

Abstract
Rationale
The collateral circulation is tissue- and life-saving in obstructive arterial disease. Disappointing outcomes in clinical trials aimed at augmenting collateral growth highlight the need for greater understanding of collateral biology.
Objective
The role of endothelial nitric oxide synthase (eNOS) in forming native (preexisting) collaterals and remodeling in obstructive disease are unknown or controversial issues, respectively.
Methods and Results
We compared the native collateral circulation in healthy tissue and collateral remodeling after femoral artery ligation (FAL) in wild-type and eNOS-knockout (KO) mice. Perfusion after FAL fell further in adult eNOS-KOs, in association with fewer native collaterals in hindlimb (confirmed in brain). This was not attributable to impaired collateral formation in the embryo-neonate, but rather from collateral loss during growth to adulthood. Compared to wild-type, eNOS-KOs evidenced reduced collateral remodeling, angiogenesis, and flow-mediated dilation of the arterial bed supplying the collaterals, resulting in lower perfusion and greater ischemic injury at all time points over 21 days following FAL. To probe the mechanism for impaired remodeling, we performed genome-wide expression profiling of isolated, remodeling hindlimb collaterals 24 hour after FAL. Upregulation of genes encoding cytokines/chemokines, inflammatory, stress response, and cell cycle proteins was evident in wild-type mice. In contrast, expression was lower in 40 of 44 cell cycle genes in eNOS-KO mice, in association with impaired proliferation of vascular wall cells.
Conclusions
Our findings suggest a novel role for eNOS in maintaining native collateral density during natural growth to adulthood and in collateral remodeling in obstructive disease, the latter through regulation of cell proliferation.
Keywords: angiogenesis, arteriogenesis, cell cycle, collateral circulation, endothelial nitric oxide synthase, nitric oxide, vascular remodeling
Obstructive arterial diseases continue to be the leading cause of morbidity and mortality in developed countries. Tissue damage caused by arterial narrowing or occlusion, eg, in acute myocardial infarction or stroke, or in chronic cardiac, brain and critical limb ischemia, is largely determined by the severity and duration of obstruction, the size of the territory supplied by the blocked artery, the density and diameters of the native (preexisting) collateral circulation, the amount of collateral growth (outward remodeling), the extent of angiogenesis, and the susceptibility of the tissue to ischemia, inflammation and thrombosis. On obstruction of a major artery, the presence of a collateral circulation connecting its downstream tree to an adjacent arterial tree serves as a natural-bypass system by providing retrograde perfusion of the tissue distal to the occlusion. Thus, the native conductance and subsequent expansion of the collateral circulation in disease have tissue/life-saving significance.1
Recent work indicates that the density and diameter of preexisting collaterals in healthy adult mice can be strongly affected by variants in genes that regulate collateral formation during development.2–4 In normal tissues at rest, flow and shear stress in the midzone of native collaterals is low-to-negligible and oscillatory, owing to little or no pressure gradient between the cross-connected arterial trees.5 Significant obstruction of a conduit artery supplying a tree with collateral connections creates a steep pressure gradient across the collaterals that induces a large increase in shear stress. If this persists, collateral outward remodeling ensues over the subsequent days-to-weeks, forming large, tortuous, conduit arteries by a process termed “arteriogenesis.”6 Collateral remodeling is inhibited by diabetes, dyslipidemia, atherosclerosis, hypertension, oxidative stress, aging and smoking.7 Although the inhibitory mechanisms have not been resolved, these diseases have in common impaired endothelial nitric oxide synthase (eNOS) activity and/or NO bioavailability, collectively termed “endothelial dysfunction.”
eNOS-derived NO is an important mediator of ischemic angiogenesis.8,9 However, unlike angiogenesis, collateral remodeling is induced by increased shear stress rather than ischemia,6,10 and involvement of eNOS/NO in this process remains controversial. Acute increase in shear stress upregulates eNOS expression and phosphorylation and promotes its intracellular translocation in endothelial cells.11,12 Increased shear also causes, through activation of eNOS, vascular smooth muscle relaxation of arteries (flow-mediated dilation),13 which then outwardly remodel in response to chronic dilation. These findings suggest eNOS may be involved in collateral remodeling. In support of this, a recent study in zebrafish embryos suggested that formation of a collateral-like network after obstruction of the aorta may be dependent on nitric oxide (NO) and myeloid cells.10 Furthermore, Yu, Sessa, and colleagues found that eNOS-KO mice exhibited impaired arteriogenesis in a hindlimb ischemia model, in association with reduced mural cell recruitment that could be reversed by local eNOS overexpression.14 Likewise, the NO donor sodium nitrite augments collateral remodeling.15 In contrast to these studies, Mees et al,16 using eNOS-KO and overexpressing mice in a hindlimb ischemia model, suggested that the eNOS/NO pathway did not impact collateral remodeling but instead regulated NO-mediated dilation of collateral vessels after arterial occlusion.
Further complicating the picture, a potential role of eNOS/NO in the formation or maintenance of the native collateral circulation has not been studied. Such an involvement could affect interpretation of previous studies, because recovery from hindlimb ischemia would be significantly impacted by reduction in the number or diameter of the collaterals present before ligation. The observations that vascular endothelial growth factor (VEGF) is important in collateral formation in the embryo and maintenance after birth3 and in remodeling in ischemia,3,17 together with the prominent position of eNOS in downstream VEGF signaling, prompted us to hypothesize that reduced eNOS expression may impair collateral formation and/or maintenance, resulting in reduced native collateral capacity in the adult.
The purpose of this study was to study the role of eNOS in native collateral formation and address the controversies regarding eNOS in collateral remodeling in obstructive disease, as well as to further define the molecular mechanisms governing collateral remodeling. We found that eNOS-KO mice have a greater loss of flow immediately after femoral artery ligation (FAL), compared to wild-type mice, in association with lower native collateral density in hindlimb before ligation, a finding confirmed in a second tissue (brain). This loss was not attributable to impaired collateral formation in the embryo or maturation in the neonate, but rather from a loss during growth to adulthood. eNOS deficiency also caused sharply lower flow and more severe ischemic injury at all time points during 3 weeks after FAL, in association with reduced collateral remodeling, angiogenesis and flow-mediated dilation of the arterial bed supplying the collaterals. Genome-wide array analysis of isolated collaterals after FAL revealed upregulation of genes encoding cytokines/chemokines, inflammatory, stress response and cell cycle proteins in wild-type and eNOS-KO mice. However, eNOS-KO mice failed to upregulate 40 of 44 cell cycle genes, in association with impaired proliferation of vascular wall cells. These findings demonstrate an important role of eNOS in maintaining native collateral density in the adult and in mediating collateral remodeling in obstructive arterial disease.
Methods
An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org.
Homozygous eNOS-KO mice and C57BL/6 wild-type were 12 to 14 week-old. FAL was by ligation proximal to the popliteal artery and distal to the lateral caudal femoral artery (LCFA) (Figure 1A, left, green arrows, less severe model)2,18 or proximal to the LCFA for more severe ischemia (Figure 1A, left, red arrow). The superior epigastric artery was ligated in both models (Figure 1A, left, blue arrow). Analyses were conducted blindly. Hindlimb perfusion was obtained using a perfusion imager modified for high resolution and depth of penetration.18,19 “Appearance” and “use” scores were obtained.2 Number of native pial collaterals interconnecting the middle and anterior cerebral artery trees was determined by imaging of yellow MicrofilP casting after heparinization, vasodilation and fixation,2,18 and in embryonic day (E)18.5 embryos postnatal day (P)1 pups by whole-mount immunohistochemistry with anti-NG2 antibody. Twenty-one days after FAL or after acute FAL in naïve mice, the abdominal aorta was cannulated, followed by maximal dilation, heparinization, fixation, and MicrofilP casting. Collaterals in the abductor/adductor were imaged either by high resolution x-ray arteriography,2 directly by successive removal of overlying muscle fibers after alcohol-methyl salicylate clearing, or by cross-section histomorphometry (see below). Intact collaterals were identified according to the Longland criteria.20 Histomorphometry for collateral diameter, capillary density and immunohistochemical staining was as detailed previously.2 Proliferation was measured by 5-bromodeoxyuridine (BrdUrd) incorporation. LCFA diameter was measured by stereomicroscope and flow velocity was measured with a Doppler microprobe. Microarray analysis of gene expression was performed on microdissected anterior and posterior gracilis collaterals 24 hour after unilateral femoral ligation and after acute contralateral ligation (control) (Figure 1A, left, black arrows). For each RNA replicate, collaterals from 15 mice (30 ligated for 24 hour and 30 acutely ligated) were pooled. Three replicates for C57BL/6 and eNOS-KO each were hybridized. Real time quantitative RT-PCR was performed for representative genes in each functional gene category identified in the array studies. All data were obtained while blinded to mouse strain.
Figure 1. Impaired recovery of perfusion in eNOS-KO mice after FAL.

A, MicrofilP-casted mouse hindlimb illustrating vasculature before (left) and 21 days after (right) FAL. 1, Proximal femoral artery. 2, LCFA. 3, Superior epigastric artery. 4 and 5, Gracilis collaterals. 6, Distal femoral artery. Green arrows and bars indicate ligation sites. Red arrow indicates higher ligation site in experiments with the second group of mice. White arrows indicate direction of flow before and after FAL. Black arrows indicate the collateral segments microdissected for RNA extraction in microarray analysis. B and C, Laser Doppler perfusion images of adductor and plantar, with region of interest marked with dotted lines (right), and summary data (left). Data in this and all subsequent figures are means±SEM. Two-way ANOVA followed by Dunn–Bonferroni t test; *P<0.05, **P<0.01, ***P<0.001.
Results
eNOS-KO Mice Have Reduced Perfusion and Greater Ischemic Injury After FAL
To induce moderate ischemia, we ligated the superior epigastric and femoral arteries distal to the LCFA and the femoral artery just proximal to the bifurcation of popliteal and saphenous arteries (Figure 1A). This induces a rapid increase in flow across the collaterals of the thigh, including the 2 readily identifiable superficial ones connecting the medial and lateral arterial trees that supply the anterior and posterior gracilis muscles and which cross-connect the LCFA and saphenous artery. We and others have shown that this model causes ischemia in the foot, calf and lateral-most thigh, but not in the collateral zone in the center of the thigh.2,3,18,19,21 This model thus separates regions of ischemic angiogenesis and arteriogenesis.
Consistent with our previous studies,2 wild-type (C57BL/6) mice recovered from FAL without significant gross ischemic injury (Figure 2A, left). However, eNOS-KO mice sustained dry toe necrosis by day-3 (Figure 2A, right) and significantly higher (worse) appearance and use-impairment scores (Figure 2B and 2C). Perfusion in the adductor collateral zone of the thigh (penetration of our Laser Doppler is ≈2 mm) and in the plantar foot (the latter correlates with overall leg perfusion18), showed greater reductions immediately after ligation and lower values at all time points over the subsequent 3 weeks in eNOS-KO mice (Figure 1B and 1C).
Figure 2. Worse ischemic injury in eNOS-KO mice after FAL.
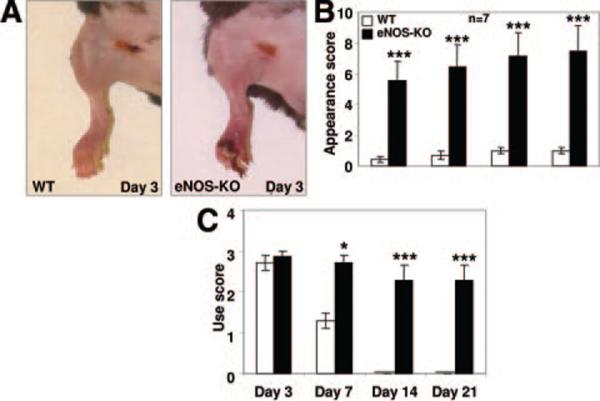
A, Representative images of mouse hindlimb after FAL. B and C, Appearance score (B) and use score (C) numerically reflect hindlimb ischemic injury after FAL. Two-way ANOVA followed by Dunn–Bonferroni t test; *P<0.05, ***P<0.001.
Decreased Collateral Density, Collateral Remodeling, and Angiogenesis in Hindlimb of eNOS-KO Mice
In eNOS-KO mice, the greater decline in perfusion immediately after ligation could reflect fewer and/or smaller diameter native collaterals, whereas the lower flow thereafter could arise from less collateral remodeling. To address hindlimb collateral number after remodeling, we performed angiography on day-21, using infusion (after maximal dilation and fixation) of an x-ray opaque casting agent.2,4,19 Collateral number according to the Longland criteria20 (see Methods), was modestly lower in eNOS-KO versus wild-type mice (Figure 3C, left; Figure 3D, second bar pair). To measure native collateral density in the unligated adductor/abductor thigh, 1) we performed x-ray angiography for a separate group of animals several minutes after acute FAL. Acute ligation in 3-month-old naïve animals, while viewing the exposed adductor, allows optimal filling of the native collaterals. We found that eNOS-KO mice have 21% fewer native collaterals detected by this method (Figure 3C, right; the first bar pair in Figure 3D). 2) We also applied an alcohol-methyl salicylate-based tissue clearing approach to further examine, according to Longland criteria, both native and remodeled collateral densities in the MicrofilP-casted hindlimb. These data closely confirmed the results with x-ray angiography (Figure 3E and 3F).
Figure 3. Impaired collateral remodeling and reduced hindlimb collateral density in eNOS-KO mice.

A, Representative images of MicrofilP-casted mouse gracilis collaterals 21 days after FAL in wild-type (left) and eNOS-KO (right) mice. Insets, Higher magnification images of boxed regions. B, Summary of lumen diameters measured from images represented in A. C and D, Representative images of x-ray angio-grams and number of hindlimb collaterals in eNOS-KO mice, and summary data. E and F, Representative images of alcohol-methyl salicylate–based tissue clearing of MicrofilP-casted hindlimb showing remodeled (left) and native (right) collaterals in eNOS-KO mice, and summary data. **P<0.01, ***P<0.001: t test compared to wild-type.
After FAL, gracilis collaterals undergo remodeling, resulting in enlarged, tortuous conduit arteries.2–4 MicrofilP casts of the hindlimb vasculature allow direct visualization and measurement of diameter of the superficial gracilis collaterals (Figure 1A, right; Figure 3A; see Methods). In eNOS-KO mice, collateral growth was diminished when measured 21 days after FAL (Figure 3B).
In addition to the less severe FAL model used above, we also ligated the femoral artery at a higher position16 (red arrow in Figure 1A, left) in a separate group of mice. This resulted in more severe ischemic injury (Online Figure I) and lower flows in eNOS-KO mice than that in wild-type (Online Figure II). Histological analysis of collaterals in the anterior and posterior gracilis muscles found that whereas baseline (native) collateral diameter was not different, collateral remodeling was inhibited by ≈55% in eNOS-KO mice (Online Figure III). Moreover, ischemia-induced angiogenesis, reflected by the capillary-to-muscle fiber number ratio in the gastrocnemius measured 21 days after FAL (Online Figure IV) was lower in eNOS-KOs, consistent with previous report8; baseline capillarity was also lower in eNOS-KOs. Overall, these data from 2 ligation models suggest that eNOS deficiency results in reduced density of native collaterals and capillaries in skeletal muscle and, after ligation, reduced remodeling and angiogenesis, which together result in greater ischemia.
Impaired Flow-Mediated Dilation of Upstream Arteries Supplying Collaterals in eNOS-KO Mice
It has been assumed that collaterals in normal tissues exhibit little or no net flow, and that after ligation the pressure drop downstream of the collateral network results in a large increase in collateral flow and shear stress. We recently confirmed this assumption, showing that both parameters rapidly increase by more than an order of magnitude after ligation.5 This causes a drop in resistance in the arterial pathway upstream of the collateral network, potentially triggering flow-mediated dilation (FMD) therein, a response well known to be largely mediated by eNOS/NO.13,22 Active FMD of an artery supplying a collateral network after acute artery occlusion has not been studied. This could contribute, along with reduced native density, to the greater drop in hindlimb perfusion immediately after ligation that was observed by Mees et al16 and confirmed by us (Figure 1). Therefore, we measured lumen diameter of the LCFA, which is the trunk of the artery tree that is upstream of a portion of the adductor collateral network including the gracilis collaterals, after FAL. We also measured flow velocity at the same point on the LCFA. Sustained FMD and increased velocity followed acute FAL in wild-type mice. This was significantly impaired in eNOS-KOs (Figure 4). In addition, topical NG-nitro-l-arginine methyl ester (L-NAME) (100 μmol/L in PBS) applied 10 minutes before ligation attenuated FMD in the wild-type, confirming dependence on NO. Because volume flow is related to diameter,4 this indicates a greatly reduced upstream “feed-forward” flow to the collateral network in eNOS-KO mice. This, along with fewer native collaterals, likely underlie the greater drop in hindlimb perfusion immediately after ligation and much greater tissue injury in eNOS-KOs seen by Mees et al16 and ourselves. These findings highlight a mechanism not measured in previous collateral studies, ie, that FMD in beds upstream of collaterals occurs immediately after ligation, predictably elevating the pressure gradient and thus flow across the collaterals. Moreover, the results show, as expected, that this is abolished when eNOS is lacking. Thus, this upstream dilation is a likely additional contributor to the reduced perfusion observed after FAL.
Figure 4. Increased Doppler-determined flow velocity and flow-mediated dilation of the LCFA, which supplies the gracilis collaterals and other adductor collaterals, in wild-type and eNOS-KO mice.

A, Representative images of LCFA (dotted lines show segment where lumen diameter and velocity were measured). B and C, eNOS-KO mice showed much reduced responses. No significant difference in diameter in eNOS-KO and wild-type mice before ligation (not shown). **P<0.01, ***P<0.001: t test compared to wild-type.
eNOS-KO Mice Lose Native Collaterals During Growth to Adulthood
Current methods, including x-ray angiography, have limited resolution to determine collateral abundance in most tissues, owing to the small size and 3D trajectory of these vessels. However, collateral density can be determined with precision in the cerebral pial circulation and values obtained there agree qualitatively with density in other tissues, including hindlimb, in gene-targeted and in-bred mice.2–4 We therefore examined pial collaterals and found significantly fewer collaterals, but not smaller diameters, in adult eNOS-KOs (Figure 5), in agreement with the hindlimb data (Figure 3). Because lower native collateral density in the adult can result from reduced formation in the embryo and/or impaired maturation (thus increased pruning) during the perinatal period,3–5 we examined earlier developmental stages. No differences were observed at embryonic day 18.5, when peak collateral formation occurs5 or during perinatal collateral remodeling and pruning which occurs during the first 3 weeks after birth (Figure 5C).3–5 Thus, eNOS does not participate in collateral formation and maturation. However, eNOS deficiency results in loss of collaterals during growth to adulthood (herein, 3- and 6-month old). To further explore the role of NO in native collateral formation and test for potential compensation by inducible (i)NOS for eNOS deficiency, we measured native pial collateral density of all progenies from parental crossings of eNOS+/−; iNOS+/−×eNOS+/−; iNOS+/− at postnatal day 28. No significant difference was found among the 9 possible genotypes (Figure 5E).
Figure 5. Cerebral cortical pial collateral number and diameter at different developmental stages and different genotypes.

A, Representative image of MicrofilP-casted adult eNOS-KO mice showing pial artery network and collaterals. Red arrowheads indicate collaterals between middle cerebral (MCA) and anterior cerebral artery (ACA) branches counted and diameters determined. B, representative image of E18.5 eNOS-KO mouse embryonic brain. C and D, Collateral number and diameter. E, No significant differences in pial collateral number in P28 pups crossed to harboring indicated alleles. *P<0.05, **P<0.01; 2-tailed t test.
Collaterals From eNOS-Deficient Mice Exhibit Impaired Activation of a Cell Cycle Gene Network During Remodeling
To explore mechanisms contributing to the impaired remodeling in eNOS-KO mice, we undertook a whole-genome microarray analysis23 in gracilis collaterals microdissected from wild-type and eNOS-KO mice 24 hour after ligation, or after acute ligation of the contralateral limb to serve as control. After setting a stringent criteria (delta=0.52) for significance analysis of microarray (SAM) analysis,24 expression of 538 gene sequences was differentially altered in wild-type mice (404 upregulated, 134 downregulated; false discovery rate=0.00%), whereas expression of 599 sequences was altered in eNOS-KOs (123 upregulated and 476 downregulated; false discovery rate of 0.03%). Functional annotation, classification, clustering and enrichment analyses of identified genes were performed using the DAVID bioinformatics resource (http://david.abcc.ncifcrf.gov/),25 along with reference to the available literature for selected genes. Real-time quantitative RT-PCR for 8 representative genes from each functional gene category identified in the microarray studies was performed to confirm the array findings (Figure 8A).
Figure 8. Real-time quantitative RT-PCR confirmation of genes identified in the microarray studies and activation of a cell cycle gene network during early collateral remodeling in wild-type mice and absence of its activation in eNOS-KO mice.

A, Quantitative RT-PCR showing expression of 8 representative genes from each functional gene category identified in the microarray studies. Fold increase in remodeling collaterals is compared to nonligated collaterals (n=3; *P<0.05, **P<0.01). B, Network identified by IPA of upregulated genes detected by arrays (Figure 7A). Color-filled symbols reflect genes in functional categories described in the legend for Figure 7 and in Results. Full-sized image of B is available in Online Figure IX.
The genes most upregulated in wild-type mice belong to 3 classes: cytokines and chemokines, inflammatory and stress response genes, and cell cycle genes (Figures 6 and 7). Many are thought to play important roles in vasculogenesis, angiogenesis and/or collateral remodeling.6,21 We found significant upregulation of 21 genes encoding cytokines/chemokines in wild-type mice (red squares, Figure 6A). Most of them were also upregulated in eNOS-KO mice, ie, upregulation of only 7 genes (33%) was abolished in eNOS-KOs (one gene was upregulated in eNOS-KO and not in wild-type mice (interleukin [IL]-17 receptor)). Likewise, only 7 of 27 (26%) of the inflammatory and stress responsive genes upregulated in wild-type were not upregulated in eNOS-KO mice (Figure 6B). We also compared “fold change” of gene expression for significant genes (Figure 6A and 6B). Only 10 of 48 genes (21%) in these 2 classes differed significantly between eNOS-KO and wild-type mice (3 genes had significantly higher relative expression levels in wild-type, whereas expression was lower in 7 other genes in wild-type) (Figure 6A and 6B). Given that the major strength of whole-genome expression profiling is class comparison, these findings suggest relatively similar gene expression profiles of cytokines/chemokines and inflammatory/stress response genes early in the collateral remodeling process in wild-type and eNOS-KO mice. Consistent with this, staining with antibodies for CD45 (pan leukocyte), CD3 (pan-T cell) and CD11b (monocytes) found comparable peri-collateral leukocyte densities in eNOS-KO and wild-type mice 7 days after ligation (Figure 6C and data not shown).
Figure 6. Comparable expression of leukocyte recruitment and inflammatory response genes during early collateral remodeling (24 hour after FAL or after acute ligation (control group)) in eNOS-KO and wild-type mice.

A and B, Comparison of upregulated chemo-attractant and cytokine receptor genes (A) and inflammatory/stress responsive genes (B). SAM indicates significantly upregulated genes identified by SAM analysis (red squares in WT or KO column); d, delta score obtained by SAM analysis; FC, fold changes of gene expression calculated on log(base 2) data. *P<0.05, ***P<0.001 for 2-tailed t test of log(base 2) expression data between eNOS and wild-type mice. Full-sized images of A and B are available in Online Figures VI and VII. C, Representative images of IHC staining for CD45+ leukocytes (brown stain), with light hematoxylin counterstaining.
Figure 7. Impaired activation of cell cycle genes in eNOS-KO mice during early (per Figure 6 legend) collateral remodeling.

A, Comparison of upregulated cell cycle regulatory genes between eNOS-KO and wild-type mice. Color-highlighted genes belong to various functional categories described in Results. Full-sized image of A is available in Online Figure VIII. B, Representative images of BrdUrd incorporation by proliferating vascular cells in remodeling collaterals 7 days after ligation. Arrows indicate BrdUrd+ cells. C, Summary of proliferating cells/collateral averaged from 10 consecutive sections of each genotype. *P<0.05, **P<0.01, ***P<0.001; 2-tailed t test.
Among the genes upregulated in wild-type mice, 41 belonged to the cell cycle gene class (Figure 7A). Ki67, a commonly used marker of cell proliferation, was among the most significantly upregulated genes. Upregulation of cyclin B, E and A, CKD1, and the CDK-regulatory protein CKS-2 (highlighted light-blue, Figure 7A) denote activation of cell cycling through G1, S, G2, and M phases. Other genes involved in cell cycling were also upregulated, eg, genes for chromosomal passenger machinery active during mitosis (highlighted green, Figure 7A),26 DNA replication complex proteins (highlighted yellow), and quality control/checkpoint surveillance genes (highlighted gray). Strikingly, none of these genes was upregulated in remodeling collaterals from eNOS-KOs. Moreover, only 4 (PSF2, tumor protein 52, BAX and splicing factor) of 123 upregulated genes in eNOS-KOs belong to cell cycle gene class. In addition to these results from SAM analysis, comparison of raw log(base 2) values for gene expression found that 25 of 41 genes had significantly higher fold-changes of expression in wild-type than in eNOS-KO mice (Figure 7A). Overall, these data suggest that eNOS-KOs have impaired activation of a cell cycle gene network early in the collateral remodeling process. Consistent with this, proliferating cells in collaterals were much lower in eNOS-KO mice (Figure 7B and 7C; BrdUrd incorporation 6 to 7 days after ligation).
In addition to DAVID functional annotation, the upregulated genes identified by SAM analysis during early arteriogenesis in wild-type and eNOS-KO mice were also subjected to Ingenuity Pathway Analysis (IPA) for further unbiased functional gene clustering (http://www.ingenuity.com/). In wild-type mice, 2 gene networks, ie, “Cell Cycle, DNA Replication and Cancer” (Figure 8B) and “Cellular Movement and Immune Cell Trafficking” (Online Figure X, right), were most prominently identified by IPA. These networks included most of the genes belonging to the cytokine/chemokine, inflammatory and stress response, and cell cycle gene classes identified by DAVID analysis (Figure 6A and 6B; Figure 7A). In particular, IL-6, which had the second highest “delta” (significance) value by SAM analysis (Figure 6A), was identified as a key stimulus of many genes in the cell cycle network (Figure 8B), eg, cyclin A and A2, aurora kinase B, Ki67, etc. However, whereas a comparably complex immune cell trafficking network was identified in both wild-type and eNOS-KO mice (Online Figure X), none of the genes in the “Cell Cycle, DNA Replication and Cancer” network detected in wild-type mice was identified by IPA in eNOS-KOs (Online Figure XI). This was despite comparable upregulation of IL-6 gene expression in both genotypes (Figures 6A and 8A). How eNOS regulates this proposed IL-6-activated cell cycle network in collateral remodeling is an important question for future investigation.
We also detected differentially upregulated genes classified as growth factors, extracellular matrix remodeling molecules and genes regulating metabolism (Online Figure XII). Functional annotation of downregulated genes by DAVID in both wild-type and eNOS-KO was less informative, resulting in diverse gene classes with low DAVID clustering scores, ie, lipid/cholesterol transport and metabolism genes (data not shown). The full microarray dataset is available at https://genome.unc.edu.
Discussion
Collateral studies often use severe ischemia models involving FAL as high as the inguinal canal. This can induce ischemia in the collateral region of the thigh, recruiting mechanisms in addition to increased shear stress that are capable of altering collateral remodeling and upstream flow-mediated dilation. Variability in histomorphometric analysis of collateral remodeling can also arise from difficulties in identifying the same collateral deep in the thigh muscles, because course of travel varies among individuals even of the same strain. Therefore, we used a moderate ischemia model that induces ischemia in and below the calf (ligation distal to the LCFA or just above it [Figure 1A, left]). This separates the region of ischemia from the midthigh where most of collateral network resides. This model also recruits the collaterals invariably present in the superficial anterior and posterior gracilis muscles, which are readily accessible for Laser Doppler flowmetry, histomorphometry and dissection for array analysis. We found that, compared to wild-type mice, eNOS-KOs had greater ischemic injury of the foot, secondary to a larger reduction in perfusion immediately after femoral ligation and lower flows thereafter. Our study identified 4 mechanisms: (1) fewer native (preexisting) collaterals; (2) impaired collateral remodeling; (3) less ischemic angiogenesis; and (4) reduced flow-mediated dilation upstream of the collateral network. The first finding is novel, and we show that it reflects a requirement of eNOS to maintain collateral density during growth to adulthood, also an intriguing new concept. The second finding addresses a point of disagreement in the literature on eNOS and remodeling. The last mechanism has not been examined previously in collateral studies. Together, these findings help clarify the controversy in the field regarding the role of eNOS in collateral function. Furthermore, they emphasize the pleiotropic importance of eNOS/NO in regulating hemodynamic mechanisms that impact the severity of obstructive disease.
Role of eNOS in Maintenance of the Native Collateral Circulation
Immediately after arterial ligation, myogenic and metabolic mechanisms favor maximal dilation in the dependent tissues. Tissue blood flow is then determined by perfusion pressure and conductance of the collateral network and its upstream and downstream circulations. Collateral conductance (the major determinant) primarily depends on density and diameter of the native collaterals, together with collateral length and blood viscosity that, however, have unitary rather than exponential effects on conductance. The lower perfusion we observed immediately after femoral ligation, which was also evident in the data reported by Mees et al,16 prompted us to investigate whether the native collateral circulation is reduced in eNOS-KO mice. Of note, arterial pressure is modestly elevated in eNOS-KO mice,27 eliminating lower pressure as a contributor to the lower perfusion after ligation. Whether eNOS-NO signaling affects native collateral formation has not been examined. We found that collateral density in the hindlimb and brain of adult eNOS-KO mice is reduced. Native collaterals diameter, however, was unaltered in both tissues. Our previous studies have found qualitative agreement between collateral densities in brain and hindlimb skeletal muscle (and intestine).2–4
Because collaterals form in the embryo and mature early after birth,3–5 and given that eNOS is expressed in the flowing embryonic vasculature,28 we examined whether eNOS deficiency affects these processes. Also, eNOS mediates some of the intracellular and extracellular actions of VEGF, and VEGF affects both native collateral formation and maturation.3 However, eNOS deficiency had no effect on these processes, ie, pial collateral number at E18.5, P1, and P21 was identical in wild-type and eNOS-KO mice. Interestingly, collaterals were lost during growth to adulthood (25% decrease at 6 months of age), suggesting that eNOS/NO impacts maintenance of the collateral circulation. Whether this is attributable to a direct role or to mild hypertension or other mechanisms, is an intriguing question. Arteriole rarefaction has been linked to hypertension in eNOS-KO mice29 which become hypertensive by 4 weeks of age. Moreover, reduced collateral density has recently been identified in aging mice, in association with decreased eNOS protein levels (H. Zhang, J.E.F., S. Epstein et al, unpublished results).
Gene Expression in Remodeling Collaterals
Collateral remodeling involves endothelial cell (EC) activation, leukocyte recruitment, matrix changes and cell proliferation.6 Although the remodeling process cannot currently be studied in vitro, increased endothelial shear stress is known to be the initiating stimulus.30 Expression arrays have identified molecular programs activated by shear stress in cultured ECs.31,32 Lee, Epstein and colleagues21 and Tirziu, Simons, and colleagues33 analyzed gene expression in RNA prepared from mouse adductor muscle containing collaterals and found that angiogenesis-, inflammation- and matrix-related genes were upregulated after FAL near the inguinal canal. Eitenmuller, Schaper, and colleagues34 hybridized cDNA from rabbit skeletal muscle containing collaterals isolated 7 days after FAL to a human microarray and identified upregulation of RhoA, cofilin, and vimentin. In the present study, we analyzed microdissected gracilis collaterals (see Methods). Cluster analysis of upregulated genes in the early phase of remodeling (24 hour after ligation) of wild-type mice identified cytokine/chemokine genes (MIP-2α, MIP-1α, MIP-1γ, MCP-1, MCP-2, MCP-3, IL6, IL-1β, TNFα, etc) as the predominant differentially expressed gene classes (Figure 6A), followed by genes involved in cell stress and inflammatory response (immune-responsive protein 1, SAA1, SAA2, SAA3, TSG-14, TSP1, HSP8, HSP70, etc) (Figure 6B). Most interestingly, 44 genes involved in cell cycle progression were upregulated in wild-type mice (Figure 7A). We confirmed several genes, linked by others using up- and down-modulating strategies to collateral remodeling, such as MCP-1,35 TNFα36 and metalloproteinase inhibitor 1.37 In addition, we identified many genes not previously linked to collateral remodeling (some evidencing striking upregulation) as targets for future investigation. This is the first study to perform genome-wide expression profiling of isolated collaterals.
Impaired Activation of Cell Cycle Gene Network, Cell Proliferation, and Collateral Remodeling in eNOS-KO Mice
In the present study, collateral remodeling was inhibited by ≈50% in eNOS-KO mice. This confirms impaired remodeling after treatment with the potent NOS inhibitor, L-NAME,34,38 and supported by a recent report showing impaired arteriogenesis in eNOS-KO and L-NAME treated wild-type mice.39 However, little is known about how eNOS/NO mediates collateral remodeling. We sought to address this by array-profiling collaterals from eNOS-KO mice. Expression was examined in collaterals taken from eNOS-KO mice 24 hour after ligation or acute ligation, and the findings were compared with the above wild-type data. Most of the enriched cytokine/chemokines, stress and inflammatory response genes were upregulated similarly in eNOS-KO and wild-type mice (Figure 6A and 6B; Online Figure X). However, several genes showed greater increases in wild-type (CXCL10, immune-responsive protein 1, SAA2) or were increased more in eNOS-KOs (IL-17R, Prmt1, metalloproteinase inhibitor 1, HSP8, c-type lectin domain family 4-n, COX-2). Peri-collateral leukocyte densities (CD45+, CD3+, and CD11b+) were comparable between wild-type and eNOS-KOs. These data agree with the array data that found little difference in expression of immune and inflammatory response genes in collaterals 24 hour after ligation in the 2 strains. In addition, as in the wild-type comparison, several new targets for future investigation were identified.
A remarkable finding was that almost none of the cell cycle genes upregulated in wild-type were upregulated in eNOS-KO mice (Figures 7A and 8). IL-6 was identified by IPA as a major regulatory node of a cell cycle network in wild-type mice. Despite a similar upregulation of IL-6 (Figure 8A), this network was not activated in eNOS-KOs (Online Figure XI). Consistent with this, vascular wall cell proliferation in collaterals from eNOS-KOs was sharply reduced (Figure 7B and 7C). Impaired vascular cell proliferation by blocking endogenous NO production during collateral remodeling was reported recently by Troidl, Schaper, and colleagues.40 These data suggest that eNOS/NO is important in activating a cell cycle network driving cell proliferation during collateral remodeling. Recent findings that VEGF contributes significantly to collateral remodeling,3,17 together with the requirement of eNOS/NO for VEGF-induced EC proliferation, support this observation.
NO has a complex dual role in cell cycle regulation. Whereas NO inhibits vascular smooth muscle cell proliferation in injury models in vivo41 and in cell culture,42 it stimulates endothelial cell DNA synthesis and proliferation via cGMP-dependent transcription43 and is a central regulator of ischemic angiogenesis,8,9 (confirmed herein). Shear stress upregulates eNOS expression in ECs44 and activates eNOS via Ser1177 phosphorylation45 in a PECAM-1–dependent manner, both in vivo12,46 and in vitro.47 Increasing evidence suggests a critical role of eNOS in mediating shear-induced vascular remodeling.38 Our findings of defective activation of cell cycle genes in eNOS-KO mice during the early phase of collateral remodeling supports the requirement of eNOS in proliferation of ECs and vascular smooth muscle cells in response to increased shear. Whereas the functions of NO in cell signaling have been extensively studied, little is known about direct involvement of eNOS/NO in cell cycle regulation. Recently, Ser1177–phosphorylated eNOS was observed to undergo rapid nuclear translocation during mitosis in mouse yolk sac arterioles (J. Luccitti et al, unpublished data). Given the rapid turnover and short half-life of NO, it is possible that translocation to the nucleus could produce local NO capable of regulating expression of cell cycle genes.
Fully dilated baseline collateral diameter was either not different (Figure 3B) or trended larger (Online Figure III) in eNOS-KO hindlimb, and it was also not different in brain (Figure 5D). Whether collaterals have tone has not been definitively resolved. However, if they do, eNOS-KO collaterals may not dilate as well as wild-type, thus shear stress may be higher in them. Because of this and/or other possible hemodynamic differences, it is possible that gene expression differences could be mediated by other shear stress transducers, independent of eNOS. Indeed, collateral remodeling was attenuated in eNOS-KOs by ≈35 to 65% (Figure 3A and Online Figure III), ie, not completely abolished.
Impaired Flow-Mediated Dilation of Upstream Collateral-Supply Vessels in eNOS-KO Mice
It is well known that increased shear stress causes endothelium-dependent flow-mediated dilation (FMD) of arteries, and that FMD depends on eNOS-generated NO.13,22 To our knowledge, no studies have examined whether increased collateral flow after acute arterial obstruction induces FMD of the upstream arterial supplying the collateral network. In the ligation model used herein, the LCFA is the upstream feed-artery to the gracilis collaterals and other deeper collaterals in the adductor. Diameter and flow velocity in the LCFA increased significantly after acute FAL in wild-type mice. In eNOS-KOs, dilation was abolished and increased flow was sharply reduced; the remaining increased flow is likely caused by lower the downstream resistance and increased pressure gradient caused by FAL. FMD of vessels supplying a collateral network, by increasing input pressure to the collaterals, likely contributes importantly to collateral-dependent flow immediately after ligation, as well as during subsequent recovery of flow. These findings introduce a new, although not unexpected concept, ie, regulation of “collateral feed-forward flow” (a mechanism that undoubtedly contributes to collateral-dependent perfusion during arterial occlusion). This will increase the pressure gradient across the collaterals, thus increasing shear and circumferential wall stress within them. These parameters have not been measured in previous collateral studies, although an early study reported an increase in collateral conductance after FAL in dog, based on pressure changes.48 Collateral feed-forward flow may be compromised by a number of risk factors related to endothelial dysfunction, ie, diabetes, dyslipidemia, atherosclerosis, hypertension, smoking and aging, and contribute to collateral insufficiency.6,7
The above findings provide an alternative explanation for the apparent defective dilation in eNOS-KOs, and opposite conclusion in eNOS transgenic mice, in the study by Mees et al.16 Recently, Isenberg et al observed higher flows in dependent tissues immediately after FAL, as well as after creation of a partial skin flap (which causes multiple small artery obstructions) in thrombospondin-1-KO mice.49 Because thrombospondin is a potent endogenous eNOS inhibitor, the authors concluded that greater NO-induced inhibition of vascular smooth muscle tone was responsible. We propose that greater collateral feed-forward flow could also contribute to their findings.
In conclusion, our results identify a new function for the remarkably busy eNOS enzyme: maintenance of the native collateral circulation during growth to adulthood. We also demonstrate that eNOS plays a prominent role in collateral remodeling and highlight a number of new molecules potentially involved in this role. In particular, based on array profiling and network analysis, we hypothesize that eNOS/NO may coordinately regulate expression of a network of genes controlling cell cycle checkpoints necessary for cell proliferation during collateral remodeling. This finding raises a fundamental question regarding how eNOS/NO regulates cell cycle progression in the collateral wall. Lastly, we propose that a novel mechanism, “collateral feed-forward flow,” which is dependent on upstream FMD, contributes to collateral perfusion of ischemic tissues immediately after arterial obstruction and during subsequent recovery of flow as collaterals remodel. This finding has implications not only in interpreting experimental studies, but also may help to explain the association of certain risk factors with poor collateral function in patients.
Supplementary Material
Novelty and Significance.
What Is Known?
The collateral circulation in tissues is an endogenous system of bypass vessels whose abundance is critical in determining ischemic injury in obstructive arterial disease.
The density of native (preexisting) collaterals and their lumen enlargement (remodeling) in obstructive disease are highly subject to differences in genetic background, and remodeling is impaired by risk factors associated with endothelial dysfunction.
The role of eNOS in collateral formation and remodeling is unknown or controversial,
What New Information Does This Article Contribute?
Endothelial nitric oxide synthase (eNOS) maintains the density of native collaterals during natural growth to adulthood and is required for full collateral remodeling by regulating a cell cycle gene network.
Collateral flow induced by arterial obstruction causes eNOS-NO-dependent flow-mediated dilation of feed arteries upstream of the collateral network, further enhancing collateral flow.
Collaterals are arteriole–arteriole anastomoses bridging adjacent arterial trees that act as bypass vessels following arterial obstruction. The density and diameter of native collaterals and their shear stress-dependent outward remodeling vary widely among individuals, possibly because of genetic and disease factors. Clinical trials aimed at augmenting collateral growth have yielded disappointing outcomes, possibly because of disease-induced impaired collateral function. In this study, we found poor hindlimb perfusion and severe ischemia after femoral artery occlusion in eNOS-deficient mice, compared with wild-type mice. We identified several contributing factors: (1) fewer native collaterals in hindlimb (and another tissue examined, brain) attributable to loss during growth to adulthood; (2) inhibition of collateral remodeling in responding to femoral artery occlusion; and (3) impaired flow-mediated dilation of the upstream arterial tree supplying the collateral network. Furthermore, genome-wide array analysis identified impaired activation of a cell cycle gene network in eNOS-deficient mice during early collateral remodeling, resulting in reduced proliferation of collateral wall cells. Pericollateral inflammatory cell recruitment was unaffected, consistent with comparable expression of chemoattractant, cytokines, and inflammatory genes. These findings highlight several new areas of investigation, including factors responsible for maintenance of the native collateral circulation and how eNOS/NO regulate cell cycle progression during collateral remodeling.
Acknowledgments
We thank Dr Hua Zhang for assistance with surgical and pial casting procedures, Dr Dan Chalothorn for assistance with the flow-mediated dilation experiments and P18 brain NG2 staining, Kirk McNaughton for histological sectioning, and William A. McFadden for Doppler measurement of flow velocity.
Sources of Funding This work was supported by NIH grants HL62584 and HL090655 (to J.E.F.). X.D. is a cardiology fellow supported by NIH T32 training grant HL083828-04 (to Dr George A. Stouffer).
Non-standard Abbreviations and Acronyms
- E
embryonic day
- EC
endothelial cell
- eNOS
endothelial nitric oxide synthase
- FAL
femoral artery ligation
- FMD
flow-mediated dilation
- FSS
fluid shear stress
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- IPA
Ingenuity Pathway Analysis
- KO
knockout
- LCFA
lateral caudal femoral artery
- L-NAME
NG-nitro-l-arginine methyl ester
- NO
nitric oxide
- P
postnatal day
- SAM
significance analysis of microarray data
- VEGF
vascular endothelial growth factor
Footnotes
Disclosures None.
References
- 1.Meier P, Gloekler S, Zbinden R, Beckh S, de Marchi SF, Zbinden S, Wustmann K, Billinger M, Vogel R, Cook S, Wenaweser P, Togni M, Windecker S, Meier B, Seiler C. Beneficial effect of recruitable collaterals: a 10-year follow-up study in patients with stable coronary artery disease undergoing quantitative collateral measurements. Circulation. 2007;116:975–983. doi: 10.1161/CIRCULATIONAHA.107.703959. [DOI] [PubMed] [Google Scholar]
- 2.Chalothorn D, Clayton JA, Zhang H, Pomp D, Faber JE. Collateral density, remodeling, and VEGF-A expression differ widely between mouse strains. Physiol Genomics. 2007;30:179–191. doi: 10.1152/physiolgenomics.00047.2007. [DOI] [PubMed] [Google Scholar]
- 3.Clayton JA, Chalothorn D, Faber JE. Vascular endothelial growth factor-A specifies formation of native collaterals and regulates collateral growth in ischemia. Circ Res. 2008;103:1027–1036. doi: 10.1161/CIRCRESAHA.108.181115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chalothorn D, Zhang H, Smith JE, Edwards JC, Faber JE. Chloride intracellular channel-4 is a determinant of native collateral formation in skeletal muscle and brain. Circ Res. 2009;105:89–98. doi: 10.1161/CIRCRESAHA.109.197145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chalothorn D, Faber JE. Differences in collateral formation in the embryo are associated with genetic variation in patterning and maturation of the cerebral cortical circulation. Arterioscler Thromb Vasc Biol. 2009;29:250. Abstract. [Google Scholar]
- 6.Schaper W. Collateral circulation: past and present. Basic Res Cardiol. 2009;104:5–21. doi: 10.1007/s00395-008-0760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kinnaird T, Stabile E, Zbinden S, Burnett MS, Epstein SE. Cardiovascular risk factors impair native collateral development and may impair efficacy of therapeutic interventions. Cardiovasc Res. 2008;78:257–264. doi: 10.1093/cvr/cvm116. [DOI] [PubMed] [Google Scholar]
- 8.Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- 10.Gray C, Packham IM, Wurmser F, Eastley NC, Hellewell PG, Ingham PW, Crossman DC, Chico TJ. Ischemia is not required for arteriogenesis in zebrafish embryos. Arterioscler Thromb Vasc Biol. 2007;27:2135–2141. doi: 10.1161/ATVBAHA.107.143990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai WJ, Kocsis E, Luo X, Schaper W, Schaper J. Expression of endothelial nitric oxide synthase in the vascular wall during arteriogenesis. Mol Cell Biochem. 2004;264:193–200. doi: 10.1023/b:mcbi.0000044388.27953.a0. [DOI] [PubMed] [Google Scholar]
- 12.Cheng C, van Haperen R, de Waard M, van Damme LC, Tempel D, Hanemaaijer L, van Cappellen GW, Bos J, Slager CJ, Duncker DJ, van der Steen AF, de Crom R, Krams R. Shear stress affects the intracellular distribution of eNOS: direct demonstration by a novel in vivo technique. Blood. 2005;106:3691–3698. doi: 10.1182/blood-2005-06-2326. [DOI] [PubMed] [Google Scholar]
- 13.Bagi Z, Frangos JA, Yeh JC, White CR, Kaley G, Koller A. PECAM-1 mediates NO-dependent dilation of arterioles to high temporal gradients of shear stress. Arterioscler Thromb Vasc Biol. 2005;25:1590–1595. doi: 10.1161/01.ATV.0000170136.71970.5f. [DOI] [PubMed] [Google Scholar]
- 14.Yu J, deMuinck ED, Zhuang Z, Drinane M, Kauser K, Rubanyi GM, Qian HS, Murata T, Escalante B, Sessa WC. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc Natl Acad Sci U S A. 2005;102:10999–11004. doi: 10.1073/pnas.0501444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar D, Branch BG, Pattillo CB, Hood J, Thoma S, Simpson S, Illum S, Arora N, Chidlow JH, Jr, Langston W, Teng X, Lefer DJ, Patel RP, Kevil CG. Chronic sodium nitrite therapy augments ischemia-induced angiogenesis and arteriogenesis. Proc Natl Acad Sci U S A. 2008;105:7540–7545. doi: 10.1073/pnas.0711480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mees BS, Wagner E, Ninci S, Tribulova S, Martin R, van Haperen, Kostin S, Heil M, de Crom R, Schaper W. Endothelial nitric oxide synthase activity is essential for vasodilation during blood flow recovery but not for arteriogenesis. Arterioscler Thromb Vasc Biol. 2007;27:1926–1933. doi: 10.1161/ATVBAHA.107.145375. [DOI] [PubMed] [Google Scholar]
- 17.Toyota E, Warltier DC, Brock T, Ritman E, Kolz C, O'Malley P, Rocic P, Focardi M, Chilian WM. Vascular endothelial growth factor is required for coronary collateral growth in the rat. Circulation. 2005;112:2108–2113. doi: 10.1161/CIRCULATIONAHA.104.526954. [DOI] [PubMed] [Google Scholar]
- 18.Chalothorn D, Zhang H, Clayton JA, Thomas SA, Faber JE. Catecholamines augment collateral vessel growth and angiogenesis in hindlimb ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H947–H959. doi: 10.1152/ajpheart.00952.2004. [DOI] [PubMed] [Google Scholar]
- 19.Chalothorn D, Moore SM, Zhang H, Sunnarborg SW, Lee DC, Faber JE. Heparin-binding epidermal growth factor-like growth factor, collateral vessel development, and angiogenesis in skeletal muscle ischemia. Arterioscler Thromb Vasc Biol. 2005;25:1884–1890. doi: 10.1161/01.ATV.0000175761.59602.16. [DOI] [PubMed] [Google Scholar]
- 20.Longland CJ. The collateral circulation of the limb. Ann R Coll Surg Engl. 1953;13:161–176. [PMC free article] [PubMed] [Google Scholar]
- 21.Lee CW, Stabile E, Kinnaird T, Shou M, Devaney JM, Epstein SE, Burnett MS. Temporal patterns of gene expression after acute hindlimb ischemia in mice: insights into the genomic program for collateral vessel development. J Am Coll Cardiol. 2004;43:474–482. doi: 10.1016/j.jacc.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 22.Cooke JP, Rossitch E, Jr, Andon NA, Loscalzo J, Dzau VJ. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. J Clin Invest. 1991;88:1663–1671. doi: 10.1172/JCI115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knapen D, Vergauwen L, Laukens K, Blust R. Best practices for hybridization design in two-colour microarray analysis. Trends Biotechnol. 2009;27:406–414. doi: 10.1016/j.tibtech.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 26.Higuchi T, Uhlmann F. Cell cycle: passenger acrobatics. Nature. 2003;426:780–781. doi: 10.1038/426780a. [DOI] [PubMed] [Google Scholar]
- 27.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teichert AM, Scott JA, Robb GB, Zhou YQ, Zhu SN, Lem M, Keightley A, Steer BM, Schuh AC, Adamson SL, Cybulsky MI, Marsden PA. Endothelial nitric oxide synthase gene expression during murine embryogenesis: commencement of expression in the embryo occurs with the establishment of a unidirectional circulatory system. Circ Res. 2008;103:24–33. doi: 10.1161/CIRCRESAHA.107.168567. [DOI] [PubMed] [Google Scholar]
- 29.Kubis N, Besnard S, Silvestre JS, Feletou M, Huang PL, Levy BI, Tedgui A. Decreased arteriolar density in endothelial nitric oxide synthase knockout mice is due to hypertension, not to the constitutive defect in endothelial nitric oxide synthase enzyme. J Hypertens. 2002;20:273–280. doi: 10.1097/00004872-200202000-00017. [DOI] [PubMed] [Google Scholar]
- 30.Pipp F, Boehm S, Cai WJ, Adili F, Ziegler B, Karanovic G, Ritter R, Balzer J, Scheler C, Schaper W, Schmitz-Rixen T. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler Thromb Vasc Biol. 2004;24:1664–1668. doi: 10.1161/01.ATV.0000138028.14390.e4. [DOI] [PubMed] [Google Scholar]
- 31.Chen BP, Li YS, Zhao Y, Chen KD, Li S, Lao J, Yuan S, Shyy JY, Chien S. DNA microarray analysis of gene expression in endothelial cells in response to 24-h shear stress. Physiol Genomics. 2001;7:55–63. doi: 10.1152/physiolgenomics.2001.7.1.55. [DOI] [PubMed] [Google Scholar]
- 32.Mack PJ, Zhang Y, Chung S, Vickerman V, Kamm RD, Garcia-Cardena G. Biomechanical regulation of endothelium-dependent events critical for adaptive remodeling. J Biol Chem. 2009;284:8412–8420. doi: 10.1074/jbc.M804524200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tirziu D, Moodie KL, Zhuang ZW, Singer K, Helisch A, Dunn JF, Li W, Singh J, Simons M. Delayed arteriogenesis in hypercholesterolemic mice. Circulation. 2005;112:2501–2509. doi: 10.1161/CIRCULATIONAHA.105.542829. [DOI] [PubMed] [Google Scholar]
- 34.Eitenmuller I, Volger O, Kluge A, Troidl K, Barancik M, Cai WJ, Heil M, Pipp F, Fischer S, Horrevoets AJ, Schmitz-Rixen T, Schaper W. The range of adaptation by collateral vessels after femoral artery occlusion. Circ Res. 2006;99:656–662. doi: 10.1161/01.RES.0000242560.77512.dd. [DOI] [PubMed] [Google Scholar]
- 35.Heil M, Ziegelhoeffer T, Wagner S, Fernandez B, Helisch A, Martin S, Tribulova S, Kuziel WA, Bachmann G, Schaper W. Collateral artery growth (arteriogenesis) after experimental arterial occlusion is impaired in mice lacking CC-chemokine receptor-2. Circ Res. 2004;94:671–677. doi: 10.1161/01.RES.0000122041.73808.B5. [DOI] [PubMed] [Google Scholar]
- 36.Hoefer IE, van Royen N, Rectenwald JE, Bray EJ, Abouhamze Z, Moldawer LL, Voskuil M, Piek JJ, Buschmann IR, Ozaki CK. Direct evidence for tumor necrosis factor-alpha signaling in arteriogenesis. Circulation. 2002;105:1639–1641. doi: 10.1161/01.cir.0000014987.32865.8e. [DOI] [PubMed] [Google Scholar]
- 37.Hillmeister P, Lehmann KE, Bondke A, Witt H, Duelsner A, Gruber C, Busch HJ, Jankowski J, Ruiz-Noppinger P, Hossmann KA, Buschmann IR. Induction of cerebral arteriogenesis leads to early-phase expression of protease inhibitors in growing collaterals of the brain. J Cereb Blood Flow Metab. 2008;28:1811–1823. doi: 10.1038/jcbfm.2008.69. [DOI] [PubMed] [Google Scholar]
- 38.Troidl K, Rüding I, Cai W-J, Mücke Y, Grossekettler L, Piotrowska I, Apfelbeck H, Schierling W, Volger OL, Horrevoets AJ, Grote K, Schmitz-Rixen T, Schaper W, Troidl C. Actin-binding Rho activating protein (Abra) is essential for fluid shear stress-induced arteriogenesis. Arterioscler Thromb Vasc Biol. 2009;29:2093–2101. doi: 10.1161/ATVBAHA.109.195305. [DOI] [PubMed] [Google Scholar]
- 39.Park B, Hoffman A, Yang Y, Yan J, Tie G, Bagshahi H, Nowicki PT, Messina LM. Endothelial nitric oxide synthase affects both early and late collateral arterial adaptation and blood flow recovery after induction of hind limb ischemia in mice. J Vasc Surg. 2010;51:165–173. doi: 10.1016/j.jvs.2009.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Troidl K, Tribulova S, Cai W-J, Ruding I, Apfelbeck H, Schierling W, Troidl C, Schmitz-Rixen T, Schaper W. Effects of endogenous nitric oxide and of DETA NONOate in arteriogenesis. J Cardiovasc Pharmacol. 2010;55:153–160. doi: 10.1097/FJC.0b013e3181c9556f. [DOI] [PubMed] [Google Scholar]
- 41.Tanner FC, Meier P, Greutert H, Champion C, Nabel EG, Luscher TF. Nitric oxide modulates expression of cell cycle regulatory proteins: a cytostatic strategy for inhibition of human vascular smooth muscle cell proliferation. Circulation. 2000;101:1982–1989. doi: 10.1161/01.cir.101.16.1982. [DOI] [PubMed] [Google Scholar]
- 42.Sarkar R, Gordon D, Stanley JC, Webb RC. Cell cycle effects of nitric oxide on vascular smooth muscle cells. Am J Physiol. 1997;272:H1810–H1818. doi: 10.1152/ajpheart.1997.272.4.H1810. [DOI] [PubMed] [Google Scholar]
- 43.Ziche M, Morbidelli L, Choudhuri R, Zhang HT, Donnini S, Granger HJ, Bicknell R. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J Clin Invest. 1997;99:2625–2634. doi: 10.1172/JCI119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balligand JL, Feron O, Dessy C. eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev. 2009;89:481–534. doi: 10.1152/physrev.00042.2007. [DOI] [PubMed] [Google Scholar]
- 45.Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA. Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- 46.Chen W, Bacanamwo M, Harrison DG. Activation of p300 histone acetyltransferase activity is an early endothelial response to laminar shear stress and is essential for stimulation of endothelial nitric-oxide synthase mRNA transcription. J Biol Chem. 2008;283:16293–16298. doi: 10.1074/jbc.M801803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fleming I, Fisslthaler B, Dixit M, Busse R. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci. 2005;118:4103–4111. doi: 10.1242/jcs.02541. [DOI] [PubMed] [Google Scholar]
- 48.Rosenthal SL, Guyton AC. Hemodyamics of collateral vasodilation following femoral artery occlusion in anesthetized dogs. Circ Res. 1968;23:239–248. doi: 10.1161/01.res.23.2.239. [DOI] [PubMed] [Google Scholar]
- 49.Isenberg JS, Pappan LK, Romeo MJ, Abu-Asab M, Tsokos M, Wink DA, Frazier WA, Roberts DD. Blockade of thrombospondin-1-CD47 interactions prevents necrosis of full thickness skin grafts. Ann Surg. 2008;247:180–190. doi: 10.1097/SLA.0b013e31815685dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.