from Invitrogen.
Sample Handling and Preparation
The water used in all procedures was purified using a Barnstead NANOpure Diamond filtration system with a 0.2 μm pore size filter. Plasmid DNA was isolated from a pUC19 transformed DH5α E. coli cell line and purified by use of a Qiagen miniprep kit following water elution from spin columns (the use of EDTA-containing buffer was avoided). The isolated DNA was quantified by absorbance measurements, divided into single-use aliquots in water, and frozen at -20 °C. Ascorbic acid solutions were prepared freshly each day by making a 100 mM ascorbic acid solution in 20 mM HEPES, 100 mM NaCl, and balancing the pH to 7.4 with 1 M NaOH. Ascorbic acid was kept on ice and diluted prior to each experiment. Solutions of H2O2 were made and used in a similar manner, but without the subsequent pH balancing, since the H2O2 concentrations used did not alter the pH of the buffer. Stock H2O2 solutions were freshly prepared from a refrigerated 30% H2O2 stock and maintained on ice prior to use. The TEMPO-9-AC radical probe was initially dissolved in DMSO and diluted into aliquots in 20 mM HEPES, 100 mM NaCl, pH 7.4. Metal-chelate (M-chelate) complexes were prepared freshly on the day of use by mixing the respective metal salt solution in water with the chelator in buffer to a final ratio of 1:1.5 and incubating for 20-30 min at RT. A slight excess of chelator was added to ensure that no free metal ion was present. Complex formation and chelator concentration were verified by UV/Vis titration. As expected,17 Fe2+ did not form stable complexes with either GGH or KGHK, while complex formation between iron and the chelators cyclam and cyclen was not observed under the conditions used in these studies (20 mM HEPES, 100 mM NaCl, pH 7.4). Accordingly, these four complexes were not included in the assays described herein.
Gel Electrophoresis Analysis of Plasmid DNA Cleavage
Reactions were run aerobically with 10 μM base pair pUC19, 100 nM M-chelate, 1 mM H2O2, and 1 mM ascorbate in 20 mM HEPES, 100 mM NaCl, pH 7.4. For each M-chelate, 8 time points were collected by incubating 8 tubes in parallel, initiating reaction at staggered time points over 6 h, and quenching simultaneously by placing on ice and adding loading dye. The quenched reactions were immediately loaded onto 1% agarose gels containing ethidium bromide, separated for 30-45 min at 120 V, and visualized using a BioRad gel doc. Supercoiled, nicked, and linearized plasmid DNA at each time point was quantified with the program ImageQuant. A correction factor of 1.47 was applied for the intensity of supercoiled DNA to account for the diminished ability of supercoiled DNA to intercalate ethidium bromide.18 Time-dependent supercoiled, nicked, and linear DNA concentration data were fit to a first order consecutive model defined by equations 1, 2, and 3, where S, N, and L correspond to the concentrations of supercoiled, nicked, and linearized plasmid, respectively, S0, N0, and L0 correspond to the respective initial concentrations, and knick and klin correspond to the observed first order rate constants of DNA nicking and linearization, respectively. Rate constants were expressed as min-1. DMSO inhibition of DNA cleavage by each M-chelate was performed as described, with the exception that only 30 min time points were used for each M-chelate with variable concentrations of DMSO (0, 10, 100, 1000 mM), and IC50 values were determined.
| (1) |
| (2) |
| (3) |
Freifelder-Trumbo Analysis of DNA Linearization
Freifelder-Trumbo analysis was performed as described previously.18,38 Briefly, the fractions of full-length linear (flin) and supercoiled (fsuper) DNA remaining at the individual time points in the electrophoresis gels used to monitor M-chelate/DNA reactions were determined. The fractions at that time are statistically related to the probable number of double strand breaks (n2) and single strand breaks (n1) by the simplified Poisson distribution defined by equation 4, and the Freifelder-Trumbo relation defined by equation 5. These fractions (flin and fsuper), and equations 4 and 5, were used simultaneously to solve for n2/n1 ratios for each M-chelate. The n2/n1 ratios were then used to assess whether DNA linearization by each M-chelate occurred by a concerted mechanism or random nicking, as described.
| (4) |
| (5) |
Determination of Overall Diffusible Radical Generation by TEMPO-9-AC Fluorescence
Reactions were run aerobically with 10 μM TEMPO-9-AC and 10 μM M-chelate, with and without 1 mM H2O2, in 20 mM HEPES, 100 mM NaCl, pH 7.4 in a black-wall 96-well plate. The increase of TEMPO-9-AC fluorescence upon reaction was monitored in real-time by excitation at 358 nm and emission at 435 nm every 4 min on a Varian Cary Eclipse fluorimeter with plate reader attachment. The overall change in fluorescence, which corresponded to the complete reaction of 10 μM TEMPO-9-AC, was used to convert units of fluorescence intensity into units of μM TEMPO-9-AC, and the rates of reaction (μM TEMPO-9-AC / min) were determined from the slopes of the kinetic plots of fluorescence intensity versus time. The Fe-TACN/H2O2/TEMPO reaction was monitored by stopped-flow techniques due to the extremely high rate. Stopped-flow measurements were made using an Applied Photophysics SpectraKinetic Monochromator, with excitation at 358 nm and an emission filer with band pass > 395 nm. Two syringes were used, one containing 20 μM TEMPO-9-AC and 2 mM H2O2, and the other containing 20 μM Fe-TACN. Data were collected only after two purge injections, and the fitted kinetic trace for the Fe-TACN/H2O2/TEMPO reaction was the average of 5 trials.
Determination of Hydroxyl Radical Generation by Rhodamine B
Reactions were run aerobically with 10 μM rhodamine B, 1 mM ascorbate and 1 μM M-chelate, with and without 1 mM H2O2 in 20 mM Na2HPO4, pH 7.4 in a clear 96-well plate. The disappearance of rhodamine B was monitored via UV/Vis at 555 nm on a Molecular Devices SPECTRA MAX M2 plate reader spectrophotometer. The overall change in absorbance, which corresponded to complete reaction of 10 μM RhB, was used to convert units of absorbance into units of μM RhB, and the initial rates (μM RhB / min) were determined from initial slopes of the kinetic plots of absorbance over time.
Ascorbic Acid Consumption
Reactions were run aerobically with 10 μM M-chelate and 1 mM ascorbic acid, with and without 1 mM H2O2, in 20 mM HEPES, 100 mM NaCl, pH 7.4 on a clear 96-well plate. The disappearance of reduced ascorbic acid was monitored via UV/Vis at 300 nm on a Molecular Devices SPECTRA MAX M2 plate reader spectrophotometer. The overall change in absorbance, which corresponded to the complete reaction of 1 mM ascorbate, was used to convert units of absorbance into units of mM ascorbate, and the initial rates (mM ascorbate / min) were determined from initial slopes of the kinetic plots of absorbance over time.
Square Wave Voltammetry
Solutions of 1 mM M-chelates were formed in 20 mM HEPES, 100 mM NaCl, pH 7.4 under anaerobic conditions at ambient temperature. M-chelate ratios used ranged from 1:1.5 to 1:2, but were adjusted to 1:1.2 for TACN to minimize the possibility of M-(TACN)2 complex formation. For each experiment, buffers, reagents, and the electrochemical cell were vacuum- and argon-purged before complex formation. Reduction potentials for each metal complex were determined via square wave voltammetry (SWV) using an EG&G Princeton Applied Research Potentiostat/Galvanostat with glassy carbon working electrode, Ag/AgCl reference electrode, and platinum auxiliary electrode, and the potential was determined by fitting to the Gaussian equation (6). Y and V are the measured variable current and variable applied potential, respectively; Y0 and VC are the fitted baseline current and fitted reduction potential, respectively; A, w, and m are the fitted peak area, peak width, and baseline current slope, respectively. Instrument parameters were established with Int. = 10 mV, Freq. = 100 Hz, I = 10-4 A. Potentials vs. Ag/AgCl were converted to potentials vs. NHE.
| (6) |
RESULTS
DNA Cleavage
Rate constants for nicking and linearization of plasmid DNA by each metal-chelate in the presence of 1 mM H2O2 and 1 mM ascorbate are summarized in Figure 1. These were obtained by fitting gel electrophoresis product intensities to a first order consecutive reaction model (defined by equations 1 to 3), as shown in Figure 2. Plasmid nicking rates above the limit of detection (Table 1) were observed for the following complexes in order of decreasing rate: Cu2+(aq) > Cu-NTA > Co-NTA > Co-EDTA > Cu-GGH > Cu-KGHK > Cu-cyclam > Fe2+(aq) > Fe-NTA > Fe-EDTA > Ni-EDTA > Cu-EDTA > Ni-DOTA > Co-cyclen > Co-KGHK > Cu-cyclen > Fe-TACN. Among metals, copper complexes generally demonstrated the highest reactivity, while NTA complexes showed the highest reactivity among chelators.
Figure 1.
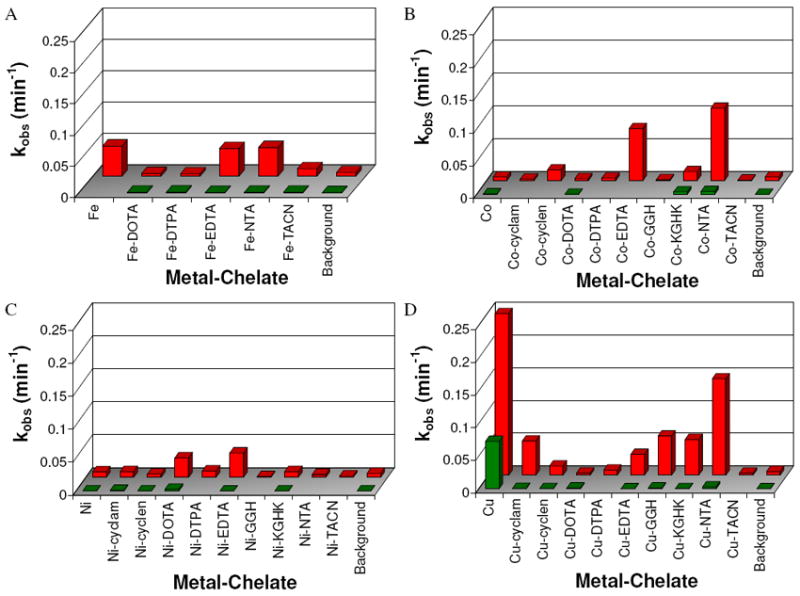
Summary of observed first-order rate constants (kobs) for consecutive DNA nicking (knick, rear) and subsequent linearization (klin, front) by each M-chelate. (A) Fe complexes, (B) Co complexes, (C) Ni complexes, and (D) Cu complexes.
Figure 2.
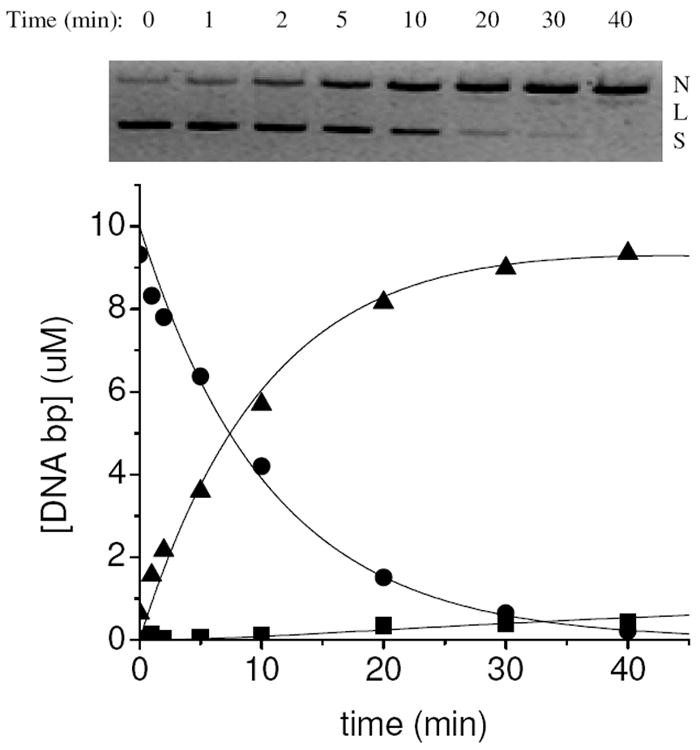
Time-dependent cleavage of plasmid DNA by Cu-NTA. (top) Conversion of plasmid DNA from supercoiled (S) to nicked (N) to linear (L) form, monitored by agarose gel-electrophoresis. (bottom) Variation of the relative concentrations of supercoiled, nicked, and linear form DNA and fitting to a first order consecutive reaction model (S = ●; N = ▲; L = ■).
Table 1.
Summary of observed rate constants for DNA nicking reactions (knick) promoted by M-chelates/H2O2/ascorbate. Initial rates for ascorbate consumption (with and without H2O2 as an added coreactant) and radical generation (again, with and without added H2O2), as well as the reduction potential for each M-chelate, are listed for comparison. Redox couples are 3+/2+ for Fe, Co, Ni-ATCUN, and Cu-ATCUN complexes and 2+/1+ for all other Ni and Cu complexes.
| complex | knick for DNA nicking (min-1) | ascorbate consumption rate for H2O2 and {O2} (μM/min) | TEMPO reaction rate for H2O2 and {O2} (μM/min) | reduction potential vs. NHE (mV) |
|---|---|---|---|---|
| Fe | 0.05 ± 0.01 | 6.4 ± 0.1 {––a} | ––a {––a} | --b |
| Fe-DOTA | ––a | ––a {––a} | 0.0601 ± 0.0005 {––a} | 39639 |
| Fe-DTPA | ––a | ––a {––a} | 0.0185 ± 0.0005 {––a} | 28039 |
| Fe-EDTA | 0.044 ± 0.009 | 80 ± 10 {6.9 ± 0.2} | 0.078 ± 0.001 {0.0222 ± 0.0006} | 39139 |
| Fe-NTA | 0.05 ± 0.01 | 130 ± 20 {4.15 ± 0.04} | 0.145 ± 0.002 {0.0449 ± 0.0005} | 46439 |
| Fe-TACN | 0.012 ± 0.006 | 17 ± 1 {5.7 ± 0.1} | 25.94 ± 0.02 {0.0099 ± 0.0001} | 175c |
| Co | ––a | ––a {––a} | 0.0319 ± 0.0009 {––a} | --b |
| Co-Cyclam | ––a | ––a {––a} | 0.0175 ± 0.0002 {––a} | 61c |
| Co-Cyclen | 0.017 ± 0.003 | ––a {––a} | ––a {0.0124 ± 0.0002} | -228c |
| Co-DOTA | ––a | ––a {––a} | ––a {––a} | 14239 |
| Co-DTPA | ––a | ––a {––a} | ––a {––a} | 111139 |
| Co-EDTA | 0.08 ± 0.02 | ––a {––a} | 0.0185 ± 0.0003 {––a} | 14639 |
| Co-GGH | ––a | 0.5 ± 0.2 {4.9 ± 0.1} | ––a {––a} | -11939 |
| Co-KGHK | 0.014 ± 0.002 | 0.5 ± 0.2 {7.1 ± 0.2} | ––a {––a} | -22839 |
| Co-NTA | 0.1 ± 0.1 | ––a {––a} | 0.075 ± 0.002 {––a} | 27439 |
| Co-TACN | ––a | ––a {––a} | ––a {––a} | -362c |
| Ni | ––a | ––a {––a} | 0.0156 ± 0.0003 {––a} | --b |
| Ni-Cyclam | ––a | ––a {––a} | ––a {––a} | -275c |
| Ni-Cyclen | ––a | ––a {––a} | ––a {––a} | -211c |
| Ni-DOTA | 0.030 ± 0.007 | ––a {––a} | ––a {––a} | -3539 |
| Ni-DTPA | ––a | ––a {––a} | ––a {––a} | --b |
| Ni-EDTA | 0.037 ± 0.002 | ––a {––a} | ––a {––a} | 17239 |
| Ni-GGH | ––a | ––a {––a} | ––a {––a} | 100039 |
| Ni-KGHK | ––a | ––a {––a} | ––a {––a} | 105539 |
| Ni-NTA | ––a | ––a {––a} | ––a {––a} | 17639 |
| Ni-TACN | ––a | ––a {––a} | ––a {––a} | 991c |
| Cu | 0.25 ± 0.05 | 80 ± 10 {12.9 ± 0.2} | 0.16 ± 0.01 {––a} | 136 |
| Cu-Cyclam | 0.053 ± 0.004 | ––a {––a} | ––a {––a} | 163c |
| Cu-Cyclen | 0.014 ± 0.004 | ––a {––a} | ––a {––a} | 280c |
| Cu-DOTA | ––a | ––a {––a} | ––a {––a} | 18039 |
| Cu-DTPA | ––a | ––a {––a} | ––a {––a} | 14839 |
| Cu-EDTA | 0.032 ± 0.003 | ––a {––a} | ––a {––a} | 4739 |
| Cu-GGH | 0.06 ± 0.01 | 2.72 ± 0.08 {––a} | ––a {––a} | 103839 |
| Cu-KGHK | 0.055 ± 0.008 | ––a {––a} | ––a {––a} | 105839 |
| Cu-NTA | 0.148 ± 0.007 | 0.72 ± 0.05 {3.46 ± 0.04} | ––a {––a} | 21539 |
| Cu-TACN | ––a | ––a {––a} | ––a {––a} | 110c |
| Background | 0.006 ± 0.001 | 0.6 ± 0.6 {0.3 ± 0.1} | 0.007 ± 0.003 {0.0039 ± 0.0007} |
Below detection limit.
Not determined.
This work.
Ascorbate Consumption
Ascorbate consumption experiments were used to measure the relative rate with which each oxidized M-chelate could be re-reduced in a multiple turnover redox cycle. The 300 nm absorbance of 1 mM ascorbate was monitored over time in reactions with each M-chelate under aerobic conditions, both with and without 1 mM H2O2. The concentration of ascorbate in each assay was initially 100-fold higher than the M-chelate concentration (10 μM), and ascorbate consumption was therefore used as a measure of the number of turnovers promoted by M-chelate reactivity. Initial rates of ascorbate consumption resulting from H2O2/M-chelate or O2/M-chelate were distinguished by subtraction (data obtained with H2O2 as a coreactant, minus data obtained in the absence of H2O2), and the resulting data points are summarized in Figure 3 and Table 1. Initial rates of ascorbate consumption resulting from H2O2/M-chelate reactivity that were greater than the limit of detection were observed for the following complexes in order of decreasing rate: Fe-NTA > Fe-EDTA > Cu2+(aq) > Fe-TACN > Fe2+(aq) > Cu-GGH. Similarly, initial rates resulting from O2/M-chelate reactivity above the limit of detection were observed for the following complexes in order of decreasing rate: Cu2+(aq) > Co-KGHK > Fe-EDTA > Fe-TACN > Co-GGH > Fe-NTA > Cu-NTA. Only Cu2+(aq), Fe-EDTA, Fe-NTA, and Fe-TACN were observed to consume ascorbate with either O2 or H2O2 as coreactant. Other complexes, Co-GGH, Co-KGHK, and Cu-NTA, were observed to consume ascorbate only with O2 as coreactant, whereas Fe2+(aq) and Cu-GGH were observed to consume ascorbate only with H2O2 as coreactant. This analysis provided a practical measure of the relative selectivity of M-chelate complexes for H2O2 and O2 coreactants under experimental conditions that were similar to those used for DNA cleavage experiments. For those complexes that were observed to consume ascorbate in the absence of added H2O2, additional tests conducted under varying levels of anaerobicity confirmed O2 as a necessary coreactant (Figure S1). The number of ascorbate turnovers promoted by each M-chelate after 6 h of incubation are listed in Table 2 (with and without added H2O2). The number of turnovers was generally higher in the presence of H2O2, although a few complexes, including Fe-EDTA, Fe-NTA, Fe-TACN, Co-GGH, Co-KGHK, Cu2+(aq), and Cu-NTA, demonstrated similar turnover activity even in the absence of H2O2, consistent with the observed initial rates of ascorbate consumption reflecting reaction with O2. Ascorbate turnover above background reactivity were observed for 21 of the 36 complexes tested in the presence of H2O2.
Figure 3.
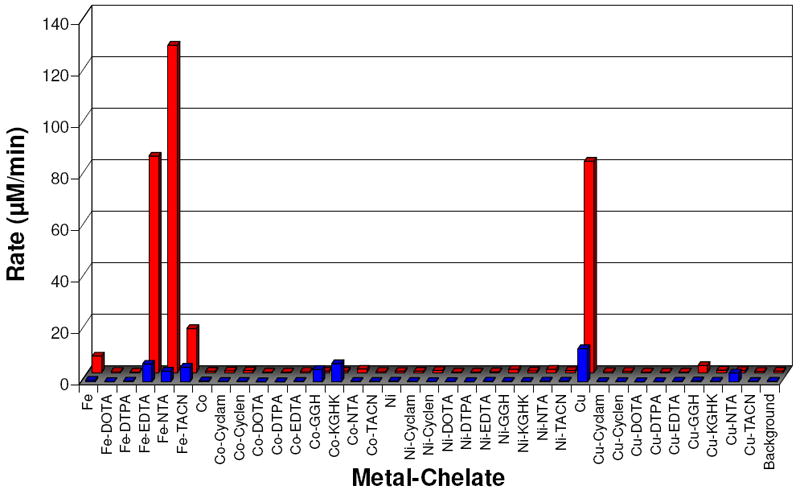
Summary of initial rates for multiple turnover consumption of ascorbate by each M-chelate/O2 (front) and M-chelate/H2O2 (rear) combination.
Table 2.
Number of turnovers observed for ascorbate (μM ascorbate consumed / 10 μM M-chelate), promoted by each M-chelate in the presence (H2O2 + O2) and absence (O2 only) of H2O2
| complex | observed number of ascorbate turnovers a | |
|---|---|---|
| with H2O2 | without H2O2 | |
| Fe | ≥ 100 | 20 ± 10 |
| Fe-DOTA | 18 ± 2 | 5 ± 1 |
| Fe-DTPA | ≥ 100 | 13 ± 2 |
| Fe-EDTA | ≥ 100 | ≥ 100 |
| Fe-NTA | ≥ 100 | ≥ 100 |
| Fe-TACN | ≥ 100 | ≥ 100 |
| Co | 31 ± 3 | 20 ± 10 |
| Co-Cyclam | 30 ± 3 | 8 ± 4 |
| Co-Cyclen | 32 ± 4 | 8 ± 3 |
| Co-DOTA | 9 ± 2 | 4 ± 1 |
| Co-DTPA | 8 ± 2 | 4 ± 1 |
| Co-EDTA | 21 ± 4 | 8 ± 2 |
| Co-GGH | ≥ 100 | ≥ 100 |
| Co-KGHK | ≥ 100 | ≥ 100 |
| Co-NTA | 54 ± 5 | 9 ± 2 |
| Co-TACN | 21 ± 4 | 8 ± 2 |
| Ni | 29 ± 3 | 10 ± 10 |
| Ni-Cyclam | 27 ± 3 | 10 ± 10 |
| Ni-Cyclen | 33 ± 4 | 6 ± 4 |
| Ni-DOTA | 9 ± 2 | 6 ± 1 |
| Ni-DTPA | 8 ± 2 | 5 ± 1 |
| Ni-EDTA | 20 ± 4 | 11 ± 2 |
| Ni-GGH | 40 ± 10 | 9 ± 2 |
| Ni-KGHK | 30 ± 8 | 9 ± 2 |
| Ni-NTA | 57 ± 5 | 12 ± 2 |
| Ni-TACN | 40 ± 10 | 12 ± 2 |
| Cu | ≥ 100 | ≥ 100 |
| Cu-Cyclam | 18 ± 3 | 6 ± 4 |
| Cu-Cyclen | 21 ± 3 | 6 ± 4 |
| Cu-DOTA | 8 ± 2 | 4 ± 1 |
| Cu-DTPA | 8 ± 2 | 5 ± 1 |
| Cu-EDTA | 15 ± 4 | 11 ± 2 |
| Cu-GGH | ≥ 100 | 17 ± 2 |
| Cu-KGHK | 44 ± 7 | 11 ± 2 |
| Cu-NTA | ≥ 100 | ≥ 100 |
| Cu-TACN | 22 ± 4 | 8 ± 2 |
| Noneb | 20 ± 3 | 20 ± 10 |
The maximum possible number of turnovers that could be observed was 100, since the concentrations of ascorbate and metal complex used were 1000 μM and 10 μM, respectively, and numbers for complexes that completely consumed 1000 μM ascorbate are listed as ≥ 100. Above-background numbers are shown in bold.
Background values were calculated the same way as for M-chelates, although no M-chelate was present.
Radical Generation
The fluorescent molecule TEMPO-9-AC contains a nitroxide radical that results in internal quenching of fluorescence.40,41 Following reaction with either hydroxyl or superoxide radicals, the nitroxide radical is converted to a non-radical species (supporting information, Figure SM7), and the fluorescence is restored.42-44 To investigate the relative rate of diffusible radical production by each M-chelate with each of the coreactants H2O2 or O2, each M-chelate was reacted with the TEMPO-9-AC radical (1:1 stoichiometry) under aerobic conditions with and without added 1 mM H2O2 and monitored by real-time fluorimetry. The steady-state rates of TEMPO conversion were thereby determined and provided a quantitative measure of diffusible radical production. As with the ascorbate consumption results, steady-state rates of TEMPO conversion resulting from H2O2/M-chelate or O2/M-chelate reactions were distinguished by subtraction (data obtained with H2O2 as a coreactant, minus data obtained in the absence of H2O2), and the resulting data points are summarized in Figure 4 and Table 1. Rates of TEMPO conversion resulting from H2O2/M-chelate activity that were greater than the limit of detection were observed for the following complexes in order of decreasing rate: Fe-TACN ≫ Cu2+(aq) > Fe-NTA > Fe-EDTA > Co-NTA > Fe-DOTA > Co2+(aq) > Fe-DTPA ~ Co-EDTA > Co-cyclam > Ni2+(aq). Similarly, initial rates resulting from O2/M-chelate reactivity above the limit of detection were observed for the following complexes in order of decreasing rate: Fe-NTA > Fe-EDTA > Co-cyclen > Fe-TACN. The complexes Fe-EDTA, Fe-NTA, and Fe-TACN were observed to produce radicals with either O2 or H2O2 as coreactants.
Figure 4.
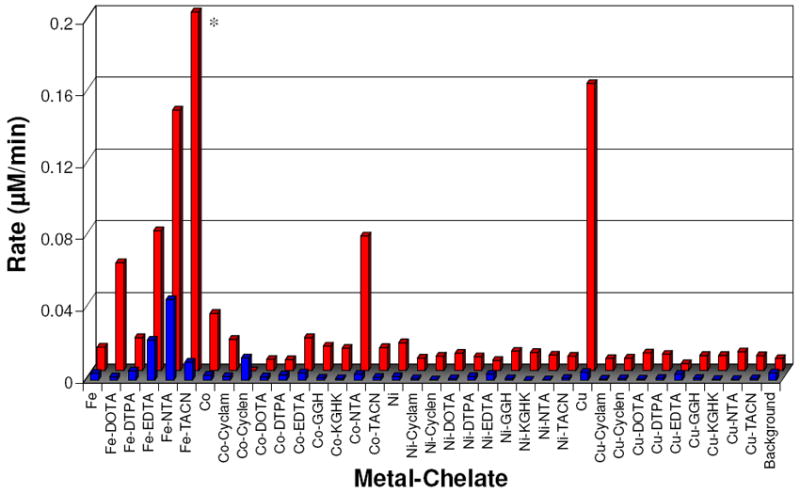
Summary of steady-state rates of TEMPO-9-AC monitored radical generation for each M-chelate/O2 (front) and M-chelate/H2O2 (rear) combination. * Fe-TACN/H2O2 was observed to promote very rapid reaction relative to other catalysts, requiring stopped-flow measurements to determine an observed rate of 25.94 ± 0.02 μM/min with the conditions used.
To differentiate between hydroxyl and superoxide radicals as reactive species, the reaction with rhodamine B (RhB), which reacts specifically with hydroxyl radicals (supporting information, Figure SM7),18,45,46 was also studied. The combination of RhB and TEMPO-9-AC reaction data allowed discrimination between hydroxyl and superoxide radicals produced by each M-chelate following reaction with 10 μM RhB and 1 mM ascorbate, with and without 1 mM H2O2. The decrease in RhB absorbance was attributed to reaction with hydroxyl radical. Rates of RhB reaction resulting from H2O2/M-chelate or O2/M-chelate reactivity were determined by subtraction (Figure S5). Reactions resulting from H2O2/M-chelate in the presence of ascorbate and determined to be above the limit of detection were observed for the following complexes in order of decreasing initial rate: Fe-NTA > Fe- EDTA > Cu2+(aq) ~ Cu-GGH. The rate observed for Cu-NTA was similar to the limit of detection. Rates resulting from reaction of O2/M-chelate in the presence of ascorbate were found to be up to 100-fold slower, with the exception of Cu2+(aq), but determined to be above the limit of detection in the order: Cu2+(aq) > Fe-EDTA > Fe-NTA.
Reduction Potentials
M-chelate reduction potentials were determined by square-wave voltammetry using similar solution conditions (pH 7.4) to those used in the DNA cleavage, ascorbate consumption, and TEMPO experiments, with the exception that these electrochemical experiments were conducted under anaerobic conditions. A summary of observed reduction potentials is listed in Table 1, and as expected, these were significantly modulated by the chelator. In general, M-chelates that were observed to cleave DNA under the reaction conditions employed demonstrated reduction potentials in the range -500 to 500 mV, with the exception of the Cu-ATCUN complexes (Cu-GGH and Cu-KGHK) with reduction potentials of 1038 mV and 1058 mV, respectively, reflecting the distinct Cu3+/Cu2+ redox couple exhibited by these two species.
DMSO Inhibition of DNA Cleavage
Both TEMPO and rhodamine experiments supported reactivity through the action of either a free diffusible or metal-associated hydroxyl radical species. M-chelate complexes observed to cleave DNA were retested for DNA cleavage in the presence of variable concentrations of DMSO, a hydroxyl radical scavenger.18,22 These DMSO inhibition experiments were performed in order to distinguish reactivity from free diffusible radicals versus a metal-associated radical that was held in close proximity to the DNA by virtue of the metal complex. That is, to distinguish between two possible pathways: namely, (1) the extent to which diffusible radicals were the active intermediates in DNA cleavage, or (2) the extent to which radical generating M-chelates were tightly associated to the DNA, since a DNA-bound M-chelate would require a shorter distance for radical migration before colliding with DNA.22 The IC50 values were measured (Figure 5) and the following active complexes were strongly inhibited by DMSO: Cu-cyclam, Co-cyclen, Co-NTA, Fe, Fe-NTA, Ni-EDTA, Fe-EDTA, Co-EDTA, Cu-cyclen, Ni-DOTA. Complexes with little to no observable DMSO inhibition included: Cu2+(aq), Cu-EDTA, Cu-GGH, Cu-KGHK, Co-KGHK, Cu-NTA, Fe-TACN. The low level of background reactivity (no complex), which was likely the result of a very low concentration of solution radicals generated by the uncatalyzed reaction of H2O2 and ascorbate, was also strongly inhibited by DMSO.
Figure 5.
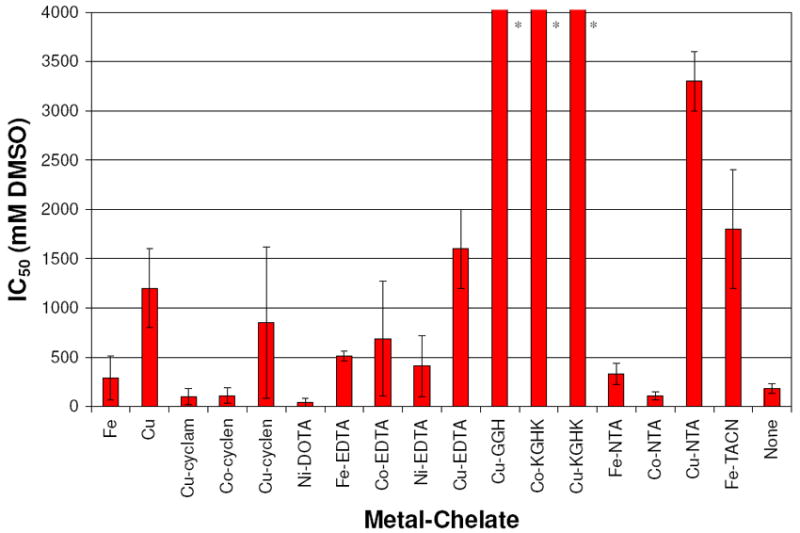
Summary of DMSO IC50 inhibition values for DNA cleavage by M-chelates/H2O2/ascorbate. * IC50 values ≫ 1000 mM.
DNA Residency Time
Relative rates of DNA linearization vs. rates of DNA nicking were assessed, and a statistical ratio of the probable number of double strand DNA cuts (n2) versus the probable number of single strand DNA nicks (n1) was established for each M-chelate following reaction with DNA. Complexes with n2/n1 < 0.01 promote DNA linearization by a random nicking pathway that results in linearization only when two random nicks on opposite strands of dsDNA occur sufficiently close together to result in linearization.18,38 Complexes with an n2/n1 ratio significantly greater than 0.01 promote linearization of DNA by a concerted mechanism, where a bound complex mediates nicking of both strands during its residency time and within the narrow locus of the binding site. This concerted pathway and subsequent fast linearization are more likely to occur for complexes that have a higher affinity for DNA and thus have a longer residency time when bound to the DNA. Among the complexes found to cleave DNA, the n2/n1 ratios were determined and are summarized in Figure 6. The degree to which nicking and linearization was concerted was found to decrease in the following order: Cu2+(aq) > Co-KGHK > Ni-DOTA > Fe-NTA ~ Fe-TACN ~ Fe-EDTA > Cu-KGHK > Cu-NTA > Cu-GGH ~ Ni-EDTA ~ Cu-EDTA > Fe2+(aq) ~ Cu-cyclam ~ Cu-cyclen ~ Co-cyclen ~ Co-EDTA ~ Co-NTA. Since a highly concerted DNA linearization reaction requires a fast linearization step following the initial nicking reaction, a correlation between the n2/n1 ratio and the ratio of the rate constants of consecutive nicking and linearization (klin/knick) was expected. Indeed, such a correlation was observed (Figure 7).
Figure 6.
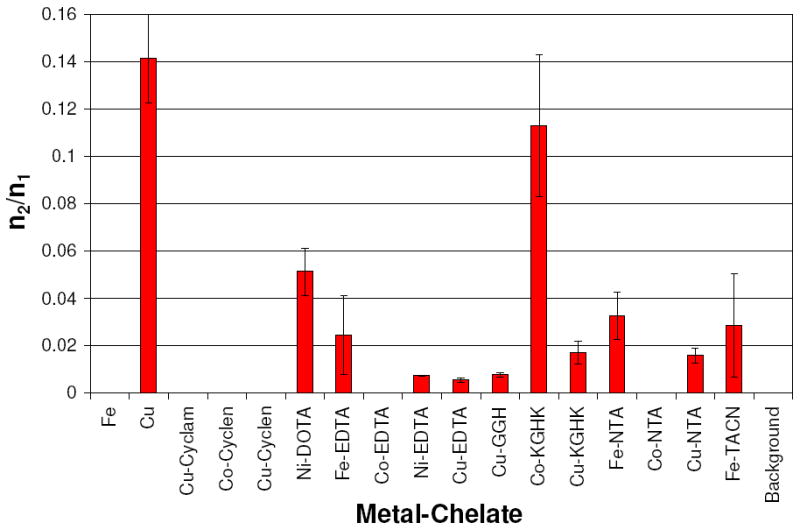
Summary of n2/n1 ratios resulting from a Freifelder-Trumbo analysis of DNA cleavage reactivity by M-chelates. M-chelates with n2/n1 = 0 had no quantifiable linearized DNA.
Figure 7.
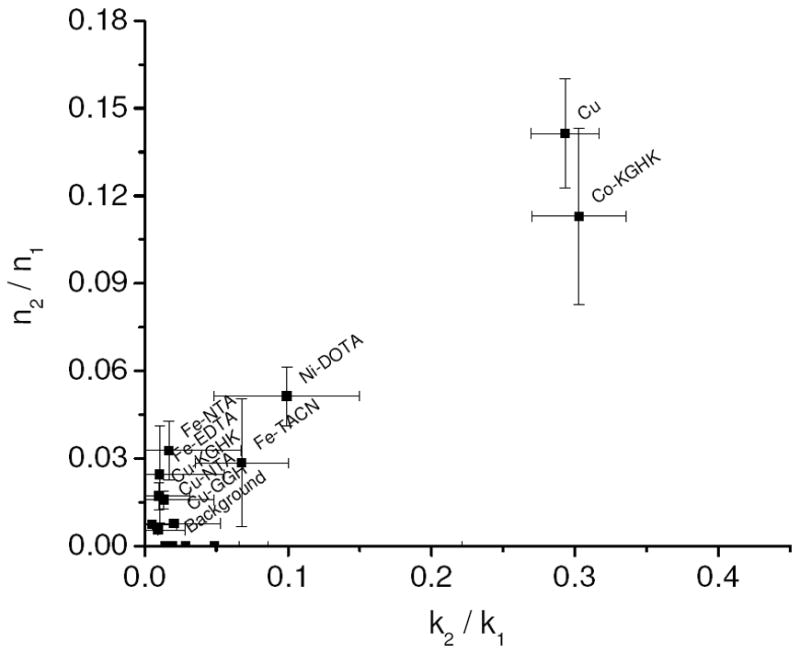
Correlation between the ratio of double and single strand breaks (n2/n1) and the ratio of rate constants for linearization and nicking (klin/knick).
DISCUSSION
DNA Cleavage and Reduction Potential
DNA cleavage by the M-chelates described herein was mediated by the generation of reactive oxygen species (ROS). The ability of M-chelates to generate reactive intermediates is directly related to their ability to switch between oxidation states and promote formation of ROS through redox chemistry with biological coreactants such as H2O2, O2, and ascorbate. Given that reported reduction potentials at neutral pH for O2/superoxide, superoxide/H2O2, H2O2/HO•, and ascorbyl radical/ascorbate are -330 mV, 890 mV, 380 mV, and -66 mV, respectively,47,48 single electron oxidation/reduction by H2O2/ascorbate is thermodynamically favored for M-chelates with reduction potentials in the range of 380 mV and -66 mV. Indeed, the most reactive M-chelates displayed reduction potentials within this range (Table 1, Figure 8), further suggesting that the most rapid rates of DNA cleavage reflected multiple turnover generation of hydroxyl radicals. We have recently observed a similar trend for targeted cleavage of HIV-1 Rev Response Element RNA by Rev-coupled M-chelates.39 M-chelate mediated reduction of dioxygen is also possible, although direct superoxide generation is less thermodynamically favored (E° = -330 mV). A few M-chelates with reduction potentials exceeding 1000 mV also demonstrated modest levels of DNA cleavage activity (Cu-GGH and Cu-KGHK), though the more moderate activity levels most likely reflect the extent to which these complexes lie outside of the optimum potential range. The reduction potentials for these ATCUN complexes are such that the oxidation of substrate is strongly thermodynamically favored but the prerequisite generation of the oxidized metal is not favored, and so these two complexes show relatively low reactivity. The relative sluggishness of the metal-ATCUN promoted redox chemistry is most likely important for the natural physiological roles of these motifs, where the slower cellular release of ROS would be advantageous.19,20,31,49-53
Figure 8.
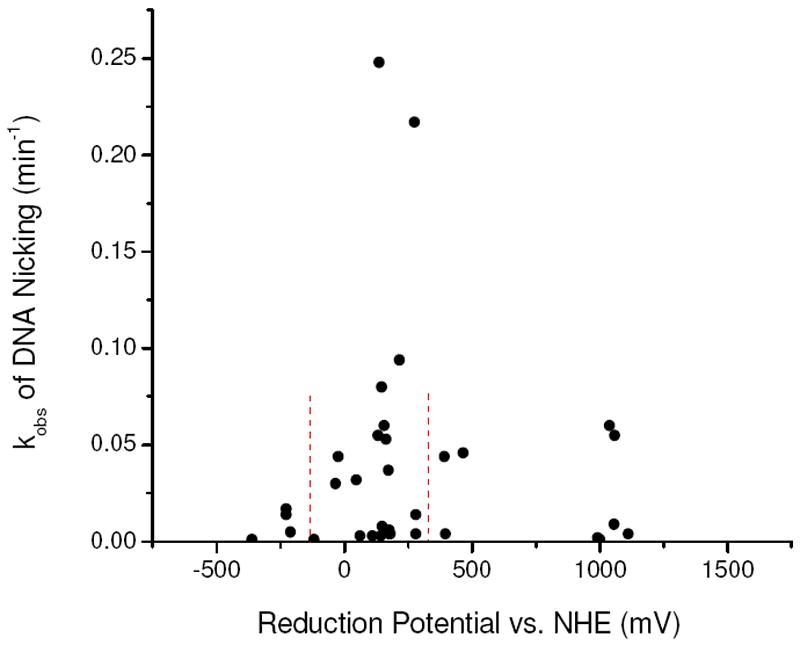
Rate constants for DNA nicking by M-chelates were observed to be highest for M-chelate complexes with reduction potentials between those of coreactants ascorbate (-66 mV) and H2O2 (380 mV), indicated by the dashed lines.
DNA Cleavage and Ascorbate Consumption
The driving force behind the observed DNA cleavage chemistry ultimately stems from the oxidants (H2O2 or O2) and the ascorbic acid reductant, while the critical catalytic role is performed by those M-chelates that catalyze the reaction between these coreactants to form metal-associated ROS, which then react with and cleave the DNA.30 In every case it was expected that the oxidant would first oxidize the M-chelate, forming an intermediate ROS with subsequent reduction of the M-chelate by ascorbate, allowing for multiple turnover cycles of oxidation and reduction.54. Indeed, with the exception of Co-GGH, every M-chelate species observed to consume ascorbate with multiple turnovers was also found to cleave DNA under the conditions used (Table 1), although there was no clear quantitative correlation between the rate of DNA cleavage and rate of ascorbate consumption. A few M-chelate complexes were observed to cleave DNA but did not undergo multiple turnover consumption of ascorbate. These complexes (Cu-cyclam, Co-cyclen, Cu-cyclen, Ni-DOTA, Co-EDTA, Ni-EDTA, Cu-EDTA, Cu-KGHK, Co-NTA) most likely operate either by a single turnover oxidative mechanism, by a multiple turnover mechanism in which DNA is directly oxidized by the M-chelate and reduction by ascorbate was unnecessary for regeneration of the reduced M-chelate, or by a hydrolytic mechanism. Consistent with these observations, Co(III)-cyclen complexes are known to hydrolyze DNA at a slow rate (kobs ~ 0.0003 min-1 with 1 mM complex),55,56 although an oxidative mechanism is also possible for [Co-cyclen]2+/3+ with coreactants. An alternative explanation for a reduction in, or the absence of, multiple turnovers in ascorbate consumption is that the M-chelate catalysts are inactivated as a result of oxidation by the ROS formed; with M-chelates that show a higher number of turnovers being less susceptible to inactivation under the conditions used.
DNA Cleavage and Radical Generation
M-chelates known to generate diffusible hydroxyl radicals in combination with H2O2 (Fe-EDTA, Fe-NTA, Cu-GGH, and Cu2+(aq)) provided significant DNA cleavage when combined with H2O2/ascorbate. They also prompt a strong response in both the TEMPO radical assay and rhodamine B consumption assays, consistent with hydroxyl radicals promoting the observed DNA cleavage by these M-chelates. However, aside from these complexes, there was very little correlation between diffusible radical generation and DNA cleavage rate, suggesting that diffusible radicals were not very efficient at mediating DNA cleavage. For example, Fe-DTPA, Fe-DOTA, Co-cyclam, Ni2+(aq), and Co2+(aq) each were observed to readily generate radicals as monitored by TEMPO-9-AC, but none of these compounds was observed to cleave DNA under similar reaction conditions. Conversely, Fe2+(aq), Cu-cyclam, Cu-cyclen, Ni-DOTA, Cu-EDTA, Ni-EDTA, Cu-KGHK, and Cu-NTA were observed to cleave DNA, but were not observed to generate radical species in standard detection assays. In order for diffusible hydroxyl radicals produced by M-chelates to effect DNA cleavage when the M-chelate is used at 100 nM concentration, it appears to be necessary for the M-chelate to possess at least minimal binding interaction with the DNA, such that the origin of each hydroxyl radical was closer to the DNA than would occur in the absence of this type of interaction. Indeed DNA cleavage by Cu-GGH, Cu2+(aq) and Fe-TACN was weakly inhibited by DMSO, relative to DNA cleavage by the low background concentration of solution radicals, in spite of the ability of DMSO to scavenge hydroxyl radicals. This is supported by a Freifelder-Trumbo analysis for DNA linearization by Fe-TACN, Fe-NTA, Fe-EDTA and Cu2+(aq) that is consistent with the existence of unique binding interactions between these complexes and DNA (Table 3). The complexes Co-cyclen, Co-EDTA, and Co-NTA promoted significant DNA cleavage and a moderate response to the TEMPO-9-AC radical assay, but did not provide a response in the rhodamine B assay, making the assignment of hydroxyl radicals as the active species more problematic. However, Co-cyclen, Co-NTA, and Co-EDTA were each strongly inhibited by DMSO, and a Freifelder-Trumbo analysis of DNA linearization/nicking by Co-cyclen, Co-NTA, and Co-EDTA indicated no association between these complexes and DNA (Table 3), suggesting that DNA cleavage by these complexes was indeed mediated by diffusible radicals. Together these data support the hypothesis that radical-mediated DNA cleavage is most efficient for M-chelates that possess binding interactions with the DNA. Among these complexes, the data also suggest that DNA cleavage by diffusible radicals is inherently less efficient than by metal-bound ROS, consistent with the expectation that metal-bound reactive oxygen intermediates would more efficiently cleave the DNA as a result of their prolonged proximity to the DNA.
Table 3.
DMSO inhibition and Freifelder-Trumbo analysis of DNA cleavage by M-chelates that exhibited cleavage rates above the limit of detection (see Table 1). A low IC50 is correlated to DNA nicking via diffusible radicals and/or weak binding of the M-chelate to DNA. Those M-chelates displaying a ratio of n2/n1 > 0.01 linearize the DNA via a concerted mechanism as a result of the higher residency time (tighter binding) for the DNA complex.
| complex | IC50 (mM DMSO) | n2/n1 |
|---|---|---|
| Cu | 1200 ± 400 | 0.14 ± 0.01 |
| Fe | 300 ± 200 | ~ 0 |
| Cu-Cyclam | 100 ± 80 | ~ 0 |
| Cu-Cyclen | 900 ± 800 | ~ 0 |
| Co-Cyclen | 110 ± 80 | ~ 0 |
| Ni-DOTA | 40 ± 40 | 0.05 ± 0.01 |
| Cu-EDTA | 1600 ± 400 | 0.01 ± 0.01 |
| Ni-EDTA | 400 ± 300 | 0.0073 ± 0.0001 |
| Co-EDTA | 700 ± 600 | ~ 0 |
| Fe-EDTA | 510 ± 50 | 0.03 ± 0.02 |
| Cu-GGH | ≫1000 | 0.01 ± 0.01 |
| Cu-KGHK | ≫1000 | 0.017 ± .005 |
| Co-KGHK | ≫1000 | 0.11 ± .03 |
| Cu-NTA | 3300 ± 300 | 0.016 ± .003 |
| Co-NTA | 110 ± 40 | ~ 0 |
| Fe-NTA | 300 ± 100 | 0.03 ± 0.01 |
| Fe-TACN | 1800 ± 600 | 0.03 ± 0.02 |
| Background | 180 ± 50 | ~ 0 |
Ascorbate Consumption, Radical Generation, and Reduction Potential
As expected, multiple turnover rates for ascorbate consumption and radical generation by M-chelates were found to be dependent on M-chelate reduction potentials (Figure 9). A similar trend was observed for DNA cleavage rates (Figure 8). Ascorbate consumption by M-chelates and the coreactant H2O2 was also observed to be fastest for those complexes with reduction potentials in the range of -66 to +380 mV (Figure 9), which correspond to the reduction potentials of ascorbyl radical/ascorbate and H2O2/HO•, respectively. As observed for native superoxide dismutase enzymes, which possess reduction potentials similarly poised between those of the relevant half-reaction redox pairs (O2/superoxide and superoxide/H2O2),57,58 reduction potentials for M-chelates between -66 and +380 mV would result in an overall favorable thermodynamic profile for oxidation by H2O2 and reduction by ascorbate, thereby promoting multiple oxidation/reduction cycles. When radical generation by M-chelate/H2O2 conditions was monitored by the TEMPO-9-AC reaction, turnover was observed to be fastest for complexes with reduction potentials less than +380 mV, such that conversion of H2O2 to hydroxyl radical was thermodynamically favored (Figure 9). Therefore, the most likely mechanism for reaction of M-chelate/H2O2/ascorbate involves Fenton-style single-electron reduction of H2O2 to form hydroxyl radicals, followed by reduction of the oxidized metal by ascorbate. The hydroxyl radicals produced by these reactions could be either diffusible or metal-bound. In that regard, the M-chelate/H2O2 reactions that generated radicals as monitored by TEMPO-9-AC most likely contain those M-chelates that produce diffusible hydroxyl radicals. Hydroxyl radical generation by Cu2+(aq)/O2 was ascorbate-dependent, as evidenced by comparison of TEMPO and RhB data.
Figure 9.
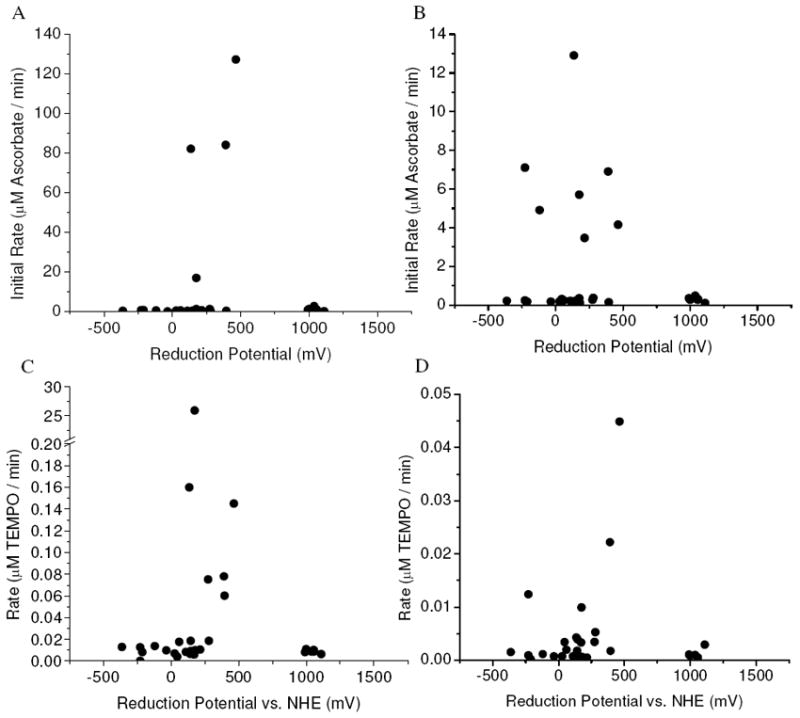
The highest rates of multiple turnover ascorbate consumption and radical generation were found for M-chelates with biologically relevant reduction potentials. (A) Ascorbate consumption by M-chelate/H2O2. (B) Ascorbate consumption by M-chelate/O2. (C) Radical generation by M-chelate/H2O2. (D) Radical generation by M-chelate/O2.
The rate of ascorbate consumption by M-chelates in the presence of the coreactant O2 provided a similar, although slightly broader dependence on reduction potentials, and the data supports reaction pathways involving either the thermodynamically unfavorable single-electron reduction of O2 (E°O2/superoxide = -330 mV) or the favorable two-electron reduction of O2 (E°O2/H2O2 = 560 mV). Of the seven M-chelates that demonstrated significant rates of ascorbate consumption driven by O2, only Fe- EDTA, Fe-NTA, and Fe-TACN were also observed to generate radicals under similar conditions in the absence of ascorbate (that is, TEMPO experiments conducted aerobically with no added ascorbate). Therefore, in the presence of both O2 and ascorbate, reactions of Fe-EDTA, Fe-NTA, and Fe-TACN most likely proceeded via single electron reduction of oxygen to form superoxide radical, with regeneration of reduced M-chelate by ascorbate. By contrast, other M-chelates (Co-GGH, Co-KGHK, Cu-NTA, Cu2+(aq)) that also show significant rates of O2 driven ascorbate consumption but do not generate radicals under similar reaction conditions in the absence of ascorbate (TEMPO experiments with O2 only), either proceed by the two-electron reduction of O2 to the peroxy state, or by single-electron reduction of O2 to form non-diffusible metal-bound superoxide. With the exception of Cu2+(aq), both pathways show consistency through the absence of detectable diffusible radicals with the O2/M-chelate/ascorbate combination. Interestingly, radical generation by the O2/Cu2+(aq) combination required the presence of ascorbate (radicals observed in the RhB/O2/ascorbate assay, but not in the TEMPO/O2 assay), and the observed radicals were most likely hydroxyl radicals (supported by the RhB assay), suggesting the stepwise 3-electron reduction of O2 to hydroxyl radical for Cu2+ (aq), in which release of the superoxide radical and peroxide intermediates did not occur to an appreciable extent.
Ascorbate consumption by M-chelates/O2 was generally found to be slower than observed for M-chelates in the presence of H2O2, with the exceptions of Co-GGH, Co-KGHK, and Cu-NTA, suggesting that under the conditions used the reaction with O2 was faster than those with H2O2 for only these three complexes. Cobalt-ATCUN complexes such as Co-GGH and Co-KGHK show relatively low reduction potentials, -119 and -228 mV, respectively, and the thermodynamically unfavorable reduction of these complexes would become favorable if coupled with two-electron reduction of O2 to H2O2 (E°O2/H2O2 = 560 mV). Alternatively, it is possible that the instability of the reduced Co-GGH and Co-KGHK species actually increased their reactivity with O2, facilitating the otherwise unfavorable formation of non-diffusible superoxide (E°O2/superoxide = -330 mV).
Effect of DNA-Binding Affinity on DNA Cleavage
It is expected that DNA cleavage rates are favorably influenced in the case of M-chelates that bind DNA with higher affinity, since ROS produced by M-chelates with higher DNA-binding affinity are on average closer to the DNA and thus are more likely to react with DNA within a given timeframe. DNA binding affinity is generally governed by one or more factors that include electrostatic attraction to the phosphate backbone, intercalation, and/or H-bonding. The variety of expected binding modes of the M-chelates, and the modest to weak levels of binding expected, makes the use of fluorescent intercalator displacement (FID) assays inappropriate for the comparison of DNA-binding affinities. Negatively-charged M-chelates, such as those formed by use of the ligands EDTA, DTPA, and DOTA, are typically thought to possess very low DNA-binding affinity due to repulsion of the negatively-charged phosphate backbone of DNA, and DNA cleavage reactions mediated by those M-chelates are most likely mediated by diffusible radicals. In fact, the choice of EDTA for typical iron mediated hydroxyl radical footprinting experiments reflects the poor binding of DNA by [Fe-EDTA]1-/2-.59 Positively-charged M-chelates, such as those formed with ligands cyclam, cyclen, and TACN (and free metal ions) have been shown to display higher affinity toward DNA as a result of attraction toward the negatively charged backbone of DNA.22 Free Cu2+(aq) has been shown to bind dsDNA nonspecifically with a dissociation constant of 42 μM, although in a few cases cooperative copper binding sites for dsDNA have been identified with significantly higher affinity.60 Trogler and coworkers have shown the complex [Fe-TACN]3+/2+ to exhibit higher DNA binding affinity than [Fe-EDTA]1-/2-,22 consistent with the results of this study. A consistent, though modest charge affect has also been documented by Huang and coworkers in the case of the tripeptide complexes [Ni-GGH]1- and [Ni-KGH]1+, with dissociation constants for DNA binding of 26 nM and 14 nM, respectively.61 Prior studies from our laboratory have demonstrated modestly faster DNA cleavage by micromolar levels of Cu-KGHK relative to Cu-GGH,18 however, at the lower concentration used in this work (100 nM rather than > 4 μM), the copper complexes of GGH and KGHK showed comparable low levels of activity. Positively-charged metal complexes of cyclam, cyclen, and TACN demonstrated moderate rates of DNA cleavage. However, several negatively-charged metal complexes with NTA and EDTA show markedly higher rates of DNA cleavage. In summary, the highly variable DNA cleavage rates and mechanisms observed for the M-chelates used in this study highlight the fact that while DNA binding affinity may somewhat skew the DNA cleavage rates, neither DNA-binding nor redox activity are solely responsible the observed nuclease activity. Rather, it is likely that specific combinations of DNA affinity, DNA alignment, and redox mechanisms are more effective than others.
Predicted Mechanisms of Observed DNA Cleavage
Mechanisms of observed DNA cleavage by M-chelates were predicted for each M-chelate based on the coreactant dependence of ascorbate consumption and radical generation, the extent of DMSO inhibition of DNA cleavage, and the extent to which DNA nicking and linearization were concerted (Freifelder-Trumbo analysis). DNA cleavage by Cu2+(aq) was most likely mediated by a combination of metal-bound hydroxyl radicals and diffusible hydroxyl radicals produced in close proximity to the DNA. Although Cu2+(aq) was observed to quickly generate diffusible hydroxyl radicals when reacted with H2O2, DNA cleavage by Cu2+(aq) was only weakly inhibited by DMSO, and linearization and nicking occurred by a notably concerted mechanism. DNA cleavage by Fe-EDTA, Fe-NTA, and Fe-TACN was most likely mediated by diffusible hydroxyl radicals produced in close proximity to the DNA, which is supported by the observation that hydroxyl radicals were produced when either Fe-NTA, Fe-EDTA, or Fe-TACN were reacted with H2O2. DMSO was found to more strongly inhibit DNA cleavage by Fe-NTA and Fe-EDTA than cleavage by Cu2+(aq) or Fe-TACN, while DNA linearization by Fe-NTA, Fe-EDTA, and Fe-TACN was less concerted than linearization by Cu2+(aq). Previous studies have indeed shown that Fe-EDTA cleaves DNA by generation of diffusible hydroxyl radicals, but also that Fe-EDTA demonstrates subtle sequence variability in its DNA reactivity, which could be a result of weak binding sites.12 Cu-GGH most likely functions by a similar mechanism, although the weaker DMSO inhibition of DNA cleavage by Cu-GGH suggests the primary involvement of a metal-bound hydroxyl radical in the observed DNA cleavage. Previous work has shown both that H2O2 is an obligatory intermediate in Cu-GGH mediated DNA cleavage by ascorbate/O2 and that DMSO inhibition studies yielded no evidence of significant involvement of diffusible hydroxyl radicals in DNA cleavage; all of which is consistent with the results presented herein.18 DNA cleavage by Cu-NTA and Co-KGHK appeared to be mediated by metal-bound superoxide radicals, which is supported by the observed consumption of ascorbate when reacted with O2 (but not H2O2), the apparent lack of diffusible radicals, the weak DMSO inhibition of DNA cleavage, and the concertedness of DNA linearization. DMSO inhibition profiles suggest that Ni-DOTA, Cu-EDTA and Cu-KGHK each cleaved DNA primarily by metal-bound ROS; although no other reactivity was observed by these complexes. Freifelder-Trumbo analysis suggests that the DNA cleavage observed for Fe2+(aq), Cu-cyclam, Cu-cyclen, Co-cyclen, Cu-EDTA, Ni-EDTA, Co-EDTA, Cu-GGH, and Co-NTA was not associated with any binding interaction between M-chelate and DNA (n2/n1 < 0.01), while other complexes that promoted DNA cleavage did appear to linearize DNA through a concerted mechanism (n2/n1 > 0.01). Indeed, previous studies of Cu-GGH and Cu-KGHK indicated that Cu-KGHK promoted linearization of DNA by a concerted mechanism, while Cu-GGH did not, and this most likely reflects the overall 1+ charge for Cu-KGHK and 1- charge of Cu-GGH.18 As expected, free Cu2+ (aq) and [Co-KGHK]+, two of the most positively-charged complexes, showed the strongest interaction with DNA as judged by n2/n1 values.
To compare the DNA nuclease activity of the catalysts studied herein, relative to that of other catalytic nucleases, second-order rate constants for DNA nicking reactions were established by use of equation 7, where kobs is the observed first order rate constant for DNA nicking (after subtraction of the observed background rate constant obtained in the absence of any M-chelate), and [M] is the concentration of M-chelate (10-7 M). Table 4 compares several second-order rate constants obtained from this study with those of several other catalysts known to cleave dsDNA. Significantly higher rates were observed for several of the M-chelates studied herein.
| (7) |
Table 4.
Comparison of second-order rate constants for DNA nicking promoted both by several M-chelate complexes, and DNA cleavage by other artificial nucleases.
| complex | substrate | k2,nick (M-1 min-1) | coreagents | conditions | ref/note |
|---|---|---|---|---|---|
| [Cu-NTA]1-/2- | dsDNA | 1420000 | ascorbate + H2O2 | pH 7.4, 37 °C | a |
| [Co-NTA]0/1- | dsDNA | 900000 | ascorbate + H2O2 | pH 7.4, 37 °C | a |
| [Cu-KGHK]2+/1+ | dsDNA | 491000 | ascorbate + H2O2 | pH 7.4, 37 °C | a |
| [Cu-KGHK]2+/1+ | dsDNA | 5580 | ascorbate | pH 7.4, 37 °C | 18 |
| [Cu-GGH]0/1- | dsDNA | 542000 | ascorbate + H2O2 | pH 7.4, 37 °C | a |
| [Cu-GGH]0/1- | dsDNA | 2340 | ascorbate | pH 7.4, 37 °C | 18 |
| [Cu-neamine]4+ | dsDNA | 7690b | none | pH 7.3, 37 °C | 25 |
| [Co-cyclen](4-5)+ | dsDNA | 13-90b | none | pH 7.0, 37 °C | 56 |
| Eu3+ | dsDNA | 108b | none | pH 7.0, 37 °C | 62 |
| [Rh-(phi)2bpy’-peptide(Zn)]0 | dsDNA | 300 | none | pH 6.0, 37 °C | 11 |
| [Co-tamen]3+ | dsDNA | 3 | none | pH 7.6, 37 °C | 63 |
| [Ce2-HXTA]3+ | ssDNA | 840 | none | pH 8.0, 37 °C | 64 |
| [Ce2-HXTA]3+ | dsDNA | 78 | none | pH 8.0, 37 °C | 64 |
This work.
Enzyme-like second order rate constants expressed as kcat/KM.
CONCLUSIONS
The goal of this study was to elucidate the reactivity patterns and mechanisms of several families of metal-chelate complexes commonly incorporated in artificial nucleases. The metal-chelate complexes studied in this work were found to provide a wide range of reactivity with both co-reactants and DNA, and this reactivity was highly dependent upon several factors including reduction potential, coordination unsaturation, charge, the ability of the metal complexes to facilitate generation of ROS, and the degree to which these ROS were either metal-coordinated or diffusible. Knowledge of these factors should allow more precise tuning of gene-targeting artificial nucleases in which metal-chelates are coupled with a gene-specific targeting sequence. The degree to which the metal-chelates from this study selectively utilize biologically available coreactants is of critical importance in drug development, since coreactants such as O2, H2O2, and ascorbate are present at widely variable concentrations in host cells, and therefore each metal-chelate might possess differing modes of reactivity depending on such factors as location within the cell (nucleus versus cytoplasm) or the state of the host cell (oxidative stress vs normal conditions). Knowledge of the fate of the ROS produced (diffusible or metal-bound) and mechanisms of DNA cleavage for each metal-chelate is also of critical importance in the context of gene targeting, because those metal-chelates which generate an excess of diffusible radicals will likely be less selective toward a targeted gene with the possibility of damage to non-targeted genes, while the metal-chelates that cleave DNA by more controlled means, such as through metal-bound ROS, will more likely be selective toward the targeted gene. In this study we found that several metal-chelates (Cu-cyclen, Ni-DOTA, Cu-EDTA, Cu-GGH, Co-KGHK, Cu-KGHK, and Cu-NTA) appear to cleave DNA primarily by metal-bound ROS, rather than diffusible ROS, and therefore, incorporation of these metal complexes in artificial nucleases should provide more controlled delivery of ROS to targeted genes. Finally, many of the metal-chelates from this study possess desirable therapeutic properties, such as the ability to perform with multiple turnovers or efficiently utilize biologically available coreactants. Compounds that alter biological targets irreversibly and with multiple turnover, such as these metal-chelates, have the potential to perform at far lower concentrations than their reversible counterparts, since catalytic drugs that promote irreversible inactivation of a target do not need to saturate the target in order to operate effectively.15,16,18,19,25 Lower dosage concentrations should result in fewer side-effects and higher efficacy.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health [HL093446 and AA016712]. Jeff Joyner was supported by an NIH Chemistry/Biology Interface training grant (T32 GM08512).
Footnotes
Supporting Information Available
The O2 dependence of ascorbate consumption; kinetic traces for each of the ascorbate consumption reaction, TEMPO-9-AC reaction, and rhodamine B reaction; a summary of rhodamine B initial reaction rates; square-wave voltammogram; structures and reaction mechanisms for TEMPO-9-AC and rhodamine B; and a summary of DNA nicking and linearization rate constants.
References
- 1.Cowan JA. Inorganic Biochemistry An Introduction. Wiley-VCH; New York: 1997. pp. 342–345. [Google Scholar]
- 2.Reedijk J. Med Inorg Chem. 2005:80–109. [Google Scholar]
- 3.Liu H-K, Sadler PJ. Acc Chem Res. 2011;44:349–359. doi: 10.1021/ar100140e. [DOI] [PubMed] [Google Scholar]
- 4.Wheate NJ, Brodie CR, Collins JG, Kemp S, Aldrich-Wright JR. Mini Rev Med Chem. 2007;7:627–48. doi: 10.2174/138955707780859413. [DOI] [PubMed] [Google Scholar]
- 5.Boger DL, Garbaccio RM. Acc Chem Res. 1999;32:1043–1052. [Google Scholar]
- 6.Minoshima M, Bando T, Shinohara K, Sugiyama H. Nucl Acids Symp Ser. 2009;53:69–70. doi: 10.1093/nass/nrp035. [DOI] [PubMed] [Google Scholar]
- 7.Baum C, von Kalle C, Staal F, Li X, Fehse B, Schmidt M, Weerkamp F, Karlsson S, Wagemaker G, Williams D. Molec Ther. 2004;9:5–13. doi: 10.1016/j.ymthe.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 8.Hacein-Bey-Abina S, et al. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 9.Magda D, Wright M, Crofts S, Lin A, Sessler J. J Am Chem Soc. 1997;119:6947–6948. [Google Scholar]
- 10.Dreyer G, Dervan P. Proc Natl Acad Sci U S A. 1985;82:968–972. doi: 10.1073/pnas.82.4.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fitzsimons MP, Barton JK. J Am Chem Soc. 1997;119:3379–3380. [Google Scholar]
- 12.Sigman D. Biochemistry. 1990;29:9097–9105. doi: 10.1021/bi00491a001. [DOI] [PubMed] [Google Scholar]
- 13.Sigman D, Bruice T, Mazumder A, Sutton C. Acc Chem Res. 1993;26:98–104. [Google Scholar]
- 14.Dervan P. Science. 1986;232:464–471. doi: 10.1126/science.2421408. [DOI] [PubMed] [Google Scholar]
- 15.Hocharoen L, Cowan JA. Chem Eur J. 2009;15:8670–8676. doi: 10.1002/chem.200900821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin Y, Cowan JA. J Am Chem Soc. 2006;128:410–411. doi: 10.1021/ja055272m. [DOI] [PubMed] [Google Scholar]
- 17.Jin Y, Cowan JA. J Biol Inorg Chem. 2007;12:637–644. doi: 10.1007/s00775-007-0221-2. [DOI] [PubMed] [Google Scholar]
- 18.Jin Y, Cowan JA. J Am Chem Soc. 2005;127:8408–15. doi: 10.1021/ja0503985. [DOI] [PubMed] [Google Scholar]
- 19.Cowan JA. Pure Appl Chem. 2008;80:1799–1810. [Google Scholar]
- 20.Jin Y, Lewis MA, Gokhale NH, Long EC, Cowan JA. J Am Chem Soc. 2007;129:8353–8361. doi: 10.1021/ja0705083. [DOI] [PubMed] [Google Scholar]
- 21.Bradford S, Kawarasaki Y, Cowan JA. J Inorg Biochem. 2009;103:871–875. doi: 10.1016/j.jinorgbio.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 22.Silver GC, Trogler WC. J Am Chem Soc. 1995;117:3983–3993. [Google Scholar]
- 23.Cooke M, Evans M, Dizdaroglu M, Lunec J. FASEB J. 2003:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 24.Inoue S, Kawanishi S. Canc Res. 1987;47:6522–6527. [PubMed] [Google Scholar]
- 25.Sreedhara A, Freed JD, Cowan JA. J Am Chem Soc. 2000;122:8814–8824. [Google Scholar]
- 26.Long EC. Acc Chem Res. 1999;32:827–836. [Google Scholar]
- 27.Ogino H, Ogino K. Inorg Chem. 1982;22:2208–2211. [Google Scholar]
- 28.Henle ES, Han Z, Tang N, Rai P, Luo Y, Linn S. J Biol Chem. 1999;274:962–971. doi: 10.1074/jbc.274.2.962. [DOI] [PubMed] [Google Scholar]
- 29.Pogozelski W, Tullius T. Chem Rev. 1998;98:1089–1107. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 30.Chiou S-H. J Biochem. 1984;96:1307–1310. doi: 10.1093/oxfordjournals.jbchem.a134951. [DOI] [PubMed] [Google Scholar]
- 31.Camerman N, Camerman A, Sarkar B. Can J Chem. 1976 [Google Scholar]
- 32.Finnen D, Pinkerton A, Dunham W, Sands R, Funk M. Inorg Chem. 1990;30:3960–3964. [Google Scholar]
- 33.Viola-Villegas N, Doyle R. Coord Chem Rev. 2009;253:1906–1925. [Google Scholar]
- 34.Mizuta T, Wang J, Miyoshi K. Inorg Chim Acta. 1995;230:119–125. [Google Scholar]
- 35.Walters MA, Vapnyar V, Bolour A, Incarvito C, Rheingold AL. Polyhedron. 2003;22:941–946. [Google Scholar]
- 36.Ren Y-W, Li J, Zhao S-M, Zhang F-X. Struct Chem. 2004;16:439–444. [Google Scholar]
- 37.Cernak J, Kuchar J, Stolarova M, Kajnakova M, Vavra M, Potocnak I, Falvello LR, Tomas M. 2010;35:737–744. [Google Scholar]
- 38.Freifelder D, Trumbo B. Biopolymers. 1969;7:681–693. [Google Scholar]
- 39.Joyner JC, Cowan JA. J Am Chem Soc. 2011;133:9912–9922. doi: 10.1021/ja203057z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pou S, Bhan A, Bhadti VS, Wu SY, Hosmane RS, Rosen GM. FASEB J. 1995;9:1085–90. doi: 10.1096/fasebj.9.11.7649408. [DOI] [PubMed] [Google Scholar]
- 41.Matko J, Ohki K, Edidin M. Biochemistry. 1992;31:703–711. doi: 10.1021/bi00118a010. [DOI] [PubMed] [Google Scholar]
- 42.Cohn CA, Simon SR, Schoonen MA. Part Fibre Toxicol. 2008;5 doi: 10.1186/1743-8977-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laight DW, Andrews TJ, Haj-Yehia AI, Carrier MJ, Änggård EE. Env Tox Pharm. 1997;3:65–68. doi: 10.1016/s1382-6689(96)00143-3. [DOI] [PubMed] [Google Scholar]
- 44.Samuni A, Goldstein S, Russo A, Mitchell JB, Krishna MC, Neta P. J Am Chem Soc. 2002;124:8719–8724. doi: 10.1021/ja017587h. [DOI] [PubMed] [Google Scholar]
- 45.Yu F, Xu D, Lei R, Li N, Li K. J Agric Food Chem. 2008;13:730–735. doi: 10.1021/jf072383r. [DOI] [PubMed] [Google Scholar]
- 46.Mishra KP, Gogate PR. Separ Purif Technol. 2010;75:385–391. [Google Scholar]
- 47.Wood P. Biochem J. 1988;253:287–289. doi: 10.1042/bj2530287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borsook H, Keighley G. Proc Natl Acad Sci U S A. 1933;19:875–878. doi: 10.1073/pnas.19.9.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lau S-J, Sarkar B. J Biol Chem. 1971;246:5938–5943. [PubMed] [Google Scholar]
- 50.Lau S-J, Kruck TPA, Sarkar B. J Biol Chem. 1974;249:5878–5884. [PubMed] [Google Scholar]
- 51.Grogan J, McKnight CJ, Troxler RF, Oppenheim FG. FEBS Lett. 2001;491:76–80. doi: 10.1016/s0014-5793(01)02157-3. [DOI] [PubMed] [Google Scholar]
- 52.Cabras T, Patamia M, Melino S, Inzitari R, Messana I, Castagnola M, Petruzzelli R. Biochem Biophys Res Commun. 2007;358:277–84. doi: 10.1016/j.bbrc.2007.04.121. [DOI] [PubMed] [Google Scholar]
- 53.Bradshaw RA, Peters T. J Biol Chem. 1969;244:5582–9. [PubMed] [Google Scholar]
- 54.Buettner GR, Jurkiewicz BA. Radiat Res. 1996;145:532–541. [PubMed] [Google Scholar]
- 55.Jeung C-S, Kim CH, Min K, Suha SW, Suha J. Bioorg Med Chem Lett. 2001;11:2401–2404. doi: 10.1016/s0960-894x(01)00439-5. [DOI] [PubMed] [Google Scholar]
- 56.Hettich R, Schneider HJ. J Am Chem Soc. 1997;119:5638. [Google Scholar]
- 57.Barrette WCJ, Sawyer DT, Fee JA, Asada K. Biochemistry. 1983;22:624–627. doi: 10.1021/bi00272a015. [DOI] [PubMed] [Google Scholar]
- 58.Azab HA, Banci L, Borsari M, Luchinat C, Sola M, Viezzoli MS. Inorg Chem. 1992;31:4649–4655. [Google Scholar]
- 59.Tullius TD, Dombroski BA. Proc Natl Acad Sci U S A. 1986;83:5469–5473. doi: 10.1073/pnas.83.15.5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sagripanti J-L, Goering PL, Lamanna A. Tox Appl Pharm. 1991;110:477–485. doi: 10.1016/0041-008x(91)90048-j. [DOI] [PubMed] [Google Scholar]
- 61.Huang X, Pieczko ME, Long EC. Biochemistry. 1999;38:2160–2166. doi: 10.1021/bi982587o. [DOI] [PubMed] [Google Scholar]
- 62.Rammo J, Hettich R, Roigk A, Schneider HJ. Chem Commun. 1996:105. [Google Scholar]
- 63.Dixon NE, Geue RJ, Lambert JN, Moghaddas S, Pearce DA, Sargeson AM. Chem Commun. 1996:1287–1288. [Google Scholar]
- 64.Branum ME, Tipton AK, Zhu S, Que LJ. J Am Chem Soc. 2001;123:1898. doi: 10.1021/ja0010103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.