

Abstract
The origin region of Vibrio cholerae chromosome II (chrII) resembles plasmid origins that have repeated initiator-binding sites (iterons). Iterons are essential for initiation as well as preventing over-initiation of plasmid replication. In chrII, iterons are also essential for initiation but over-initiation is prevented by sites called 39-mers. Both iterons and 39-mers are binding sites of the chrII specific initiator, RctB. Here, we have isolated RctB mutants that permit over-initiation in the presence of 39-mers. Characterization of two of the mutants showed that both are defective in 39-mer binding, which helps to explain their over-initiation phenotype. In vitro, RctB bound to 39-mers as monomers, and to iterons as both monomers and dimers. Monomer binding to iterons increased in both the mutants, suggesting that monomers are likely to be the initiators. We suggest that dimers might be competitive inhibitors of monomer binding to iterons and thus help control replication negatively. ChrII replication was found to be dependent on chaperones DnaJ and DnaK in vivo. The chaperones preferentially improved dimer binding in vitro, further suggesting the importance of dimer binding in the control of chrII replication.
INTRODUCTION
Binding of proteins, called initiators, to the origin of replication is generally the event that initiates duplication of the genome (1). In bacterial origins, the presence of multiple initiator binding sites is the norm and they are used to form a specialized nucleoprotein structure that unwinds the origin, a crucial step that precedes DNA synthesis (2–4). In Escherichia coli, where initiator binding has been studied in most detail, great strides have been made to understand the opening process but the mechanism of unwinding still remains speculative, primarily because only partial structural information about the nucleoprotein complexes is available (5). Multi-site binding provides more regulatory opportunities to prevent untimely or over initiation of replication. Understanding the details of the initiator–origin interaction is therefore basic to understanding genome maintenance.
The replication origin of E.coli, oriC, contains high and low affinity sites for binding the initiator DnaA. DnaA binds to these sites in either ATP or ADP bound forms. The high affinity sites are used to nucleate binding to low affinity sites, some of which must be occupied by DnaA-ATP (4). Over-initiation of replication is prevented primarily by limiting binding to the low affinity sites (6,7).
Multi-site initiator binding to the origin is also the norm in replication of lambdoid phages and in a large group of plasmids of E.coli (3). Here, the sites are direct repeats of nearly identical sequences, called iterons, which bind cognate initiators with high affinity. Limiting initiator binding is also used as one of the mechanisms to prevent over-initiation of plasmid replication, although not of lambdoid phages (8–10).
Multiple mechanisms are used to limit synthesis, activity and availability of initiators. In E.coli, initiator synthesis is limited by transcriptional auto-repression and by sequestering the initiator promoter for a significant part of the cell cycle (6). The initiator activity is restrained by a process called the regulatory inactivation of DnaA (RIDA), which accelerates conversion of DnaA-ATP to DnaA-ADP. Finally, initiator availability is reduced by titration of the protein by hundreds of sites distributed throughout the chromosome. Iteron-carrying plasmids use basically the same strategies to limit their initiators except that promoter sequestration and ATP binding are not involved: initiators are inactivated simply by dimerization since only monomers have significant affinity for iterons (11).
Replication of plasmids and chromosomes differ in an important respect: whereas plasmids replicate throughout the cell cycle, a fundamental feature of chromosomes is that they replicate at a particular time of the cell cycle (12). Vibrio cholerae provides an opportunity to understand how the timing of replication can be changed from random to specific. Vibrios have two chromosomes (chrI and chrII) (13,14). The replication origin of chrI is nearly identical to that of the E.coli chromosome, and the chrII origin is similar to that of plasmids with iterons (15). ChrII, however, is a bona fide chromosome because it carries essential genes while plasmids do not, and because it replicates at a specific time of the cell cycle (16).
The control of chrII replication initiation is more involved than that of plasmid replication. While plasmid initiators bind only to iterons, the chrII initiator, RctB, binds additionally to sites that we call 39-mers (17) (Figure 1). The 39-mers are the key inhibitors that prevent chrII over-replication, whereas in plasmids this is done by the iterons. ChrII iterons have an internal Dam methylation site, GATC, and methylation of the adenine residues of GATC is required for RctB binding (18). The plasmid iterons lack GATC sites. The added features of chrII replication have provided some clues as to how the replication could be restricted to a specific stage of the cell cycle (17).
Figure 1.

Replication of oriII plasmids in E.coli. The top diagram is a schematic representation of the region responsible for chrII replication. The region marked as oriII suffices for autonomous replication in E.coli when the initiator RctB is provided. The adjacent region, incII, negatively regulates the activity of oriII. The region has two kinds of binding site for RctB: 39-mers (black rectangles) and iterons (white and striped arrowheads represent respectively 11- and 12-mers in their relative natural orientations). The significance of iteron orientations is not known. The region has two promoters: prctA that controls RctB binding to the downstream 39-mer (19) and prctB that expresses the initiator gene (20). Two versions of oriII plasmids (low- and high-copy) were used in the presence of three RctB proteins (WT, ΔC157 and F378S) in two different hosts (dnaKJ+ and ΔdnaKJ). The high-copy plasmid was derived my mutating RctB binding sites of incII (crossed sites) of the low-copy plasmid. WT, ΔC157 or F378S RctB was supplied constitutively from pGD16, pGD24 or pGD28, respectively. The stars indicate cases where oriII plasmid copy number could not be measured because the plasmid failed to transform the host. Error bars here and elsewhere represent 1 SD. We note that the intensity of low-copy plasmid band in both dnaKJ+ and ΔdnaKJ cells with WT RctB was below the detection limit (<d.l.) and assumed as zero. In contrast, its intensities in the presence of ΔC157and F378S mutants were 0.74 ± 0.25 and 0.63 ± 0.04, respectively. The mean was from values of three independent colonies, all of which were above the detection limit (estimated as 0.1 ± 0.1).
To get a better understanding of the role of the two kinds of the RctB binding site in the control of chrII replication initiation, here we have isolated mutant RctBs that are more proficient in supporting chrII replication. We have characterized two of the mutants and show that both the mutants are defective in binding, in vivo and in vitro, to a replication inhibitory 39-mer site. This finding provides a reasonable explanation for how the mutants could promote replication. In vitro, RctB bound to a 39-mer as monomer and an iteron as monomer and as dimer. Dimer binding was less with both the mutants, suggesting that it might be inhibitory to replication initiation. We also show that chaperones DnaJ and DnaK are required for efficient chrII replication in vivo and they improve all RctB binding in vitro, particularly the dimer binding. This was unexpected from plasmid studies that showed that mainly monomers bind iterons and the chaperones promote monomer binding by converting dimers to monomers (21,22). These results suggest that the binding of RctB dimers to iterons is yet another mechanism to control chrII replication. The dimer binding could be a potential negative feedback mechanism to control over-initiation of replication, as will be discussed.
MATERIALS AND METHODS
Bacterial strains and plasmids
Vibrio cholerae and E.coli strains, and plasmids used in this study are listed in Table 1. The plasmids constructed in this study are as follows.
Table 1.
Bacterial strains and plasmids used in this study
| Genotype/relevant characteristics | Source/figure | |
|---|---|---|
| Strains | ||
| BR2846 | K-12 recA1 Δ (argF-lac)U169; Strain for cloning in pMLB1109 | (23) |
| BR4391 | BR4501 Δ (srlR-recA)306::Tn10 4501 = MG1655 thi-3178::Tn10Kan | Figure 1 and Supplementary Figure S1 |
| BR4392 | BR3660 Δ (dnaK-J)::miniKan Δ(srlR-recA)306::Tn10 | Figure 1 and Supplementary Figure S1 |
| BR8706 | =Stbl2 Δ(lac-proAB) Δ(araFGH) ΔaraEp PCP18-araE; araE under constitutive CP18 promoter | (24)/Figure 2 and Supplementary Figure S2 |
| CVC205 | V. cholerae El Tor N16961 lacZ::res::tet::res | (20) |
| CVC553 | DH5Δlac(λpir), where λpir = λDKC370; strain for maintaining R6Koriγ plasmids | (17) |
| BL21(DE3) | Expression of recombinant protein in E.coli | Stratagene |
| DH5α | Strain for cloning | Invitrogen |
| Plasmids | ||
| pACYC177 | P15Aori;ApR, KnR | Fermentas |
| pAS1 | PrctB with the 29-mer (nt 1049–1133) in pMLB1109; pSC101ori; ApR | (25)/Figure 1 |
| pBEND2 | pBRori; ApR | (26) |
| pGD14 | pET22b where bla gene was replaced by aadA7 gene; MCS has a KpnI site and the BamHI site is now between EcoRI and KpnI; pBRori; SpR | This study |
| pGD16 | rctB (nt 1118–3115) in pGD14 vector; pBR ori; SpR | Figure 1 and Supplementary Figure S1 |
| pGD24 | rctB (nt 1118–2621) in pGD14 vector; pBR ori; SpR | Figure 1 and Supplementary Figure S1 |
| pGD25 | Site directed mutagenesis on pGD16 to create F378Y RctB; SpR | Supplementary Table S3 |
| pGD28 | Site directed mutagenesis on pGD16 to create F378S RctB; SpR | Figure 1, Supplementary Figure S1 |
| pGD43 | Site directed mutagenesis on pTVC16 to create F378S RctB; SpR | Figures 3 and 7 |
| pJJ02 | rctB (nt 1118–2621) under PBAD control in pSC101ori; SpR | Figure 2 and Supplementary Figure S2 |
| pJJ03 | rctB (nt 1118–2621) under pT7 control in pTXB1; pBRori; ApR | Figures 3 and 5 |
| pJJ16 | 1 × 12-mer (nt 785–823) in pSP5; pUC ori; ApR | Figure 4 |
| pJJ17 | 1 × 39-mer (nt 439–498) in pSP5; pUC ori; ApR | Figure 4 |
| pJJ56 | rctB (nt 1118–3115) under Ptac control in pMAL-c2X; pBR ori; ApR | Figure 5 |
| pJJ58 | pTVC11 with the mutation creating F378S; pSC101ori; SpR | Figure 2 and Supplementary Figure S2 |
| pMLB1109 | Source of promoter-less lacZ gene; pSC101ori; ApR | M. Berman |
| pSP5 | pUC ori; ApR | This study |
| pTVC11 | rctB (nt 1118–3115) under PBAD control in pSC101ori; SpR | (20)/Figure 2 and Supplementary Figure S2 |
| pTVC12 | PBAD in pSC101ori; SpR | (20)/Figure 2 |
| pTVC16 | rctB (nt 1118–3115) under pT7 control in pTXB1; ApR | This study |
| pTVC61 | pBRori. Digestion with EcoRV and HpaI leaves 300 bp flanks on either side of MCS; CmR | (19) |
| pTVC126 | PrctA with 2 × 11-mers up to the beginning of the rctA annotated start codon (nt 377–249) in pMLB1109; ApR | (25)/Figure 2 |
| pTVC148 | 2 × 12-mers before the oriII IHF site (nt 879–952) in pTVC61; CmR | Figure 8 |
| pTVC174 | 1 × 39-mer (nt 449–487) in pBEND-2; ApR | (17)/Figures 3 and 5; Supplementary Figure S5 |
| pTVC195 | 1 × 12-mer (nt 565–602) in pBEND-2; ApR | (17)/Figures 3, 5 and 6; Supplementary Figures S4 and S5 |
| pTVC228 | 6 × 12-mers (nt 788–934) in pTVC243; CmR | (17)/Figure 8 |
| pTVC234 | PrepA from P1 (nt 556–600) in pMLB1109; ApR | (17)/Figure 2 |
| pTVC243 | pTVC61 shorter by 400 bp. Digestion with EcoRV and HpaI leaves 100 bp flanks on either side of MCS; CmR | (17) |
| pTVC248 | 3 × 11-mer (nt 291–445) in pTVC243; CmR | (7)/Figure 7 |
| pTVC251 | oriII (nt 441–1133) in R6Koriγ; ApR | (17)/Figure 1 and Supplementary Figure S2 |
| pTVC336 | pTVC251 with mutated 39-mer, 12-mer, and 11-mer; ApR | (17)/Figure 1 and Supplementary Figure S2 |
| pTVC500 | pAS1 whose 29-mer is replaced by a 39-mer (nt 1049–1133); ApR | Figure 2 |
pAS1 was constructed by annealing complementary oligonucleotides [Supporting Information in (19)] (Supplementary Table S1).
pGD12 is same as pET22b (Stratagene, La Jolla, CA, USA) except its bla gene was replaced with the aadA7 gene by PCR. aadA7 was amplified from pTVC11 using primers GD4 and GD7, and the backbone of pET22b without the bla gene was amplified using primers GD5 and GD9. The two PCR products were mixed and used as template for amplification with primers GD6 and GD16. The final PCR product was digested with PstI and self-ligated, generating pGD12. Next, the multi-cloning site between the NdeI and XhoI sites of pGD12 was replaced with annealed oligonucleotides GD1 and GD2, generating pGD14. pGD16 is a clone of rctB with an internal BamHI site (created by silent mutations within the open reading frame) in pGD14. The N-terminal part of rctB was amplified from pTVC14 using the primers GD30 and GD14, the C-terminal part of rctB was amplified from pTVC14 using the primers GD31 and GD13. The two PCR products were mixed and used as template with the primers GD30 and GD31. The final PCR product was digested by NdeI and XhoI, and cloned in NdeI and XhoI digested pGD14. The resulting plasmid is called pGD16. To clone the rctBΔC157 gene, the relevant region was amplified from pGD16 using the primers GD10 and GD27, the product digested with EcoRI and KpnI and cloned in pGD14 digested with the same enzymes, generating pGD24. pGD28 and pGD43 are identical to pGD16 and pTVC16, respectively, except for the F378S change in the rctB ORF. The change was introduced by site directed mutagenesis using primers GD23 and GD24 and QuikChange II XL Kit (Agilent Technologies, La Jolla, CA, USA).
pJJ02 was constructed by cloning the PCR product using the Phusion High-Fidelity polymerase (NEB, Beverly, MA, USA), N16961 (CVC205) DNA as template, and JJ02 and BNHE1 as primers, into pTVC11, after digesting both with NheI and NotI. pJJ03 was constructed similarly except that the primers were JJ01 and JJ60, and the PCR product was cloned into pTXB1 (NEB), after digesting both with NdeI and XhoI. pJJ17 were constructed by cloning complementary oilgonucleotides in pSP5 at the EcoRI site. pJJ56 was constructed by cloning the PCR product using N16961 DNA as template and JJ115 and JJ70 primers, in pMAL-c2X vector (NEB), after digesting both with EcoRI and BamHI. pJJ58 is same as pTVC11 except for the F378S change in the rctB ORF. The change was introduced by site-directed mutagenesis as before using primers JJ36 and JJ64.
pTVC148 was constructed by cloning an oligonucleotide fragment with NotI and XhoI ends into the transcription-free region of pTVC61, after digesting the vector with the same enzymes. pTVC500 was constructed similarly to pAS1, except that the EcoRI oligonucleotide fragment (chrII nt 1049–1133), upstream of the promoter-less lacZ gene in pMLB1109, contained a 39-mer, precisely replacing the 29-mer of PrctB.
pSP5 is derived from pBR322 by destroying its EcoRI site (by end filling and ligation) and cloning two copies of the multi-cloning site cassette of pUC13 as inverted repeats at the HindIII site. The cloning sites are arranged in the order HindIII–EcoRI–HindIII.
Mutagenesis of rctB and selection mutants conferring chaperone-independence
The rctB mutant library was generated by PCR, using the Genemorph II Random Mutagenesis Kit (Agilent Technologies). The mutagenesis conditions were expected to generate up to 10 mutations per kb. The whole rctB gene was amplified using primers GD30 and GD31. The PCR product was digested with EcoRI and KpnI, and cloned at the same sites of pGD16 (SpR). The resulting library was introduced in DH5α cells by electroporation. A total of 2800 transformants were pooled, their plasmid DNA was isolated and electroporated into the E.coli ΔdnaKJ strain (BR4392). The transformants thus obtained were pooled, made chemically competent and further transformed with either pTVC22 (mini-oriII; ApR) or pACYC177 (positive control; ApR). The transformants were selected after overnight incubation at 37°C on LB agar plate containing ampicillin (100 µg/ml) and spectinomycin (40 µg/ml).
Mutagenesis of incII sites
The mini-oriII plasmid, pTVC251, has in addition to the minimal origin a part of the adjoining negative control locus, incII (Figure 1). The incII region includes a 39-mer, an 11-mer and a 12-mer, which are all binding sites for the initiator RctB (17). Using overlap extension PCR, a part of the 39-mer sequence, 5′-CGGAAGCATG-3′, was changed to 5′-GCCTTCGTAC-3′, and a part of the 11- and 12- mers, 5′-GATC-3′ to GTAG (27). These changes significantly decreased RctB binding in vitro [figure S7 of ref. (17) and figure S1C of ref. (18)] and replication control activity in vivo [figure 3 of ref. (17)].
Protein purification
To purify WT RctB and the ΔC157 and F378S mutants, the corresponding genes were cloned into pTXB1 vector (NEB), and the derivate plasmids: pTVC16, pJJ03 and pGD43, respectively, were used to transform BL21(DE3). The transformed colonies were inoculated into 5 ml of LB medium with ampicillin (100 µg/ml) and grown overnight at 37°C. Two ml of this culture was added to 2 l of the same medium and grown to OD600 of 0.6 at 37°C. The expression of the cloned protein was induced with 0.4 mM IPTG and the culture was incubated overnight at 16°C. Cells were harvested and resuspended in 20 ml of lysis buffer [25 mM Tris–HCl (pH 8.0), 500 mM NaCl, 10% glycerol (v/v) and 1 mM EDTA] and lysed using French Press. The lysate was spun at 13 000 rpm for 20 min (Biofuge fresco, Heraeus, UK) and the supernatant was loaded onto a Bio-Rad Poly-Prep column (Bio-Rad, Hercules, CA, USA) with 1 ml of packed chitin beads (NEB). The column was washed with 20 volumes of the lysis buffer, and the bound proteins were reacted with 1 ml of 50 mM DTT in the same buffer at 4°C inside the column. After overnight incubation, the released protein was collected by gravity flow. The process was repeated twice with fresh DTT solution but with 1 h incubation each time. The released proteins were pooled and concentrated using Amicon Ultra (30 K) filters (Millipore Corp., Billerica, MA, USA) at 10 000 rpm for 5 min. The concentrated protein (≤500 µl) was further purified by gel filtration using a Superdex 200 column equilibrated with RctB buffer [25 mM Tris–HCl (pH 8.0), 300 mM NaCl, 5% glycerol (v/v) and 1 mM EDTA] and as described in the legend to Supplementary Figure S3. The fractions containing RctB was concentrated about 5-fold using Amicon filters as above and dialyzed against a buffer [25 mM Tris–HCl (pH 8.0), 300 mM NaCl, 10% glycerol (v/v) and 1 mM EDTA] and stored at −80°C at a final concentration of about 1.5 mg/ml. The protein concentration was measured by Bradford reagent (Fermentas, Glen Burnie, MD, USA), using Bovine serum albumin (BSA) as standard. No visible contaminating band was detected in 10% SDS–PAGE upon loading 6 µg of RctB and staining with SimplyBlue™ SafeStain (Invitrogen, Carlsbad, CA, USA), where a band of 250 ng RctB could be easily detected. The purity of the protein was thus considered >96%.
Mal-RctB fusion protein expressed from pJJ56 was purified similarly, except that the induction was done in the presence of 0.2 mM IPTG, and cells were lysed by freeze-thaw in buffer A [20 mM Tris–HCl (pH 7.4), 200 mM NaCl, 10% glycerol and 1 mM EDTA], followed by sonication in a Digital Sonifier (Branson, Danbury, CT, USA) at 30% power level for 20 s, thrice with 2-min intervals. The lysate supernatant was loaded onto a column containing the maltose resin (NEB). The column was washed with 20 volumes of buffer A and the bound protein was eluted with 10 mM maltose in buffer A. The eluted protein was dialyzed against buffer A and stored at −80°C.
EMSA and handcuffing assay
Plasmids (5 µg) were digested with appropriate restriction enzymes (EcoRV + HpaI or EcoRV) followed by de-phosphorylation of the ends with SAP (Promega, Madison, WI, USA). The desired fragments were gel purified and radio-labeled with 30 units of polynucleotidyl kinase (NEB) and 50 µCi of adenosine 5′-[γ32P]triphosphate (Perkin-Elmer, Waltham, MA, USA). The labeled fragments were purified through G-50 columns (Roche Diagnostics Corporation, Indianapolis, IN, USA). EMSA and handcuffing assay were done as described (19,28).
β-Galactosidase assay
These were done from late log phase cultures (OD = 0.4–0.5) of BR8706 and its derivatives as described (29).
Plasmid copy-number measurement
Host cells for plasmids were BR4391 (dnaKJ+), BR4392 (ΔdnaKJ) in Figure 1, and BR8706 (where araE is expressed constitutively from the chromosome) in Supplementary Figure S2. The plasmids carrying either the wild-type rctB gene or its mutant variants were pGD16, pGD24 and pGD28 (Figure 1), and pTVC11, pJJ02 and pJJ58 (Supplementary Figure S2). Cells with these RctB source plasmids were further transformed with oriII-plasmids. The oriII plasmids contained a second origin (R6K oriγ), which was utilized during plasmid construction, maintenance, and isolation, using a host (CVC553) that supplied the cognate initiator (π protein). When measurement of oriII activity was desired, cells had a source of RctB instead of the π protein. In these experiments, LB plates had lower drug (50 instead of 100 µg/ml of ampicillin) to improve growth of cells with mini-oriII plasmids. After overnight growth the colonies were collected by washing the plates and plasmid copy number was determined as described (17). Even with these modifications, the low-copy oriII plasmid band could not be detected in some cases (Figure 1 and Supplementary Figure S2).
Refolding of RctB in vitro
WT, ΔC157 and Mal-fused RctB proteins, either singly or in pair-wise combination in ≤ 2 µl, were added to 50 µl of 8 M urea to a final concentration of 2.67 µM and dialyzed against 100 ml of the RctB buffer containing 8 M urea for 2 h. For refolding, the urea concentration in the dialysis buffer was reduced stepwise by adding fresh RctB buffer. Urea concentration was first reduced to 6 M and dialysis continued for 1 h. This process was repeated with 4, 2 and 1 M urea in the dialysis buffer, except the last dialysis in 1 M urea was for 2 h. Finally, dialysis was continued for another 6 h with RctB buffer without urea. These proteins were used within a day for EMSA.
RESULTS
Isolation of RctB mutants that cause over-replication
Characterization of initiator mutants that cause over-initiation of plasmid replication (copy-up mutants) has contributed greatly to our understanding of plasmid replication control. For low-copy iteron-carrying plasmids, such as P1 and F, which depend on chaperones for efficient replication, the majority of the mutants isolated on the basis of chaperone-independent replication, also showed the copy-up phenotype (30–32). Plasmids carrying the chrII origin, oriII, replicate efficiently in E.coli, when the chrII-specific initiator, RctB, is provided (15,33). Here, we show that this is not the case when E.coli is deleted for chaperones DnaK and DnaJ (ΔdnaKJ; BR4392). When an oriII plasmid (pTVC22) was used to transform ΔdnaKJ cells containing a RctB source plasmid (pGD16), no transformants were seen after overnight incubation (Supplementary Table S2). This opened the possibility for isolation of copy-up mutants of RctB following the chaperone-independent replication phenotype.
In order to isolate RctB mutants that might allow oriII to function in ΔdnaKJ cells, a plasmid library of rctB mutants was generated by error prone PCR. The library was used en masse to transform ΔdnaKJ cells. The transformants were pooled and further transformed with either an oriII plasmid (pTVC22) or a control plasmid (pACYC177). When the presence of both the resident rctB plasmid and the incoming control plasmid was selected, about 2000 colonies were obtained. When an equal amount of oriII plasmid replaced the control plasmid, the number of colonies was 32. From 10 of these colonies, the rctB plasmid was isolated and sequenced. In each case, the rctB gene showed multiple changes (Supplementary Table S3). Mutants were created with single changes in some of these positions by site-directed mutagenesis of the wild-type gene present in pGD16, and their ability to replicate the oriII plasmid (pTVC22) in ΔdnaKJ cells was tested. Only mutations causing substitutions F233I, F378L, F378S and K502ochre conferred replication proficiency. We chose the mutants K502ochre and F378S for further study. The ochre mutant was interesting as it was replication proficient in spite of lacking 157 C-terminal amino acids. The F378S mutant was chosen because the position 378 was changed in two of the above mutants and in a third mutant (F378Y), found in a separate screen (Supplementary Table S3). The mutant with the serine change was chosen because it showed a stronger replication phenotype than the other two. Hereafter, the mutants will be referred to as ΔC157 and F378S.
The replication phenotype was tested quantitatively by measuring copy number of oriII plasmids. Two oriII plasmids were used. One (pTVC251) carried the minimal oriII and a part of the adjoining negative control locus, incII (Figure 1). This plasmid was chosen because here the negative control operates near maximally and severely restricts oriII activity (19). In the other (pTVC336), the three RctB binding sites of incII were mutated, leaving only the oriII region intact (17). In the latter plasmid, oriII activity is found to be near maximal. These will be referred to as low- and high-copy oriII plasmids, respectively.
The oriII-plasmids were electroporated into cells that already had an established plasmid supplying RctB. In ΔdnaKJ cells, the mutant RctB proteins supported replication of both the oriII plasmids at copy numbers higher than those obtained with WT RctB (Figure 1). The copy number hierarchy of the two oriII plasmids was maintained, indicating that the mutants remain capable of mediating incII-mediated negative control. The copy numbers increased further when dnaKJ+ cells were used, indicating that the mutants were still dependent on chaperones for full activity. The copy number of the low-copy oriII plasmid increased more in the presence of F378S compared to ΔC157, indicating that F378S is more dependent on chaperones than ΔC157. The increase in copy number was not due to changes in initiator concentration either in dnaKJ+ or ΔdnaKJ cells, as determined by western blotting (Supplementary Figure S1).
Unexpectedly, dnaKJ+ cells supplying mutant RctB proteins could not be transformed with the high-copy oriII plasmid. This was not due to significant changes in RctB supply (Supplementary Figure S1). We considered the possibility that too high a rate of replication of the oriII plasmid could be the reason for the failure to obtain transformants, as plasmid over-replication is known to arrest cell growth (33). Since the rate of oriII firing depends upon RctB supply (20,34), we tried to reduce the rate by reducing RctB supply. This was achieved by a lower copy number vector and limiting induction of rctB transcription (Supplementary Figure S2A). Under these conditions, the high-copy oriII plasmid could transform and replicate in dnaKJ+ cells in the presence of either of the RctB mutants. In summary, it appears that although selected as chaperone-independent, copy-up is the major phenotype of both the RctB mutants.
RctB mutants are altered in DNA Binding in vivo
To understand the basis of the copy-up phenotype, we determined the binding of the mutant proteins to iterons and to a 39-mer, the two kinds of sites to which RctB specifically binds. We used promoter-repression as reporter of binding in vivo. In the origin region of chrII, there are two promoters, one overlapping an iteron (P-rctA) and the other overlapping a 29-mer (P-rctB) (17) (Figure 1). The 29-mer is similar in sequence and structure to a 39-mer and binds RctB. In any event, we replaced the 29-mer with the 39-mer from the middle of incII and this did not alter the promoter activity. The promoters were fused to a promoter-less lacZ gene present in a plasmid and these reporter plasmids were introduced into E.coli. Promoter activities were then determined in the presence of an inducible plasmid source of RctB (Figure 2).
Figure 2.

Binding of RctB to two kinds of site in vivo. The binding was tested by measuring activity of promoters which are naturally repressed by RctB. RctB was supplied from an arabinose-inducible promoter, present in a pSC101-derived plasmid. Plasmids supplying WT, ΔC157 and F378S RctB proteins were pTVC11, pJJ02 and pJJ58, respectively. The empty vector carrying the inducible promoter but no rctB (pTVC12) was used as a negative control (the column marked ‘None’). Three target promoters were used: PrepA carrying two iterons of plasmid P1 (unrelated to chrII iterons) as negative control (A), PrctA for iteron binding, as it has in its vicinity two 11-mer iterons (B), and PrctB for 39-mer binding, as it contains naturally a 29-mer, a site similar to the 39-mer present in the middle of incII (Figure 1) (C). The results were similar when the 29-mer was exchanged with the 39-mer (data not shown). The promoters were fused to a promoter-less lacZ gene (transcriptional fusions; cartoons to the right of the bar diagrams) of a pBR322-derived plasmid, pMLB1109, which resulted in plasmids pTVC234, pTVC126 and pTVC500, respectively. These plasmids were introduced into E.coli (BR8706) harboring the rctB-plasmids for measuring lacZ activity (in Miller units). The white bars represent activity without induction of rctB, and the gray bars, after induction with 0.2% arabinose. RctB synthesis was confirmed by western blotting (Supplementary Figure S2B). n.s. = not significantly different by t-test with α = 0.01; when the t-test indicates significant difference between the measures, the P-values are shown.
Assuming promoter repression to be diagnostic of RctB binding, the results indicate that compared to the WT protein, ΔC157 is defective in 39-mer binding but is improved in iteron binding. Since 39-mer binding inhibits initiation, and iteron binding promotes initiation, the reduced 39-mer binding and increased iteron binding provides a ready explanation for the copy-up phenotype. F378S behavior was similar to that of ΔC157 in that it also bound with lower affinity to a 39-mer and greater affinity to iterons compared to WT RctB but ΔC157 exhibited a greater degree of discrimination. These results and the results of copy-number measurements (Figure 1) suggest that the copy-up phenotype of the two mutants have common but not identical bases.
RctB mutants are altered in DNA binding in vitro
The DNA fragment used in these studies carried either a single 12-mer or a single 39-mer. WT RctB bound to both kinds of site at similar protein concentrations but maximal binding was significantly more to the 39-mer compared to the iteron (∼90% versus ∼20%; Figure 3). The opposite was the case for the ΔC157 mutant: it bound negligibly to the 39-mer but efficiently to the iteron (∼2% versus ∼80%). The F378S binding pattern was different but it bound the 39-mer less avidly than the WT protein (∼45% versus ∼90%). The 39-mer binding defect seen in vivo is thus supported in vitro for both the mutants. However, while both the mutants were improved in iteron binding in vivo, this was obvious only for the ΔC157 mutant in vitro. The two mutants thus appear to be different in their binding characteristics in vitro as well.
Figure 3.

EMSA of RctB binding to fragments carrying a 12- or a 39-mer. EMSA was performed with end-labeled (32P) DNA fragments carrying a single copy of either a 12-mer or a 39-mer with 55 bp of vector sequences at both flanks (obtained from plasmids pTVC195 or pTVC174, respectively). RctB was either WT or mutants ΔC157 and F378S, and used in the concentration range of 0.67–33 nM. The binding was analyzed using a 5% polyacrlyamide gel. Arrows show two retarded bands of the 12-mer fragment. The binding profiles are shown at the bottom. The data points were fitted to equation 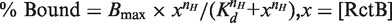 , nH = Hill slope and Bmax = maximum %bound. The values of nH for the WT, ΔC157 and F378S were 1.8, 1.2 and 1.4 for the 12-mer, and 1.7, 1 and 5 for the 39-mer.
, nH = Hill slope and Bmax = maximum %bound. The values of nH for the WT, ΔC157 and F378S were 1.8, 1.2 and 1.4 for the 12-mer, and 1.7, 1 and 5 for the 39-mer.
Binding to the iteron in vitro appeared to be complex: two retarded bands were reproducibly seen both for the WT and F378S proteins, although the fragment used had a single iteron. The faster migrating band was more intense in the case of the F378S mutant protein, opposite to the case of the WT protein. The ΔC157 mutant mainly showed one retarded band that corresponded to the faster migrating band with the WT protein. The finding that both the copy-up mutants are enriched in the faster migrating band suggests that the form of the protein present in this band is more proficient in promoting replication initiation.
RctB binds to an iteron both as monomer and dimer, and to a 39-mer only as monomer
The molecular weight (Mw) of RctB monomer is 75 kDa, calculated from its amino acid sequence. The Mw appeared to be ∼160 kDa in gel filtration studies, indicating that the protein can dimerize, like the initiators of iteron-carrying plasmids (Supplementary Figure S3). Plasmid initiators, however, generally bind to iterons as monomers. Dimers can also bind but only to one-half of an iteron sequence, and this half-site binding is considerably weaker (28). Dimers do bind efficiently when the half-iteron site is naturally present as an inverted repeat (35,36). The chrII iterons differ from plasmid iterons in having an internal dyad-symmetric element, TGATCA, and these bases are the only bases of the iterons that are fully conserved. The element is however asymmetrically placed within the iteron. These features prompted the possibility that RctB might bind to single iterons either as monomer or as dimer, which could explain the two retarded bands seen by EMSA (Figure 3).
We used Ferguson analysis to estimate the Mw of the two retarded bands of the iteron-carrying fragment and the single retarded band of the 39-mer carrying fragment. In this analysis, the Mw of a protein–DNA complex is determined by comparing its electrophoretic mobility with that of known protein standards in a series of native gels of increasing concentrations. It is assumed that mobility of each species is influenced solely by its size, shape, and net charge. Initially, migration of each standard along with the migration of DNA and DNA–protein complexes is plotted against gel concentrations (Figure 4A) and, from the slope, the retardation coefficient (Kr) is determined. The Mw of unknown species is determined from their Kr (Figure 4C) using the plot of the standards (Figure 4B). We used a 60 bp EcoRI fragment carrying either a centrally located iteron (from pJJ16) or a 39-mer (from pJJ17). Assuming the Mw of a single base pair to be 660 kDa, the Mw of the DNA fragment alone is calculated to be 40 kDa. This value compares well with 43.3 ± 1.3 kDa, deduced from Figure 4B. Accounting for the contribution of DNA, the Mw of the protein bound to 39-mer corresponded to 73 ± 2 kDa, and the protein bound to iteron corresponded to 81 ± 2 kDa and 142 ± 4 kDa, for the lower and the upper retarded bands, respectively. Since the calculated Mw of RctB monomer is 75 kDa, these results are consistent with RctB binding to 39-mer as monomer and to iteron as monomer and as dimer.
Figure 4.

Molecular weight of DNA–protein complexes by Ferguson gel analysis. EMSA was performed using a 60 bp DNA fragment that included either a centrally located 12-mer or a 39-mer. The fragments were derived from pJJ16 or pJJ17, respectively. (A) Ferguson plots of gel concentration versus log mobility of protein standards; the lines are best fits from linear regression. Protein standards (Sigma) used were carbonic anhydrase (29 kDa), chicken egg albumin (45 kDa), BSA (monomer, 66 kDa), BSA (dimer, 132 kDa), and β-amylase (200 kDa). For clarity, Ferguson plots of free DNA and DNA–protein complexes (a–d) are shown separately in panel (C). (B) A plot of retardation coefficient (−Kr) versus molecular weight (Mw) of protein standards. Kr values are the slopes of the regression lines of the Ferguson plots (A). The data were fit by linear regression analysis. (D) The Mw of free DNA and DNA–protein complexes. These values were determined from the plot in panel (B), using the Kr values from panel (C). Mw of proteins present in DNA–protein complexes (fifth column) were obtained by subtracting the Mw of free DNA from the Mw of the complexes (fourth column). Theoretical Mws (sixth column) were deduced from sequences of free DNA or RctB monomer. The error values are one standard deviation of the mean, deduced from two independent experiments.
RctB heterodimers bind iterons
To confirm dimer binding to iterons independently, we constructed derivates of RctB of different Mws to form heterodimers. The heterodimers, being of Mw intermediate to those of the two parental homodimers, are expected to migrate in between the two homodimers, when analyzed by EMSA. The appearance of such uniquely migrating species can be taken in support of dimer binding. Three proteins were used: WT RctB, a truncated version, the ΔC157 mutant, and a larger protein, a Mal-RctB fusion. These three proteins were unfolded and refolded separately, and in pair-wise combinations: WT + ΔC157, WT + Mal-RctB, and ΔC157 + Mal-RctB (further details are in the ‘Materials and Methods’ section). The binding patterns of refolded WT, ΔC157, and Mal-RctB proteins were similar to those obtained with their native counterparts (Figure 3; data not shown for Mal-RctB). When refolded protein combinations were used, two new bands were observed in the case of WT + Mal-RctB, although one band was expected (Figure 5A). The positions of the new bands were intermediate between the putative dimer binding bands of the two starting proteins. The observation that no new retarded bands were seen when ΔC157 was combined with the other two proteins suggests that ΔC157 is inefficient either in dimer formation (at the assay concentration) or in binding to DNA as dimer. Together with the results of Figure 3, the suggestion is that the single retarded band seen with ΔC157 represents monomer binding, and the two retarded bands seen with full length RctB represent monomer and dimer binding.
Figure 5.

EMSA of RctB hetero-dimer binding to a 12-mer. Proteins used are described at the top. They were unfolded and refolded either individually or by pair-wise combinations before use in EMSA. Binding of proteins to a 12-mer carrying fragment is shown in panel (A) and to a 39-mer carrying fragment (B). The fragments of 150 bp were obtained from pTVC195 and pTVC174, respectively. Retarded bands are bracketed on the left of relevant lanes. Note the new retarded species (bracket on the right) only in the WT + Mal-RctB lane (A). The refolded proteins when tested for binding to the 39-mer, did not show any new retarded species (B).
Evidence for heterodimer binding was also obtained by coexpressing WT RctB and RctB fused in frame to chitin-binding domain (RctB-CBD). In these experiments, the two proteins were over-produced in the same cell and crude extracts were mixed with the 12-mer fragment and the binding determined by EMSA (Supplementary Figure S4).
The refolded proteins when tested for binding to the 39-mer, did not show any new retarded species (Figure 5B). This result is expected if RctB binds to 39-mer as monomer, as was also the suggestion from the results of Figure 3.
Chaperones promote binding of RctB both as monomer and as dimer
The chaperones DnaJ and DnaK were routinely used in studies by EMSA described above. When the chaperones were omitted, the binding was less in all cases, indicating that the chaperones promote RctB binding both to iterons and to the 39-mer; both the mutant proteins were similar to the WT RctB in this respect (Supplementary Figure S5). These results are consistent with the results of copy number measurements that indicated that the replication of the mutants has remained largely chaperone dependent (Figure 1).
In the case of iteron-carrying plasmids, the chaperones remodel the initiator protomers in a way that disfavors dimerization (37,38). The increase of monomeric species leads to increased iteron binding. Since RctB seems to bind iterons both as monomer and dimer, we expected that the chaperones would preferentially increase monomer binding. However, both monomer and dimer binding increased, and the results were same whether the protein was WT RctB or the F378S mutant (Figure 6). Although the structural basis of chaperone action on RctB remains to be studied, it appears that the chaperones refold the RctB protomer in a way that they remain competent in dimerization.
Figure 6.

EMSA showing the effect of chaperones on RctB binding to 12-mer. RctB was either WT or the F378S mutant. Binding reactions were performed in two sets identically except that from one set the chaperones DnaJ and DnaK were omitted. DNA shifted by RctB monomer is marked by gray arrow and by RctB dimer by black arrow. Other details are same as in Figure 3.
Handcuffing by RctB mutants
One of the properties of plasmid initiators is that they can pair iterons by forming protein bridges, a process called handcuffing. Participation of the origin iterons in handcuffing either in cis or in trans, is believed to inhibit replication by causing steric hindrance to origin function. Copy-up mutants of plasmid initiators have often been found to be defective in handcuffing, indicating the importance of the mechanism in lowering of plasmid copy number (11). Since RctB can also handcuff iterons (17), reduction of handcuffing could be a mechanism underlying the copy-up phenotype of the RctB mutants. To test this hypothesis, the handcuffing efficiency of the mutants was determined using a fragment with three iterons (the 11-mers of incII). (Fragments with lower number of iterons handcuff poorly and the 39-mer by itself does not handcuff (17).) When normalized for binding, ΔC157 was severely defective in handcuffing, consistent with its copy-up phenotype. In contrast, F378S was at least as efficient as the WT RctB in handcuffing, suggesting that handcuffing is unlikely to be reason for its copy-up phenotype (Figure 7).
Figure 7.

Handcuffing of three 11-mer carrying fragments by RctB. A fragment with three tandem 11-mers of incII (Figure 1) was obtained from pTVC248 and reacted with varying RctB concentrations. The proteins tested were WT, ΔC157 and F378S. One half of the binding mixture was loaded onto 5% polyacrylamide gel to monitor bound DNA by EMSA (bracket B) (top panel). The position of the free probe is marked shown by ‘F’. The other half of the binding mixture was treated with ligase, and after deproteinization, loaded onto 1.2% agarose gel to monitor ladder (L) and monomer circle (C) formation (middle panel). The bottom panel represents graphically the efficiency of ladder formation for the WT and the two mutants. Percentage of ladder for each lane was calculated by taking intensity of the area (marked L) containing all bands above free (F) DNA and monomer circles (C), after subtracting the intensity of the similar area in lane 1 (without RctB) and further dividing by the total intensity of bands.
RctB binding to iterons is cooperative
In our experimental condition, WT RctB bound to single iterons poorly, and majority of the probe remained unbound (Figures 3 and 6). In contrast, when we used multiple iterons, it was possible to achieve saturation of binding (Figure 8A, lane 3). This indicates binding to one site increases the affinity of binding to a neighboring site in cis (positive cooperativity). In contrast to the WT protein, ΔC157 was found to be significantly defective in cooperative binding (Figure 8A; lanes 3 versus 6). Although ΔC157 bound to single iterons more efficiently than WT RctB, it required more protein than the WT to saturate binding to multiple sites, which is also indicative of a defect in cooperative binding (Figure 8A, lanes 3 versus 7). The degree of cooperativity was quantified by estimating cooperativity factor, τ (39). A τ value of greater than 1.4 indicates positive cooperativity, and only WT RctB exceeded this value (Figure 8B).
Figure 8.

Cooperative binding of RctB to the six 12-mers of oriII. (A) The DNA fragment (346 bp) was obtained from pTVC228, and was used at 20 pM. RctB concentrations are shown in the top of the auto-radiograms. The cooperativity factor (τ) was determined following the equation (4 × unbound species) (fully bound species)/(intermediate species)2, and the τ values were plotted in panel (B). All three lanes were used for each protein, and the histograms represent mean values from three gels. (C) Cooperativity was also determined from the Hill plot. In these experiments, binding was performed with a fragment containing two 12-mer sites derived from pTVC148. The Hill coefficient (nH) was determined from the slope of the plot of Log [Free DNA/bound DNA] and Log (RctB concentration). RctB concentrations ranged from 3.3 nM to 17 nM (data not shown).
Cooperativity was also quantified by determining the Hill coefficient, nH. The value of nH > 1 indicates positive cooperativity. For ease of quantification, a fragment with only two iterons was used. The nH value for WT RctB was found to ∼2, confirming that the binding is positively cooperative (Figure 8C). The nH value for ΔC157 was ∼1, indicating that the binding is not cooperative, and for F378S was ∼1.4, indicating that the mutant has lost some of the cooperative binding property. Loss of positive cooperativity thus might be another reason for the copy-up phenotype of the mutants.
DISCUSSION
In bacteria, the control of chromosomal replication is mediated by DnaA. In the three most studied cases, E.coli, Bacillus subtilis and Caulobacter crescentus, the control mechanisms seem to differ significantly among the three, although they are all centered on DnaA (6). The chrII of V. choleare provides a fresh perspective on chromosomal replication control, as it is controlled by the chrII-specific initiator RctB. Nonetheless, here also initiator–origin interaction plays a central role in initiation of DNA replication, as it does in other organisms (40). In this study, we have shown that RctB binds specifically to iterons in two forms, as monomer and dimer, the latter binding being more efficient. Monomer binding, in contrast, increases in mutant initiators that promote replication. This implies that dimer binding could be a previously unrecognized negative regulatory mechanism for chrII replication initiation. In the iteron-family of plasmids, whose replication region has many similarities to that of chrII, the initiators bind to iterons as monomer, although there is an isolated evidence of dimer binding as well (36). The avid dimer binding in the case of chrII suggests that it could be a significant regulatory mechanism, which is not generally the case with iteron-carrying plasmids.
Dimers of plasmid initiators, although they do not bind iterons, they do contribute to the control of replication in a major way. Dimerization reduces monomer concentration and therefore initiator binding, since monomers only bind iterons (35). In fact, in many members of the iteron family of plasmids, chaperones such as DnaJ and DanK, are required to dissociate dimers so that enough monomers are available for initiation (21,22). The chaperones remodel the initiators in a way that reduces dimerization (37,38,41). We also show here that plasmids driven by the chrII origin (oriII) depend on chaperones for optimal replication but the chaperones improve both monomer and dimer binding in vitro, the latter more efficiently (Figure 6 and Supplementary Figure S5), the opposite of what was expected from plasmid studies (28). This suggests that reducing dimerization is not the role of chaperones in chrII replication. Our results lead instead to the following model of specific DNA-binding activity of RctB:
 |
The model also helps to explain the apparent contradictory findings that the chaperones increase dimer binding, a putative inhibitory mechanism, and yet promote replication. At low RctB concentration, the chaperone-mediated increase of active monomers might promote replication since RctB is limiting for replication (20,34). However, if RctB concentration overshoots, the consequent preferential increase in dimer concentration might prevent over-replication. Dimer binding thus can be viewed as a negative feedback mechanism for initiation control. It should be noted that our results do not rule out the possibility that chaperones make only active monomers, as in plasmids, and the monomers dimerize upon interacting with DNA (42). In any event, the negative feedback would apply equally well.
We have shown recently that prevention of over-replication of chrII is mediated not by iterons alone, as in plasmids, but primarily by a second kind of RctB binding site, the 39-mers (17). This was inferred by mutating those binding sites. Here, we have come to the same conclusion by mutating RctB. Both RctB mutants characterized here that allowed over-replication of plasmids driven by oriII, are defective in 39-mer binding. These studies further establish the importance of 39-mer in controlling chrII replication.
Although the mutant initiators confer over-replication, they are still control proficient. The copy number of oriII plasmids decreased when RctB binding sites of incII were added, irrespective of whether the initiator was WT or mutant (Figure 1). The added incII region included two iterons and a 39-mer. Relative to the WT protein, both the mutants exhibit decreased affinity for the 39-mer but increased affinity for the iterons (Figure 2). The increased binding apparently allowed the incII iterons to play a more prominent inhibitory role. This proposal is based on our earlier finding that the iterons of incII become more potent inhibitors when the 39-mers are mutated (17).
Another potential regulatory mechanism revealed in this study is the involvement of cooperative binding of the initiator to the iterons of the origin. It is also intriguing that both the mutants are reduced in cooperative binding. Most of the plasmid initiators appear to bind to iterons in a non-cooperative manner, although under some restricted conditions exceptions have been reported (43). The significance of cooperative binding in the control of replication remains to be studied.
The copy-up phenotype of the two mutants was conferred by two very different changes: one is a deletion of C-terminal 157 amino acids and the other a substitution of a single amino acid far from the C-terminal. In a recent study, selection of regulatory mutants by a different strategy also revealed, among others, these two types of mutant (44). Of the two mutants, ΔC157 is expected to confer a higher copy number: it is more defective in 39-mer binding and more proficient in iteron binding as monomer (Figures 2 and 3). ΔC157 is also more defective in handcuffing, which is believed to be the major mechanism that inhibits plasmid replication. However, F378S seems to be a better initiator in vivo (Figure 1 and Supplementary Figure S2A). It apparently binds to iteron and 39-mer in a sigmoidal fashion, the significance of which remains to be understood. Whether the reduced 39-mer binding fully accounts for the proficiency of the F378S mutant in promoting chrII replication remains to be determined.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–3, Supplementary Figures 1–5 and Supplementary Reference [45].
FUNDING
Funding for open access charge: Intramural Research Program; Center for Cancer Research; National Cancer Institute; National Institutes of Health.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Michael Yarmolinsky for thoughtful comments, Paul Morrison for help with statistical analysis and Subrata Pal for supplying pSP5.
REFERENCES
- 1.Jacob F. The replicon: thirty years later. Cold Spring Harb. Symp. Quant. Biol. 1993;58:383–387. doi: 10.1101/sqb.1993.058.01.045. [DOI] [PubMed] [Google Scholar]
- 2.Dodson M, Echols H, Wickner S, Alfano C, Mensa-Wilmot K, Gomes B, LeBowitz J, Roberts JD, McMacken R. Specialized nucleoprotein structures at the origin of replication of bacteriophage lambda: localized unwinding of duplex DNA by a six-protein reaction. Proc. Natl Acad. Sci. USA. 1986;83:7638–7642. doi: 10.1073/pnas.83.20.7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bramhill D, Kornberg A. A model for initiation at origins of DNA replication. Cell. 1988;54:915–918. doi: 10.1016/0092-8674(88)90102-x. [DOI] [PubMed] [Google Scholar]
- 4.Rozgaja TA, Grimwade JE, Iqbal M, Czerwonka C, Vora M, Leonard AC. Two oppositely oriented arrays of low-affinity recognition sites in oriC guide progressive binding of DnaA during Escherichia coli pre-RC assembly. Mol. Microbiol. 2011;82:475–488. doi: 10.1111/j.1365-2958.2011.07827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duderstadt KE, Chuang K, Berger JM. DNA stretching by bacterial initiators promotes replication origin opening. Nature. 2011;478:209–213. doi: 10.1038/nature10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katayama T, Ozaki S, Keyamura K, Fujimitsu K. Regulation of the replication cycle: conserved and diverse regulatory systems for DnaA and oriC. Nat. Rev. Microbiol. 2010;8:163–170. doi: 10.1038/nrmicro2314. [DOI] [PubMed] [Google Scholar]
- 7.Leonard AC, Grimwade JE. Regulation of DnaA assembly and activity: taking directions from the genome. Annu. Rev. Microbiol. 2011;65:19–35. doi: 10.1146/annurev-micro-090110-102934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Das N, Valjavec-Gratian M, Basuray AN, Fekete RA, Papp PP, Paulsson J, Chattoraj DK. Multiple homeostatic mechanisms in the control of P1 plasmid replication. Proc. Natl Acad. Sci. USA. 2005;102:2856–2861. doi: 10.1073/pnas.0409790102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murotsu T, Matsubara K. Role of an autorepression system in the control of lambda dv plasmid copy number and incompatibility. Mol. Gen. Genet. 1980;179:509–519. doi: 10.1007/BF00271740. [DOI] [PubMed] [Google Scholar]
- 10.Mensa-Wilmot K, Carroll K, McMacken R. Transcriptional activation of bacteriophage lambda DNA replication in vitro: regulatory role of histone-like protein HU of Escherichia coli. EMBO J. 1989;8:2393–2402. doi: 10.1002/j.1460-2075.1989.tb08369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paulsson J, Chattoraj DK. Origin inactivation in bacterial DNA replication control. Mol. Microbiol. 2006;61:9–15. doi: 10.1111/j.1365-2958.2006.05229.x. [DOI] [PubMed] [Google Scholar]
- 12.Leonard AC, Helmstetter CE. Replication patterns of multiple plasmids coexisting in Escherichia coli. J. Bacteriol. 1988;170:1380–1383. doi: 10.1128/jb.170.3.1380-1383.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, et al. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000;406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okada K, Iida T, Kita-Tsukamoto K, Honda T. Vibrios commonly possess two chromosomes. J. Bacteriol. 2005;187:752–757. doi: 10.1128/JB.187.2.752-757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egan ES, Waldor MK. Distinct replication requirements for the two Vibrio cholerae chromosomes. Cell. 2003;114:521–530. doi: 10.1016/s0092-8674(03)00611-1. [DOI] [PubMed] [Google Scholar]
- 16.Rasmussen T, Jensen RB, Skovgaard O. The two chromosomes of Vibrio cholerae are initiated at different time points in the cell cycle. EMBO J. 2007;26:3124–3131. doi: 10.1038/sj.emboj.7601747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Venkova-Canova T, Chattoraj DK. Transition from a plasmid to a chromosomal mode of replication entails additional regulators. Proc. Natl Acad. Sci. USA. 2011;108: 6199–6204. doi: 10.1073/pnas.1013244108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demarre G, Chattoraj DK. DNA adenine methylation is required to replicate both Vibrio cholerae chromosomes once per cell cycle. PLoS Genet. 2010;6:e1000939. doi: 10.1371/journal.pgen.1000939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venkova-Canova T, Srivastava P, Chattoraj DK. Transcriptional inactivation of a regulatory site for replication of Vibrio cholerae chromosome II. Proc. Natl Acad. Sci. USA. 2006;103:12051–12056. doi: 10.1073/pnas.0605120103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pal D, Venkova-Canova T, Srivastava P, Chattoraj DK. Multipartite regulation of rctB, the replication initiator gene of Vibrio cholerae chromosome II. J. Bacteriol. 2005;187:7167–7175. doi: 10.1128/JB.187.21.7167-7175.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wickner S, Hoskins J, McKenney K. Monomerization of RepA dimers by heat shock proteins activates binding to DNA replication origin. Proc. Natl Acad. Sci. USA. 1991;88:7903–7907. doi: 10.1073/pnas.88.18.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishiai M, Wada C, Kawasaki Y, Yura T. Replication initiator protein RepE of mini-F plasmid: functional differentiation between monomers (initiator) and dimers (autogenous repressor) Proc. Natl Acad. Sci. USA. 1994;91:3839–3843. doi: 10.1073/pnas.91.9.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simons RW, Houman F, Kleckner N. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene. 1987;53:85–96. doi: 10.1016/0378-1119(87)90095-3. [DOI] [PubMed] [Google Scholar]
- 24.Fekete RA, Chattoraj DK. A cis-acting sequence involved in chromosome segregation in Escherichia coli. Mol. Microbiol. 2005;55:175–183. doi: 10.1111/j.1365-2958.2004.04392.x. [DOI] [PubMed] [Google Scholar]
- 25.Venkova-Canova T, Saha A, Chattoraj DK. A 29-mer site regulates transcription of the initiator gene as well as function of the replication origin of Vibrio cholerae chromosome II. Plasmid. 2012;67:102–110. doi: 10.1016/j.plasmid.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J, Zwieb C, Wu C, Adhya S. Bending of DNA by gene-regulatory proteins: construction and use of a DNA bending vector. Gene. 1989;85:15–23. doi: 10.1016/0378-1119(89)90459-9. [DOI] [PubMed] [Google Scholar]
- 27.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 28.Das N, Chattoraj DK. Origin pairing (‘handcuffing’) and unpairing in the control of P1 plasmid replication. Mol. Microbiol. 2004;54:836–849. doi: 10.1111/j.1365-2958.2004.04322.x. [DOI] [PubMed] [Google Scholar]
- 29.Miller JH. A Short Course In Bacterial Genetics. A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- 30.Sozhamannan S, Chattoraj DK. Heat shock proteins DnaJ, DnaK, and GrpE stimulate P1 plasmid replication by promoting initiator binding to the origin. J. Bacteriol. 1993;175:3546–3555. doi: 10.1128/jb.175.11.3546-3555.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawasaki Y, Wada C, Yura T. Mini-F plasmid mutants able to replicate in the absence of sigma 32: mutations in the repE coding region producing hyperactive initiator protein. J. Bacteriol. 1991;173:1064–1072. doi: 10.1128/jb.173.3.1064-1072.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishiai M, Wada C, Kawasaki Y, Yura T. Mini-F plasmid mutants able to replicate in Escherichia coli deficient in the DnaJ heat shock protein. J. Bacteriol. 1992;174:5597–5603. doi: 10.1128/jb.174.17.5597-5603.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nordström K. Plasmid R1–replication and its control. Plasmid. 2006;55:1–26. doi: 10.1016/j.plasmid.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Duigou S, Knudsen KG, Skovgaard O, Egan ES, Lϕbner-Olesen A, Waldor MK. Independent control of replication initiation of the two Vibrio cholerae chromosomes by DnaA and RctB. J. Bacteriol. 2006;188:6419–6424. doi: 10.1128/JB.00565-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chattoraj DK. Control of plasmid DNA replication by iterons: no longer paradoxical. Mol. Microbiol. 2000;37:467–476. doi: 10.1046/j.1365-2958.2000.01986.x. [DOI] [PubMed] [Google Scholar]
- 36.Kunnimalaiyaan S, Krüger R, Ross W, Rakowski SA, Filutowicz M. Binding modes of the initiator and inhibitor forms of the replication protein pi to the gamma ori iteron of plasmid R6K. J. Biol. Chem. 2004;279:41058–41066. doi: 10.1074/jbc.M403151200. [DOI] [PubMed] [Google Scholar]
- 37.Dibbens JA, Muraiso K, Chattoraj DK. Chaperone-mediated reduction of RepA dimerization is associated with RepA conformational change. Mol. Microbiol. 1997;26:185–195. doi: 10.1046/j.1365-2958.1997.5691920.x. [DOI] [PubMed] [Google Scholar]
- 38.Giraldo R, Fernández-Tresguerres ME. Twenty years of the pPS10 replicon: insights on the molecular mechanism for the activation of DNA replication in iteron-containing bacterial plasmids. Plasmid. 2004;52:69–83. doi: 10.1016/j.plasmid.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 39.Fekete RA, Frost LS. Characterizing the DNA contacts and cooperative binding of F plasmid TraM to its cognate sites at oriT. J. Biol. Chem. 2002;277:16705–16711. doi: 10.1074/jbc.M111682200. [DOI] [PubMed] [Google Scholar]
- 40.Nielsen O, Lϕbner-Olesen A. Once in a lifetime: strategies for preventing re-replication in prokaryotic and eukaryotic cells. EMBO Rep. 2008;9:151–156. doi: 10.1038/sj.embor.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pak M, Wickner S. Mechanism of protein remodeling by ClpA chaperone Acad. Sci. USA 1997; 94,10485] Proc. Natl Acad. Sci. USA. 1997;94:4901–4906. doi: 10.1073/pnas.94.10.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim B, Little JW. Dimerization of a specific DNA-binding protein on the DNA. Science. 1992;255:203–206. doi: 10.1126/science.1553548. [DOI] [PubMed] [Google Scholar]
- 43.Urh M, York D, Filutowicz M. Buffer composition mediates a switch between cooperative and independent binding of an initiator protein to DNA. Gene. 1995;164:1–7. doi: 10.1016/0378-1119(95)00493-p. [DOI] [PubMed] [Google Scholar]
- 44.Yamaichi Y, Gerding MA, Davis BM, Waldor MK. Regulatory cross-talk links Vibrio cholerae chromosome II replication and segregation. PLoS Genet. 2011;7:e1002189. doi: 10.1371/journal.pgen.1002189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khlebnikov A, Datsenko KA, Skaug T, Wanner BL, Keasling JD. Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiology. 2001;147:3241–3247. doi: 10.1099/00221287-147-12-3241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.