

Abstract
Pulmonary hypertension remains an important cause of morbidity and mortality. Although there is currently no cure, descriptions of defective intracellular trafficking and protein misfolding in vascular cell models of pulmonary hypertension have been recently reported. We tested the hypothesis that activation of the unfolded protein response (UPR) would be associated with the development of severe PH. We investigated activation of the UPR in archival tissues from patients with severe PH, and in the monocrotaline-induced rat model of severe PH. We tested the ability of a pharmacologic agent capable of modulating the UPR to prevent and reverse pulmonary hypertension. We found evidence of an active UPR in archival tissue from humans with PH, but not in control lungs. Similarly, monocrotaline-treated rats demonstrated a significant difference in expression of each of the major arms of the UPR compared to controls. Interestingly, the UPR preceded the appearance of macrophages and the development of lung vascular remodeling in the rats. Treatment of monocrotaline rats with salubrinal, a modulator of the PERK arm of the UPR, attenuated PH and was associated with a decrease in lung macrophages. In culture, pulmonary artery smooth muscle cells with UPR induction produced IL-6 and CCL-2/MCP-1, and stimulated macrophage migration. These effects were abolished by pretreatment of cells with salubrinal. These data support the hypothesis that the UPR may play a role in the pathogenesis of inflammatory vascular remodeling and PH. As such, understanding the functional contributions of the UPR in the setting of PH may have important therapeutic implications.
Keywords: pulmonary hypertension, unfolded protein response, vascular remodeling, monocrotaline
Pulmonary hypertension (PH) is distinguished by an increase in pulmonary arterial pressure and resistance. It is defined as pulmonary artery pressure greater than 25 mmHg at rest or pressure greater than 35 mmHg during exercise. This insidiously progressive disease can be familial or sporadic, and may arise alone or secondary to other disease processes.[1] The incidence of idiopathic primary pulmonary hypertension (IPAH) is approximately six cases per million, and the disease presents more frequently in young women.
The vascular changes associated with severe PH have been well documented. Resistance pulmonary arteries remodel all three layers. The smooth muscle cells within the medial layer hypertrophy, and the number of peripheral arteries decreases.[2] Occlusive neointimal lesions form in smaller pulmonary arteries, and are composed of vascular smooth muscle cells, endothelial cells, and immune cells.[3] In addition, there is increasing evidence of larger pulmonary artery change with respect to wall compliance and stiffening.[4] Imbalances in vasodilators and vasoconstrictors, particularly endothelin-1,[5] genetic mutations,[6–8] extracellular matrix perturbation,[9–10] and a pro-inflammatory milieu[11–12] all contribute to a complex remodeling of the pulmonary vasculature leading to hypertrophy of the right ventricle and cor pulmonale.
The origins of the insult(s) that serve as the genesis of PH pathobiology have long been under intense investigation. Ingestion of diet pills, hypoxia, viruses, and chronic thromboembolic disease have all been implicated.[13–16] We hypothesized that, regardless of etiology, the lung vasculature is severely and chronically stressed under these conditions. In many circumstances of cell or tissue stress, the unfolded protein response (UPR) is activated and plays a number of important roles in both normal and disease processes.[17] When proteins are created in a cellular environment, they are transmitted to the endoplasmic reticulum (ER) to be folded before they are secreted. In response to a cellular stressor resulting in the accumulation of misfolded proteins, the ER stress response has four components: translational attenuation; enhanced expression of ER chaperones; enhanced expression of the components of endoplasmic reticulum associated degradation (ERAD); and induction of apoptosis.[18] When excessive amounts of abnormally folded proteins accumulate within the ER and cytosol of cells, aggregation occurs and cell death pathways are ultimately activated.[19] ER stress has been investigated in various disease contexts and has been linked to the pathobiology of neurodegenerative disorders, atherosclerosis, alcoholic liver disease, viral infection, rheumatoid arthritis, idiopathic pulmonary fibrosis,[20–22] and most recently, severe pulmonary artery hypertension.[23–25]
We hypothesized that one pathobiological mechanism underpinning severe PH could be an underlying protein folding derangement. We therefore investigated whether the UPR is activated in human IPAH and in the monocrotaline (MCT)-induced rat model of severe PH. We first sought to identify whether UPR activation is evident in tissue derived from human PH patients. We found significant evidence of UPR activation in adult PH patient lung sections particularly in areas of vascular remodeling. Using the rat model, we found a similar UPR activation that was robustly concentrated in and around areas of vascular remodeling. Disturbances in the three primary pathways of the UPR were observed early (Week 1) in the disease process, preceded the development of remodeling, and was associated with influx of CD68+ macrophages. Pharmacologic modulation of UPR by salubrinal, in both prevention and reversal experiments, resulted in decreased plasma ET-1, decreased plasma pro-inflammatory cytokines, decreased pulmonary artery pressure and vascular remodeling, and a sharply decreased macrophage influx. UPR activation in cultured rat pulmonary artery smooth muscle cells was sufficient to induce a pro-inflammatory phenotype capable of inducing macrophage migration, an effect blocked by salubrinal. We conclude that UPR activation precedes vascular remodeling associated with severe PH, and UPR modulation prevents and attenuates PH by protection against cell stress and by diminution of macrophages in the pulmonary vasculature.
MATERIALS AND METHODS
Human subjects
Lung samples were obtained from surgical lung biopsies performed for evaluation of pulmonary arterial hypertension. Lung biopsies were evaluated from patients with idiopathic pulmonary artery hypertension (IPAH) and with familial pulmonary artery hypertension (fPAH). Control lung sections (n ≥ 7) were obtained from four subjects and demonstrated normal lung parenchyma. Hemodynamic and demographic data confirming PAH for these patients (and lack of PAH in controls) has already been published elsewhere (personal communication, JE Loyd, Vanderbilt University Medical Center, Nashville, Tenn.). Tissue sections were a kind gift from JE Loyd. We examined tissues (n ≥ 4) from seven patients with IPAH and from eleven patients with FPAH. Bone morphogenetic protein receptor type 2 (BMPR2) mutation status was confirmed for all FAPH.
Animal model of PH
The Institutional Animal Care and Use Committee of the institution approved this study. The characterization of the MCT rat model has been previously summarized[26] and utilized male rats with a single subcutaneous injection of 60 mg/kg at 4–6 weeks of age. Wistar and Lewis strains were used in separate experiments, and no strain differences in response to MCT or salubrinal could be discerned (data not shown).
Lung parenchyma from five animals was obtained for each of the 0-, 1-, 2-, and 3-week time-points. For salubrinal treatment, rats were given MCT as described,[27] plus salubrinal at 1 mg/kg in ≤ 1mL neat DMSO, delivered intraperitoneal, twice weekly commencing either at the time of MCT injection, or two weeks post-MCT. This dose and regimen were selected based on a published regimen for rats[28] and on our own dose—response experiments. The mean right ventricular systolic pressure (mmHg) was 79 ±5, 59 ±4, 35 ±4, 35 ±6 in rats treated with MCT and then salubrinal twice weekly at 0, 0.5, 1.0, and 5.0 mg/kg body weight, respectively. A salubrinal-only control group was included in all subsequent experiments.
Rat hemodynamics and tissue processing
After completion of the treatment period, rats were anaesthetized and the right ventricular systolic pressure was determined by pressure transducer catheter using a subxiphoid approach. The mean right ventricular systolic pressure (mmHg) was 22 ±3, 31 ±4, 45 ±4, 73 ±6 in rats treated with MCT for 0, 1, 2, and 3 weeks, respectively. The chest was then opened, the right and left cardiac atria were dissected from the heart, and the free wall of the heart was isolated and the ratio of right ventricular weight/left ventricle plus septum weight was determined. The Fulton index scores (mass right ventricle/[mass left ventricle + mass septum]) were 0.26±0.03, 0.29±0.03, 0.35±0.03, 0.59±3 for rats treated with MCT for 0, 1, 2, and 3 weeks, respectively. Freshly excised lungs from all animals were inflated with 0.5% low-melt agarose at a constant pressure of 25 cm H2O. Each lung was sectioned transversally through the hilum and included intermediate and peripheral lung tissue. A second sagittal section through the peripheral lung parenchyma was also made. Lung sections were taken from the same lung regions in both the treated and control groups. Frozen and paraffin-embedded tissues were sectioned and prepared for histological analysis.
Immunohistochemistry
Human and rat tissues were sectioned at 5 μm for either hematoxylin and eosin staining or immunohistochemical staining. Activation of UPR was assessed by antibody reactivity to activating transcription factor 6 (ATF6, Imgenex-273 1:100) and C/EBP homologous protein (CHOP, Abcam Ab11419 1:100). Apoptotic cells were identified by immunoreactivity to cleaved caspase-3 antibody (Abcam Ab47131 1:200). The cleaved caspase-3-positive cells were counted and divided by the total number of cells in three randomly chosen high-power field (×400) for each section from each of three animals per group. The mean percentage of caspase-3+ cells for each animal was then obtained by averaging the results of the countings. Macrophages were identified by staining with antibodies against CD68 (AbBiotec 250594 1:200). Secondary antibodies conjugated to fluorescent dyes (Invitrogen), or biotin combined with immunoperoxidase/avidin-biotin, were as per the manufacturer protocol (Vectastain ABC kit, Vector Labs), and either hematoxylin or Nuclear Fast Red (Vector Labs) was used as a counterstain. Avidin/biotin blocking kits (Vector Labs) were used to block endogenous enzyme activity. Antibody isotype negative controls were included with each sample group. Images were acquired at room temperature using a Zeiss Axiovert S100 fitted with Zeiss 20×0.4 numerical aperture (n.a.) and 10×0.3 n.a. objectives and Axiocam camera. Acquisition of images was by Axiovision 4.6 software (Zeiss).
RNA analyses of rat lung
Total RNA was isolated from each rat group (n=≥5) by RNeasy (Qiagen). RNA concentration and relative purity were measured by A260/A280 ratio using a Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies, Inc.) according to the manufacturer's protocols. cDNA was transcribed with the Superscript III First-Strand Synthesis System (Invitrogen). cDNA yield was determined by measuring A260 on the Nanodrop ND-1000. For real-time-PCR of rat lung, primer sequences were used as previously described,[29–30] for the following gene targets: x-box binding protein-1 (XBP1) spliced (Forward 5’-GACTCCGCAGCAGGTG-3’, Reverse 5’-GCGTCAGAATCCATGGGA-3’), XBP1 unspliced (Forward 5’-CAGACTACGTGCGCCTCTGC-3’, Reverse 5’-CTTCTGGGTAGACCTCTGGG-3’), ATF6 (Forward 5’-GGAAGTTACCAAGGCTTCTTTGAC-3’, Reverse 5’-TGGGTGGTAGCTGGTAATAGCA-3’), growth arrest and DNA damage-inducible protein (GADD34, Forward 5’-TTTCTAGGCCAGACACATGG-3’, Reverse 5’-TGTTCCTTTTTCCTCCGTGG-3’), and hypoxanthineguanine phosphoribosyltransferase (HPRT, (Forward 5’-AAGCTTGCTGGTGAAAAGGA-3’, Reverse 5’-CAAGGGCATATCCAACAACA-3’).
Immunoblot analyses
Lysates from rat tissues (n= ≥5 from each rat group) were isolated by addition of either an ice-cold TB or RIPA buffer in the presence of protease, kinase, and phosphatase inhibitors (HALT, Pierce-Thermo Scientific). Proteins (25 μg) were subjected to electrophoresis on 4–12% gradient Bio-Tris gels (NOVEX, Invitrogen) and transferred to PolyScreen PVDF Transfer membrane (NEN Life Science Products) in a Tris-glycine buffer (NOVEX, Invitrogen) containing 10% methanol. Prestained molecular mass marker proteins (Bio-Rad) were used as standards for the SDS-PAGE. Western blots were visualized using Western Blot Chemiluminescence Reagent (GE Amersham). Densitometric band normalization of ATF6, CHOP, phospho-eIF2alpha (Cell Signaling 119A11), and total eIF2alpha (Cell Signaling 9722) relied on beta-actin (Novus Biologicals NB600-501B) loading control blots.
Rat ET-1 ELISA
Plasma was prepared by centrifugation of whole blood collected in heparinized tubes. The concentration of ET-1 was then measured according to the manufacturer's instructions (ADI-900-020A, Enzo Life Sciences).
Rat inflammatory cytokine arrays
Plasma was prepared by centrifugation of whole blood collected in heparinized tubes. The concentrations of cytokines were then measured according to the manufacturer's instructions (Multi-Analyte ELISArray MER-004A, SABiosciences).
Resistance vessel thickness measurements
Images were acquired from smooth muscle actin immunostained lung tissue sections as described above. Morphometry was conducted on digital images using ImageJ software, http://rsb.nih.gov/ij/) as previously described.[31] Measurements included external and lumen diameter derived from circumference and maximal smooth muscle and intimal thickness. Smooth muscle thickness included both medial and intimal smooth muscle and was normalized by expressing as a percentage of the external diameter. Three sections of each rat lung from five animals per group were analyzed.
Cell culture
Pulmonary artery smooth muscle cells (PASMC) and pulmonary artery adventitial fibroblasts (PAF, not shown) were isolated from control rats as previously described.[25] Immunofluorescent staining of endothelium (von Willebrand Factor Dako A0082 1:250) and smooth muscle cells (smooth muscle actin Sigma A2547 1:100) was used to assess cell identity. Macrophages were differentiated from peripheral blood with recombinant human macrophage colony-stimulating factor (M-CSF, eBioscience 14-8769-80) at 10 ng/mL as previously described.[32] UPR induction by tunicamycin (Sigma T7765) was performed essentially as described at 1 μg/mL in phosphate buffered saline vehicle for 24 hours.[33] Salubrinal was applied as previously described[34] at 25 μM in dimethylsulfoxide (Sigma D8418) and media for 30 minutes prior to introduction of tunicamycin or vehicle. Rat macrophage migration assays were performed as previously described using a modified Boyden chamber (Corning).[35]
Statistical analyses
All experiments were performed in triplicate, and data were expressed as means ±SE and analyzed by unpaired Student's t-test for comparison between two groups or one-way ANOVA with post hoc analysis for multiple comparisons. A value of P <0.05 was considered significant.
RESULTS
Induction of the unfolded protein response in patients with PH
The pathogenesis of PH has been linked to hypoxia, viral infection, genetic mutation, and inflammation. The UPR has been shown to play pivotal roles in all of these processes.[18] To establish whether the UPR is activated in lungs from patients with IPAH, we performed immunohistology using antibodies specific for activated ATF6 and the pro-apoptotic UPR protein CCAAT/-enhancer-binding protein homologous protein (CHOP). We found ATF6 predominantly in medial and adventitial layers of large (>500 μm internal diameter) and medium sized pulmonary vessels (Fig. 1C) in lung sections from patients with IPAH but not in controls (Fig. 1A). Furthermore, we found ATF6 expressed in plexiform lesions (Fig. 1E) and in muscularized small pulmonary arterioles in IPAH lung (Fig. 1F). CHOP was similarly expressed in medium to small pulmonary vessels (Fig. 1D) from PH lungs, but not in control lung (Fig. 1B). In some IPAH lung sections, airway epithelium strongly expressed ATF6, as did intraluminal white blood cells (Fig. 1G). No differences in UPR expression or localization were observed between patients with IPAH and FPAH.
Figure 1.
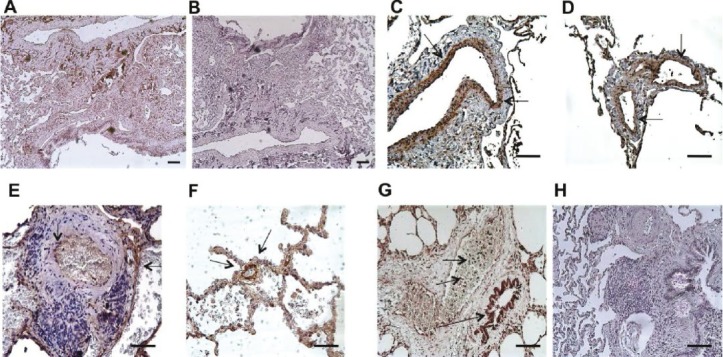
(A-B) Bronchovascular region of control lung biopsy sections showing scant immunopositivity for ATF6 (A) or CHOP (B). (C-D) Adult pulmonary hypertension lung sections show pronounced medial layer expression of ATF6 (C) and, to a lesser extent, CHOP (D). (E) Plexiform lesion with ATF6+ cells. (F) Small remodeled vessel with ATF6+ medial cells. (G) A number of PAH lung sections showed significant macrophage (short arrows) and airway epithelium (longer arrow) expression of ATF6. (H) Negative control using isotype matched antibody. Bar=50 μm. Antigen DAB, arrows; hematoxylin counterstain.
The monocrotaline-induced rat model of PH demonstrates activation of the UPR in lung cells and in macrophages
The MCT rat model of PH is associated with widespread lung cell apoptosis and an acute perivascular inflammation that develops into chronic inflammation with extensive vascular remodeling.[26–27] To determine if the MCT-induced rat model of PH demonstrates activation of the UPR, we repeated the human immunohistological analysis using rat-specific antibodies and also performed whole lung expression analysis by immunoblot and qPCR. In doing so, we closely examined the time course of the development of PH. We found that by two days after MCT injection, expression of ATF6 and CHOP were localized to medial and adventitial layers of pulmonary vessels and to airway epithelium (Figs. 2C and D), compared to a low level of expression in control lungs (Figs. 2A and B). At 21 days post-MCT, we found exuberant expression of ATF6 in the media and adventitia of medium-to-small bronchovascular regions and less so in airway epithelium (Fig. 2E). CHOP was largely absent by three weeks (localized protein, Fig. 2F; whole lung mRNA, Fig. 2I). Using qPCR on whole lung lysates, we found increased transcripts for ATF6, sXBP-1, CHOP, and GADD34 in lungs of MCT treated rats as early as seven days after injection compared to controls (Fig. 2I). The number of active caspase-3 positive cells in PH lungs increased by Week 1, prior to the appearance of remodeling (Fig. 2J). CD68+ macrophages became increasingly organized and restricted to the adventitial layers of medium-to-large bronchovascular areas (Figs. 3A—D). These data indicate acute lung cell activation in the MCT rat model of PH of two arms of the UPR by protein analysis and three arms of the UPR by transcriptional analysis. Furthermore, the pronounced UPR activation and caspase-3 processing we observed occurred prior to any significant macrophage influx, lung vascular remodeling, or hemodynamic change.
Figure 2.
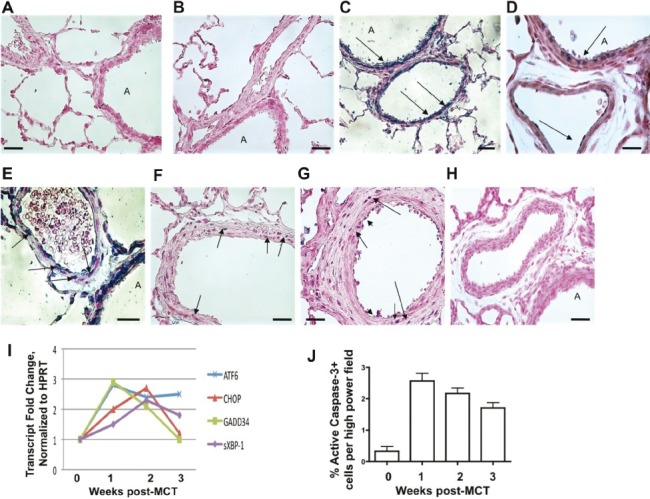
Control rats display limited immunoreactivity for (A) ATF6 and (B) CHOP. Airway epithelial cells and endothelium immunopositive for (C) ATF6 and (D) CHOP appear 2 days postmonocrotaline (arrows). At 3 weeks postmonocrotaline, smooth muscle cells and fibroblasts express (E) ATF6 and (F) CHOP (arrows), and less so in airway epithelium. (G) Active caspase-3 (blue) is evident through all three layers of a pulmonary artery from a rat after 1 week of monocrotaline (arrowheads = endothelial cells, medium arrows = smooth muscle cells, long arrows = adventitial cells). (H) Representative example of negative control using isotype matched antibody. “A” = airway. Black bar = 50 μm. Images are representative of analysis of five fields each from n≥7 lung sections obtained from five rats per group. For all images: Antigen Vector Blue, arrows; fast red counterstain. (I) Monocrotaline rats transcriptionally engage the major arms of the UPR as evidenced by increased ATF6, sXBP-1, GADD34, and also CHOP. Whole lung ATF6 and GADD34 gene expression peaks at Week 1, while CHOP and sXBP-1 levels peak at Week 2. ATF6 and sXBP-1 mRNAs remain elevated after 3 weeks postmonocrotaline, while GADD34 and CHOP levels abate to control levels by Week 3. (J) The % of active caspase-3 positive lung cells increases by 7 days postmonocrotaline, and remains diminishingly so for 3 weeks.
Figure 3.
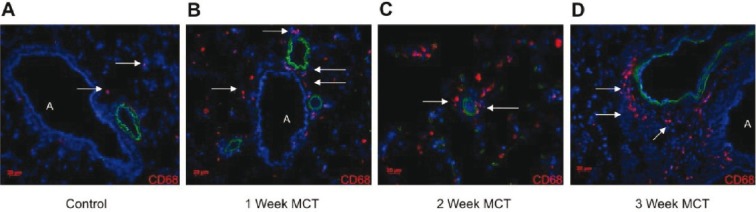
Accumulation of CD68 positive cells into PH rat lung. CD68+ cells (A-D, arrows, Cy3, DAPI nuclei) are sparsely dispersed throughout control lung, but steadily increase with monocrotaline beginning at Week 1. CD68+ cells localize to adventitia of medium to large vessels (B, D) and to media of smaller vessels (C). Note the absence of bronchovascular remodeling in Week 1 compared to Week 3, despite the influx of CD68+ cells. “A” = airway. Red bar = 20 μm. Images are representative of analysis of five fields each from n≥7 lung sections obtained from five rats per group.
Modulation of UPR prevents and partially reverses monocrotaline-induced PH
Based on our immunohistological and qPCR data, we tested the idea that modulation of the UPR could affect the inflammation and potentially the vascular remodeling observed in MCT-induced PH. We treated rats with the ER stress protectant salubrinal, which modulates the selective translation component of the UPR.[36] We performed dose—response experiments and found that intraperitoneal injections of salubrinal at 1.0 mg/kg twice weekly resulted in a statistically significant reduction in right ventricular systolic pressure in MCT rats in both prevention and reversal regimens (Fig. 4A). These findings were corroborated by evidence of significant attenuation of pulmonary vascular remodeling and reduced vessel thickening by salubrinal compared to MCT alone (Fig. 4B). Salubrinal was associated with reduced ET-1 plasma levels (Fig. 4C), which may partially account for the decreased pulmonary artery pressure by virtue of decreased vasoconstriction. Interestingly, MCT rats given salubrinal for prevention displayed decreased lung airway and vascular cell expression of ATF6, CHOP, but not the reversal group (all images taken at 4 weeks post-MCT injection, Figs. 5A–F). CD68+ cells and UPR activation were much less abundant in salubrinal treatment groups compared to MCT alone (Figs. 5G and H). The increased number of active caspase-3 positive cells in PH lungs was greatly attenuated by both prevention and reversal regimens of salubrinal treatment (Fig. 5I). Immunoblot analysis of MCT rat lung lysate revealed temporal increases in both ATF6 and CHOP and a decrease in peIF2alpha, all of which were reversed in rats given salubrinal (Fig. 6A). These findings were consistent with the immunofluorescence results in Figure 5. Salubrinal treated MCT rats had decreased lung transcripts of ATF6 (Fig. 6B) and sXBP-1 (Fig. 6C) compared to MCT alone.
Figure 4.
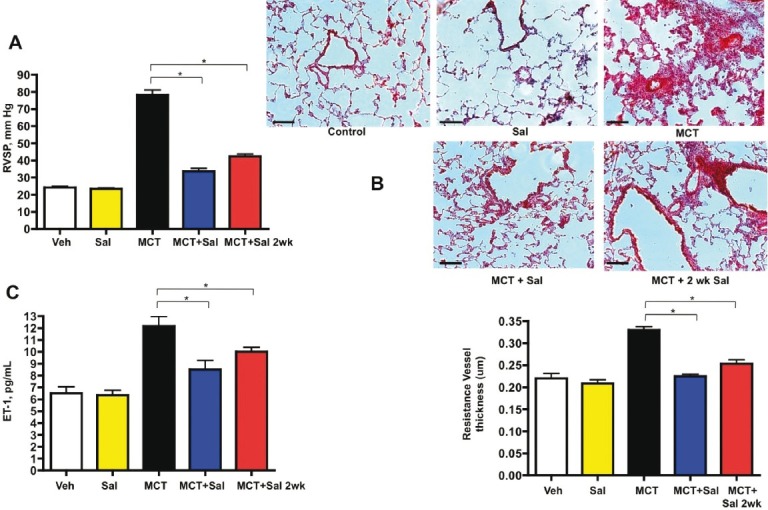
Pulmonary hypertension and vascular remodeling are prevented and partially reversed by the ER stress protectant salubrinal. (A) Right ventricular systolic pressure is significantly reduced by administration of salubrinal at Day 0 or Day 14 postmonocrotaline injection. (B) Hematoxylin and eosin staining of frozen tissue sections shows reduction in bronchovascular remodeling and resistance vessel thickening (bar graph) in salubrinal-treated PH rats compared to monocrotaline alone. (C) Plasma ET-1 levels decrease in salubrinal treated rats compared to monocrotaline alone. Sal = salubrinal given just prior to MCT injection; Sal 2wk = salubrinal given at 2 weeks post-MCT injection. Black bar = 50 μm. * P=<0.05.
Figure 5.
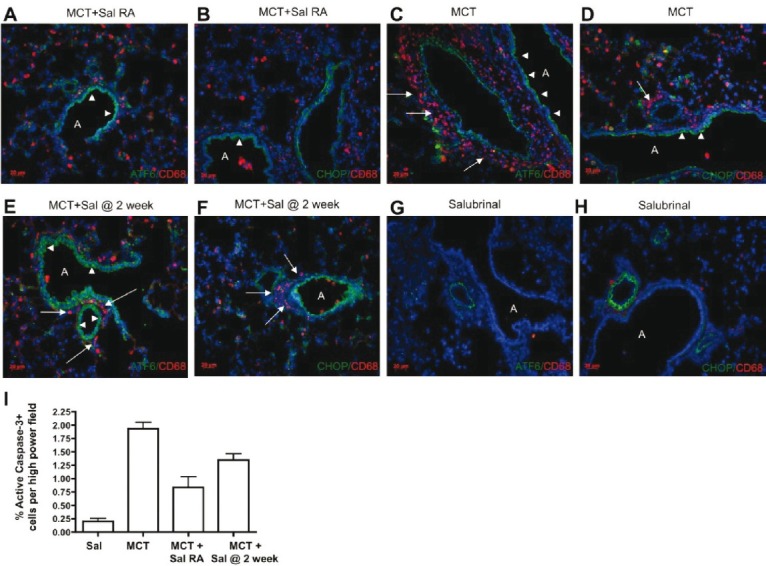
Treatment with the UPR modulator salubrinal decreases UPR localization in bronchovascular cells (arrowheads) and impairs lung influx of macrophages (arrows) in the monocrotaline-induced rat model of PH. Salubrinal was administered twice per week commencing at either Day 0 (Sal right away {RA} or 14 days after (2 weeks) MCT injection. Rat lung sections were tested for immunopositivity for CD68 and ATF6 (A, C, E, G), or CD68 and CHOP (B, D, F, H). Salubrinal largely prevented the appearance of macrophages, and attenuated numbers of ATF6 and CHOP immunopositive cells when given to rats prior to established PH. When given after 2 weeks of established PH, salubrinal treatment was associated with decreased macrophages but with no difference in ATF6 or CHOP positive cells. (G-H) Representative lung staining of control rat given salubrinal for 4 weeks but without monocrotaline. No bronchovascular remodeling or macrophage influx is apparent. No increases in ATF6 or CD68 positivity were noted. For all images, antigen Cy3 or Alexa 488 as indicated, DAPI nuclei. “A” = airway. Red bar = 20 μm. Images are representative of analysis of five fields each from n≥7 lung sections obtained from five rats per group. (I) The % of active caspase-3 positive lung cells postmonocrotaline is diminished by salubrinal treatment, either as a prevention, or (less so) as a “treatment.”
Figure 6.
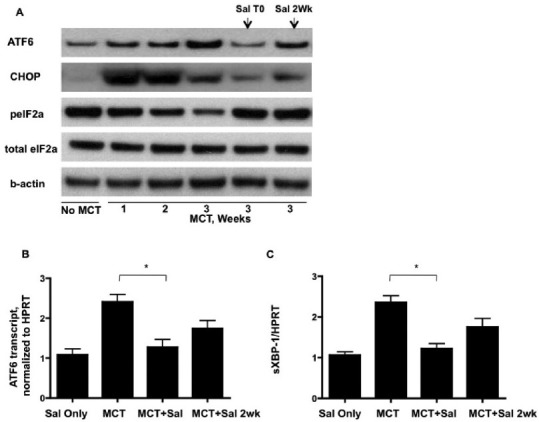
Lung gene expression associated with the unfolded protein response is modulated by systemic administration of salubrinal. (A) Immunoblot analysis of UPR protein expression. ATF6 and pro-apoptotic CHOP increase with progression of monocrotaline-induced PH, but are decreased in salubrinal treated rats. Salubrinal increases the phosphorylation of eIF2alpha, reversing the observed decreases as PH progresses from Weeks 1 to 3. (B) Real-time PCR of rat whole lung reveals that control of the PH-associated increase in ATF6 also occurs at the transcriptional level and is sensitive to salubrinal treatment. (C) The spliced form of XBP-1 increases with PH, but is decreased with salubrinal treatment. * P=<0.05
Increases in plasma pro-inflammatory cytokines in the monocrotaline-induced rat model of PH are attenuated by UPR modulation.
UPR has been linked to inflammation in a variety of pathological settings. Recent studies have shown that pharmacologic modulation of the UPR reduces inflammation and subsequent disease.[37–38] We speculated that plasma from MCT-treated rats would be associated with a pro-inflammatory pattern of cytokines that could be reduced by salubrinal. We found that MCT rats had elevated plasma chemokine (c-c motif) ligand 2/monocyte chemotactic protein-1 (CCL2/MCP-1), granulocyte-macrophage colony-stimulating factor (GM-CSF), CCL5/regulated upon activation, normal T-cell expressed, and secreted (RANTES), interleukin (IL)-6, IL-1b, and IL-2 (Fig. 7). The levels of CCL2/MCP-1, GM-CSF, CCL5, IL-1b, and IL-6 cytokines were reduced in both the prevention and reversal groups of salubrinal treated MCT rats, whereas IL-2 levels remained unchanged.
Figure 7.
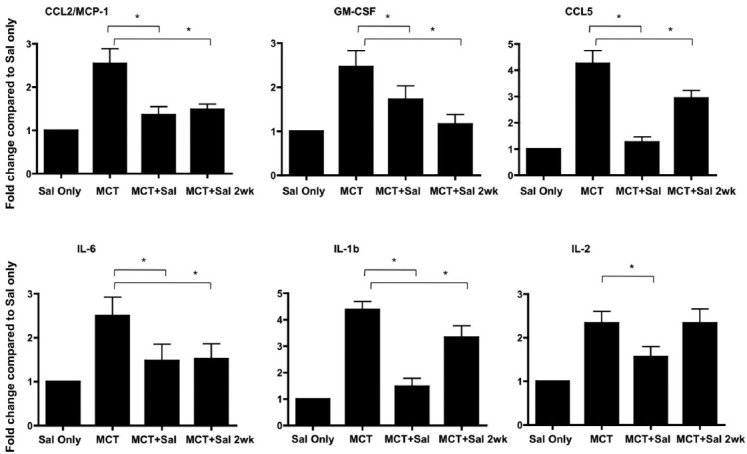
Plasma inflammatory cytokines CCL2/MCP-1, GM-CSF, CCL5, IL-6, IL-1b, and IL-2 are increased in monocrotaline treated rats. Cytokines were measured after salubrinal injecitons alone (Sal only) or after 3 weeks of MCT injection with and without the indicated salubrinal treatments (Sal = salubrinal given just prior to MCT injection; Sal 2wk = salubrinal given at 2 weeks post-MCT injection). Levels of these cytokines (with the exception of IL-2 measured after MCT treatment with salubrinal commencing after 2 weeks) decrease in rats treated with salubrinal in prevention and reversal experiments. * P=<0.05.
UPR activation in pulmonary artery smooth muscle cells and adventitial fibroblasts promotes a pro-inflammatory phenotype capable of recruiting macrophages
To confirm the target cell(s) associated with the prevention and attenuation of PH by salubrinal-mediated UPR modulation, we performed culture assays using pulmonary artery smooth muscle cells (PASMC) and pulmonary artery adventitial fibroblasts (PAF, not shown). We chose these cell types for several reasons: (1) the macrophage influx into the lung following MCT injection was most pronounced in the adventitia around medium-to-large bronchovascular structures (Fig. 3D); (2) because of the interest in the pulmonary artery media and adventitia vis-à-vis inflammation and pulmonary vascular remodeling; and (3) the UPR immunostaining signature was pronounced in these two cell types in human and rat PH lung sections. We induced the UPR using 1 μg/mL tunicamycin, a dose that did not cause widespread cell death (detailed in materials and methods, data not shown). We cultured PASMC with 25 μM salubrinal for one hour prior to treatment with tunicamycin for 24 hours, a regimen based on the literature[34] and our own dosing experiments (data not shown). We found that tunicamycin-treated PASMC (isolated from control rat lungs) significantly increased the migration of rat peripheral blood-derived macrophages compared to control treated PASMC (Fig. 8A). Pretreatment with salubrinal prevented the increase in UPR-associated macrophage migration. Lysates from tunicamycin treated PASMC showed increases in ATF6 and CHOP, with concomitant diminution of peIF2alpha (Figs. 8B and C). Pretreatment with salubrinal abolished the tunicamycin-induced ATF6, CHOP, and peIF2alpha changes. Using ELISA, we measured IL-6 and CCL2/MCP-1 levels in the PASMC supernatants and found a significant increase in production of both in the tunicamycin-treated cells compared to salubrinal-only treated cells (Fig. 8D). Similar results with regard to cytokine and chemokine production as well as macrophage migration were observed when we used rat PAF, and when we used either PAF or PASMC isolated from rats with MCT-induced PH (data not shown). These results confirm that pulmonary vascular cells are cellular targets for induction of the UPR, and when engaged, may promote inflammatory processes of cytokine and chemokine production and secretion, as well as increased macrophage migration.
Figure 8.
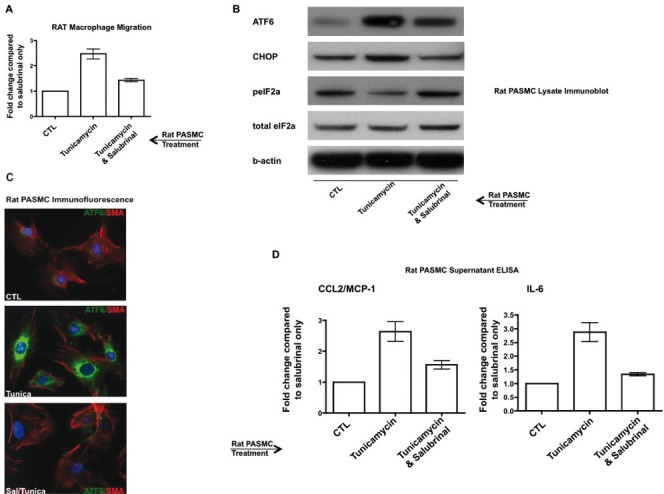
Activation of the UPR in rat pulmonary artery cells can stimulate rat macrophage migration and production of inflammatory cytokines and chemokines, all of which is reversible by salubrinal. (A) Migration of rat peripheral blood macrophages is increased secondary to tunicamycin-UPR induction in pulmonary artery smooth muscle cells (PASMC), and is prevented by salubrinal. The number of macrophages migrating in minimal media was used as a control and arbitrarily set as 1 (CTL). (B-C) UPR induction in PASMC by tunicamycin as assessed by (B) immunoblot, and (C) immunofluorescent staining for ATF6 (green) and smooth muscle actin (SMA, red). Note that salubrinal treatment increases peIF2alpha expression (B). (D) Tunicamycin-treated PASMC secrete pro-migratory and pro-inflammatory cytokines, the production of which is reversible by salubrinal. DAPI nuclei; original magnification = 100×; tunicamycin was used at 1 μg/mL for 24 hours.
DISCUSSION
In this study, we demonstrate an active unfolded protein response in lung sections from patients with IPAH and those derived from the MCT-induced rat model of PH. Our finding of UPR activation prior to widespread macrophage influx, pulmonary vascular remodeling, and the development of pulmonary hypertension strongly suggests that UPR activation is an early pathobiological event, at least in the MCT-induced rat model of PH. Furthermore, modulation of one arm of the UPR by salubrinal experimentally prevented PH and attenuated well-established PH by mechanisms that may be associated with lung influx of macrophages. Finally, we provide evidence that UPR modulation in lung fibroblasts and smooth muscle cells may contribute to the inflammatory process observed in the MCT rat model of PH, since administration of salubrinal abolishes increased levels of pro-inflammatory cytokines and chemokines, both in vivo and also using rat PAF and PASMC in culture.
Our results in the MCT rat model raise the possibility that derangement of ER homeostasis in the lung may help to establish and maintain the inflammation observed in patients with PH.[39] Admittedly, in this study we did not directly confront the definition of “derangement” of the UPR in the context of MCT-induced lung injury, other than documenting a substantial activation of the system. The unfolded protein response can be triggered by a wide variety of stimuli including hypoxia, oxidative stress, gene mutation (and subsequent protein misfolding and/or transport dysregulation) and glucose deprivation,[40–48] most of which have been implicated in PH. In the MCT rat model of PH, we found early expression of both ATF6 and CHOP in the pulmonary vasculature and similar results were obtained in primary cell culture. This is consistent with previous reports in this model of detection of apoptosis in whole lung tissue[49] and pulmonary artery endothelial and smooth muscle cells,[50,51] as well as UPR activation in alveolar epithelial and pulmonary artery endothelial cell cells treated with MCT-pyrrole.[52] We noted that ATF6 was predominantly localized to pulmonary artery smooth muscle cells and airway epithelium (Figs. 2C and E), while CHOP localized to nuclei and cytoplasm of airway epithelium and pulmonary artery endothelial cells and smooth muscle cells (Figs. 2D and F). This may reflect an as yet unidentified differential sensitivity to UPR-mediated apoptosis in airway versus vascular compartments in this model, and should be investigated further. Indeed, expression of both ATF6 and CHOP in the airways of rats two days after MCT injection (and prior to macrophage influx) suggests that ER stress and homeostatic disturbance of nonvascular lung cells may contribute to the disease process in the vasculature. An accumulation of misfolded proteins can trigger a cellular survival response in the endoplasmic reticulum, as have been observed in rheumatoid arthritis synoviocytes.[53] The possibility that UPR is a contributing pathway towards the establishment of antiapoptotic/pro-survival lung cell phenotypes in PH is intriguing.
Why would there be such a robust UPR activation in PH and how might the UPR participate in PH disease mechanisms? Using time course analysis of lung tissue from the MCT rats, we found that the UPR is engaged prior to vascular remodeling, which suggests at least the potential for causal roles for the UPR. MCT is a toxin that causes pulmonary hypertension and multiorgan pathology in the rat, and has been recently suggested to be a poor model of angio-obliterative pathobiological components of the disease.[54] In vascular cell culture, MCT causes activation of IRE1,[52] an endoribonucleolytic effector of XBP-1, which we too found robustly spliced in the MCT rat lung tissue compared to controls (Figs. 2H and 6C). Furthermore, in both the MCT and hypoxia rat models of PH compared to controls, proteomic analyses reveal that increased expression of UPR family proteins ERp29 and ERp57 are part of a lung protein pattern change characterized by differential expression of proteins governing vasoconstriction and vascular remodeling.[55] Apart from MCT or hypoxia, transient expression of mutant bone morphogenetic receptors (using mutations sequenced from patients with PH) fused to green fluorescent protein results in dysregulated trafficking and golgi retention.[24] Taken together with our work here, these studies collectively point to a central role for the endoplasmic reticulum and the UPR in the development of PH in so far as vascular cell insult and cell death (MCT), deranged oxygen tension and oxidative stress (hypoxia), and expression of mutant receptors.
In the MCT rats, we were surprised to find cells expressing ATF6 and/or CHOP that had morphology reminiscent of macrophages (Figs. 1G and 5D). Indeed, in lung sections from MCT rats, we found early appearance and later abundance of CD68+ cells (Fig. 3). UPR in macrophages can be induced by nitric oxide,[56] neuroendocrine hormones,[57] and increased trafficking of cholesterol[58], all of which correlate with immunostimulatory activities. We are currently quantitatively and qualitatively assessing phagocytic cells by flow cytometry in both peripheral blood and lung single cell suspensions, (not shown). Interestingly, UPR-activated macrophages in advanced atherosclerotic lesions accumulate large amounts of unesterified cholesterol which leads to both cell death and to lesional necrosis, promoting plaque instability[58]. It has previously been shown that clodronate-mediated depletion of circulating phagocytic cells in hypoxic rats prevented pulmonary vascular remodeling.[59] Similarly, when we administered salubrinal to MCT rats, we observed a dramatic decrease in the number of CD68+ cells to the lung bronchovasculature in the context of lowered right ventricular systolic pressure, reduced plasma ET-1, and blunted vascular remodeling (Figs. 4 and 5). This was associated with increased lung peIF2alpha, decreased ATF6, CHOP, and XBP-1, and a reduction in plasma chemokines and cytokines (Figs. 6 and 7). These results collectively suggest that UPR engagement may directly link inflammation to the vasoconstriction and vascular remodeling associated with PH, perhaps by both resident and nonresident phagocytic cell activities. It has been previously reported that high levels of ET-1 can induce the UPR in pulmonary artery smooth muscle cells and that such stimulation leads to a pro-inflammatory/pro-adhesive phenotype competent to recruit inflammatory cells.[25] Further studies should delineate the mechanistic details of such a paradigm.
In conclusion, our study establishes the UPR as an important associative finding in PH. We also provide the first data to suggest that the UPR might be playing important mechanistic roles in macrophage recruitment in PH and perhaps the establishment of a persistent pro-inflammatory environment in the bronchovasculature. Further investigations that more mechanistically describe the contributions of the UPR in specific lung cell types might lead to therapeutic strategies for PH patients that could potentially be combined with current modalities.
Footnotes
Source of Support: This work was supported by National Institutes of Health Specialized Centers of Clinically Oriented Research Grant HL-084923–02 and National Institutes of Health Program Project Grant HL-014985–35 (K.R.S.), the Brigid Hope Research Fund, and the Leah Bult Pulmonary Hypertension Research Fund.
Conflict of Interest: None declared.
REFERENCES
- 1.Humbert M. Update in pulmonary hypertension 2008. Am J Respir Crit Care Med. 2009;179:650–6. doi: 10.1164/rccm.200901-0136UP. [DOI] [PubMed] [Google Scholar]
- 2.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S20–31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2008;118:2372–9. doi: 10.1172/JCI33452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lammers SR, Kao PH, Qi HJ, Hunter K, Lanning C, Albietz J, et al. Changes in the structure-function relationship of elastin and its impact on the proximal pulmonary arterial mechanics of hypertensive calves. Am J Physiol Heart Circ Physiol. 2008;295:H1451–9. doi: 10.1152/ajpheart.00127.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhodes CJ, Davidson A, Gibbs JS, Wharton J, Wilkins MR. Therapeutic targets in pulmonary arterial hypertension. Pharmacol Ther. 2009;121:69–88. doi: 10.1016/j.pharmthera.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Davies RJ, Morrell NW. Molecular mechanisms of pulmonary arterial hypertension: Role of mutations in the bone morphogenetic protein type II receptor. Chest. 2008;134:1271–7. doi: 10.1378/chest.08-1341. [DOI] [PubMed] [Google Scholar]
- 7.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26:81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 8.Yeager ME, Halley GR, Golpon HA, Voelkel NF, Tuder RM. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res. 2001;88:E2–E11. doi: 10.1161/01.res.88.1.e2. [DOI] [PubMed] [Google Scholar]
- 9.Papakonstantinou E, Kouri FM, Karakiulakis G, Klagas I, Eickelberg O. Increased hyaluronic acid content in idiopathic pulmonary arterial hypertension. Eur Respir J. 2008;32:1504–12. doi: 10.1183/09031936.00159507. [DOI] [PubMed] [Google Scholar]
- 10.Chapados R, Abe K, Ihida-Stansbury K, McKean D, Gates AT, Kern M, et al. ROCK controls matrix synthesis in vascular smooth muscle cells: coupling vasoconstriction to vascular remodeling. Circ Res. 2006;99:837–44. doi: 10.1161/01.RES.0000246172.77441.f1. [DOI] [PubMed] [Google Scholar]
- 11.Tuder RM, Voelkel NF. Pulmonary hypertension and inflammation. J Lab Clin Med. 1998;132:16–24. doi: 10.1016/s0022-2143(98)90020-8. [DOI] [PubMed] [Google Scholar]
- 12.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 104:236–44. doi: 10.1161/CIRCRESAHA.108.182014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voelkel NF, Cool CD, Flores S. From viral infection to pulmonary arterial hypertension: A role for viral proteins? AIDS. 2008;22:S49–53. doi: 10.1097/01.aids.0000327516.55041.01. [DOI] [PubMed] [Google Scholar]
- 14.MacLean MR. Pulmonary hypertension, anorexigens and 5-HT: pharmacological synergism in action? Trends Pharmacol Sci. 1999;20:490–5. doi: 10.1016/s0165-6147(99)01389-9. [DOI] [PubMed] [Google Scholar]
- 15.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ Res. 2006;99:675–91. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 16.Sacks RS, Remillard CV, Agange N, Auger WR, Thistlethwaite PA, Yuan JX. Molecular biology of chronic thromboembolic pulmonary hypertension. Semin Thorac Cardiovasc Surg. 2006;18:265–76. doi: 10.1053/j.semtcvs.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- 18.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 19.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–62. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–58. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 21.Tsukano H, Gotoh T, Endo M, Miyata K, Tazume H, Kadomatsu T, et al. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2010;30:1925–32. doi: 10.1161/ATVBAHA.110.206094. [DOI] [PubMed] [Google Scholar]
- 22.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–46. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sehgal PB, Mukhopadhyay S, Xu F, Patel K, Shah M. Dysfunction of Golgi tethers, SNAREs, and SNAPs in monocrotaline-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1526–42. doi: 10.1152/ajplung.00463.2006. [DOI] [PubMed] [Google Scholar]
- 24.Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, et al. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11:1517–25. doi: 10.1093/hmg/11.13.1517. [DOI] [PubMed] [Google Scholar]
- 25.Yeager ME, Belchenko DD, Nguyen CM, Colvin KL, Ivy DD, Stenmark KR. Endothelin-1, the unfolded protein response, and persistent inflammation-role of pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2012;46:14–22. doi: 10.1165/rcmb.2010-0506OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–32. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 27.Hill KR. Hepatic veno-occlusive disease produced experimentally in rats by the injection of monocrotaline. Lancet. 1958;1:623. doi: 10.1016/s0140-6736(58)90874-2. [DOI] [PubMed] [Google Scholar]
- 28.Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, et al. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci. 2007;27:901–908. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu HY, Minamino T, Tsukamoto O, Sawada T, Asai M, Kato H, et al. Overexpression of endoplasmic reticulum-resident chaperone attenuates cardiomyocyte death induced by proteasome inhibition. Cardiovasc Res. 2008;79:600–10. doi: 10.1093/cvr/cvn128. [DOI] [PubMed] [Google Scholar]
- 30.Jayanthi S, McCoy MT, Beauvais G, Ladenheim B, Gilmore K, Wood W, 3rd, et al. Methamphetamine induces dopamine D1 receptor-dependent endoplasmic reticulum stress-related molecular events in the rat striatum. PLoS One. 2009;4:e6092. doi: 10.1371/journal.pone.0006092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Achcar RO, Yung GL, Saffer H, Cool CD, Voelkel NF, Yi ES. Morphologic changes in explanted lungs after prostacyclin therapy for pulmonary hypertension. Eur J Med Res. 2006;11:203–7. [PubMed] [Google Scholar]
- 32.Tamaki Y, Sasaki K, Sasaki A, Takakubo Y, Hasegawa H, Ogino T, et al. Enhanced osteolytic potential of monocytes/macrophages derived from bone marrow after particle stimulation. J Biomed Mater Res B Appl Biomater. 2008;84:191–204. doi: 10.1002/jbm.b.30861. [DOI] [PubMed] [Google Scholar]
- 33.Gess B, Hofbauer KH, Wenger RH, Lohaus C, Meyer HE, Kurtz A. The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1-Lalpha. Eur J Biochem. 2003;270:2228–35. doi: 10.1046/j.1432-1033.2003.03590.x. [DOI] [PubMed] [Google Scholar]
- 34.Chinta SJ, Poksay KS, Kaundinya G, Hart M, Bredesen DE, Andersen JK, et al. Endoplasmic reticulum stress-induced cell death in dopaminergic cells: effect of resveratrol. J Mol Neurosci. 2009;39:157–68. doi: 10.1007/s12031-008-9170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sacerdote P, Massi P, Panerai AE, Parolaro D. In vivo and in vitro treatment with the synthetic cannabinoid CP55, 940 decreases the in vitro migration of macrophages in the rat: Involvement of both CB1 and CB2 receptors. J Neuroimmunol. 2000;109:155–63. doi: 10.1016/s0165-5728(00)00307-6. [DOI] [PubMed] [Google Scholar]
- 36.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–9. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 37.Nakka VP, Gusain A, Raghubir R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox Res. 2010;17:189–202. doi: 10.1007/s12640-009-9110-5. [DOI] [PubMed] [Google Scholar]
- 38.Verfaillie T, Garg AD, Agostinis P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2010 Aug 21; doi: 10.1016/j.canlet.2010.07.016. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 39.Voelkel NF, Cool C, Lee SD, Wright L, Geraci MW, Tuder RM. Primary pulmonary hypertension between inflammation and cancer. Chest. 1998;114:225S–230S. doi: 10.1378/chest.114.3_supplement.225s. [DOI] [PubMed] [Google Scholar]
- 40.Sawada N, Yao J, Hiramatsu N, Hayakawa K, Araki I, Takeda M, et al. Involvement of hypoxia-triggered endoplasmic reticulum stress in outlet obstruction-induced apoptosis in the urinary bladder. Lab Invest. 2008;88:553–63. doi: 10.1038/labinvest.2008.21. [DOI] [PubMed] [Google Scholar]
- 41.Gregersen N, Bross P. Protein misfolding and cellular stress: An overview. Methods Mol Biol. 2010;648:3–23. doi: 10.1007/978-1-60761-756-3_1. [DOI] [PubMed] [Google Scholar]
- 42.Maitra M, Wang Y, Gerard RD, Mendelson CR, Garcia CK. Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J Biol Chem. 2010;285:22103–13. doi: 10.1074/jbc.M110.121467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shang J, Gao N, Kaufman RJ, Ron D, Harding HP, Lehrman MA. Translation attenuation by PERK balances ER glycoprotein synthesis with lipid-linked oligosaccharide flux. J Cell Biol. 2007;176:605–16. doi: 10.1083/jcb.200607007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen J, Prywes R. ER stress signaling by regulated proteolysis of ATF6. Methods. 2005;35:382–9. doi: 10.1016/j.ymeth.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 45.Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol. 2001;3:24–9. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- 46.Iwakoshi NN, Lee AH, Glimcher LH. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol Rev. 2003;194:29–38. doi: 10.1034/j.1600-065x.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- 47.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–4. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 48.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–9. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allen JR, Carstens LA. Pulmonary vascular occlusions initiated by endothelial lysis in monocrotaline-intoxicated rats. Exp Mol Pathol. 1970;13:159–71. doi: 10.1016/0014-4800(70)90003-1. [DOI] [PubMed] [Google Scholar]
- 50.Jones PL, Rabinovitch M. Tenascin-C is induced with progressive pulmonary vascular disease in rats and is functionally related to increased smooth muscle cell proliferation. Circ Res. 1996;79:1131–42. doi: 10.1161/01.res.79.6.1131. [DOI] [PubMed] [Google Scholar]
- 51.Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6:698–702. doi: 10.1038/76282. [DOI] [PubMed] [Google Scholar]
- 52.Mukhopadhyay S, Sehgal PB. Discordant regulatory changes in monocrotaline-induced megalocytosis of lung arterial endothelial and alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1216–26. doi: 10.1152/ajplung.00535.2005. [DOI] [PubMed] [Google Scholar]
- 53.Yoo SA, You S, Yoon HJ, Kim DH, Kim HS, Lee K, et al. A novel pathogenic role of the ER chaperone GRP78/BiP in rheumatoid arthritis. J Exp Med. 2012;209:871–86. doi: 10.1084/jem.20111783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, et al. The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol. 2012;302:L363–9. doi: 10.1152/ajplung.00212.2011. [DOI] [PubMed] [Google Scholar]
- 55.Laudi S, Steudel W, Jonscher K, Schöning W, Schniedewind B, Kaisers U, et al. Comparison of lung proteome profiles in two rodent models of pulmonary arterial hypertension. Proteomics. 2007;7:2469–78. doi: 10.1002/pmic.200600848. [DOI] [PubMed] [Google Scholar]
- 56.Renna M, Faraonio R, Bonatti S, De Stefano D, Carnuccio R, Tajana G, et al. Nitric oxide-induced endoplasmic reticulum stress activates the expression of cargo receptor proteins and alters the glycoprotein transport to the Golgi complex. Int J Biochem Cell Biol. 2006;38:2040–8. doi: 10.1016/j.biocel.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 57.Yang C, Zhou JY, Zhong HJ, Wang HY, Yan J, Liu Q, et al. Exogenous norepinephrine correlates with macrophage endoplasmic reticulum stress response in association with XBP-1. J Surg Res. 2011;168:262–71. doi: 10.1016/j.jss.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 58.Kedi X, Ming Y, Yongping W, Yi Y, Xiaoxiang Z. Free cholesterol overloading induced smooth muscle cells death and activated both ER- and mitochondrial-dependent death pathway. Atherosclerosis. 2009;207:123–30. doi: 10.1016/j.atherosclerosis.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 59.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–69. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]