

Abstract
The treatment of high-grade tumours must consider a tumour environment dominated by cells that support cancer growth. In addition to directing angiogenesis and invasion, alternatively activated macrophages in the tumour provide protection from adaptive immunity and permit tumour growth. Agonist antibodies to the tumour necrosis factor receptor family member OX40 are an effective therapy for cancer in a range of murine models; however, as with many immune therapies, αOX40 therapy is less effective as the tumour grows and develops an immune suppressive environment. We demonstrate that αOX40 directly activates T cells and that this T-cell activation alters macrophage differentiation in the tumour environment. We demonstrate that macrophages in the tumour limit the efficacy of αOX40 therapy, and that combining αOX40 therapy with inhibitors of arginase significantly enhances survival of tumour-bearing mice. These data demonstrate that macrophages in the tumour environment limit the effectiveness of OX40-based immunotherapy, and combination therapies that target both the cell-mediated immune response and the suppressive tumour environment will be required for translation of effective immunotherapies to patients with established tumours.
Keywords: arginase, interleukin-12, interleukin-18, macrophage, OX40
Introduction
As cancers develop, an array of non-cancer cells are co-opted or recruited into the tumour environment. The persistent selective pressure of immunosurveillance suggests that a critical point in the evolution of a tumour to the stage where it presents as a growing symptomatic mass in patients probably involves the ability to redirect immune processes, and particularly to suppress adaptive immune responses. The process by which cancer cells control their environment is critical to their evolution into invasive tumours, and results in a tumour environment infiltrated with non-cancer cells that support cancer growth. The tumour macrophage is one of the more important cells in this mix because it influences many aspects of the tumour environment. Tumour macrophages initiate neoangiogenesis,1 remodel the basement membrane and extracellular matrix to allow growth and invasion,2 and also regulate immune responses in the tumour.3
Despite local immune suppression, tumour antigen-specific T cells can be isolated from many tumours, amplified in vitro and restored to full cytolytic function.4,5 Hence, even in the tumour-bearing host, some effector cells are initially expanded and can reach the tumour site. Nevertheless, the cytokine and chemokine environment of the tumour is not optimal for the attraction of activated effector cells;6 furthermore, the tumour environment both creates7 and attracts8 suppressive regulatory T cells. In these ways, the tumour environment exhibits a pattern of inflammatory resolution. Inflammatory onset is associated with expression of pro-inflammatory cytokines such as tumour necrosis factor-α (TNF-α) and inflammatory mediators such as inducible nitric oxide synthase (iNOS) as part of a general M1 differentiation pattern, whereas inflammatory resolution is associated more with expression of interleukin-10 (IL-10), arginase I and general M2 differentiation.9,10 Unfortunately for the development of T-cell immunotherapies for cancer, a tumour environment of inflammatory resolution does not support effective adaptive immune responses.11
Our laboratory has studied the use of agonistic antibodies to the TNF receptor superfamily member OX40 (CD134) to overcome a lack of adjuvant within the tumour environment in vivo. OX40 is transiently up-regulated following antigen receptor signalling on CD4 and CD8 T cells, and ligation of OX40 by antibodies or its natural ligand (OX40L/CD252) results in improved expansion, effector function and long-term survival of CD4 and CD8 T cells.12–16 Expression of the ligand for OX40 is tightly associated with acute inflammation, and has been demonstrated at sites of psoriasis,17 rheumatoid arthritis18 and autoimmune demyelination.19 Provision of agonistic antibodies to OX40 (αOX40) significantly enhances antigen-specific T-cell responses to soluble antigen12,14 and to antigens expressed in cancer cell lines.13 This suggests that the lack of OX40 signalling may be a limiting factor to generate optimal antigen-specific immune responses in vivo. Treatment of an array of tumour types with αOX40 results in significantly enhanced survival and complete tumour regression in some mice.20 In this way, the αOX40 reagent is one of a new class of immunotherapeutic adjuvant antibodies, including antibodies to 4-1BB (CD137) and GITR, which provide a positive signal to antigen responsive T cells, and antibodies to CTLA4 (CD152) and PD1 (CD279), which block negative signals to activated T cells. Such agents can overcome the limitations on antigen-presenting cells within the tumour by directly intervening in the regulatory signals that control T-cell activation.
OX40 agonist antibody therapy is effective at treating small tumours (within 3–7 days of tumour challenge), but in common with antibodies to GITR, and CTLA4, αOX40 therapy is much less effective if tumours have had time to establish their immune suppressive environment (10 days after tumour challenge).21–23 Therefore, we hypothesize that the development of a mature tumour environment limits the efficacy of αOX40 therapy, by establishing inflammatory resolution in the tumour. We demonstrated that following αOX40 therapy of established tumours there is an influx of CD8 T cells to the tumour, associated with a transient control of tumour growth, which was accompanied by an altered profile of tumour macrophages. To understand why this tumour control ends and these tumours eventually recur following αOX40 therapy, we examined the gene expression pattern of tumour macrophages. We identified an increase in expression of the M2 macrophage marker arginase I following αOX40 therapy and demonstrate that blockade of arginase enhanced the efficacy of αOX40 therapy. We demonstrate that extension of acute inflammation by treatment with IL-12 and IL-18 enhances the effect of αOX40 therapy on established tumours. In view of these data, we propose that αOX40 therapy causes a transient T-cell-mediated acute inflammation in the tumour. However, tumour macrophages can terminate the adaptive immune response in part through expression of arginase. These results describe a potentially limiting factor in the use of T-cell-targeted immunotherapy for treatment of established tumours, and identify approaches to overcome immune resolution in the tumour.
Materials and methods
Animals, cell lines and in vivo antibodies
Six to eight week old C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, MA). OX40-Cre C57BL/6 mice have recently been described,24,25 and these mice were crossed with B6.129(Cg)-Tg(CAG-Bgeo/GFP)21Lbe/J obtained from The Jackson Laboratory (Bar Harbor, ME) and F1 Cre+ GFP+ mice or control single-positive littermates were used in experiments. These experiments used the MCA205 sarcoma cell line as previously described.21 Control RatIg antibody was purchased from Sigma (St Louis, MO) and the rat anti-OX40 antibody (OX86) was produced in the laboratory from hybridomas and affinity-purified over protein G columns. All animal protocols were approved by the Institution’s Animal Care and Use Committee.
Isolation of tumour-infiltrating cell populations
C57BL/6 mice were challenged with 1 × 106 MCA205 tumour cells subcutaneously in the right flank, which were allowed to establish for 10–14 days. Mice were treated with 250 μg αOX40 or control antibody intraperitoneally, and 7 days later the tumour was harvested. Isolation of tumour-infiltrating cells was performed as previously described.21 Briefly, the excised tumour was dissected into 1-mm pieces using crossed scalpels, then digested for 1–2 hr with agitation at room temperature in 1 mg/ml collagenase (Invitrogen, Carlsbad, CA), 100 μg/ml hyaluronidase (Sigma) and 20 mg/ml DNase (Sigma) in PBS. The resultant preparation was filtered through 100-μm nylon mesh and density gradient centrifugation was performed by layering the cell suspension over Ficoll. The resultant buoyant cell layer of tumour-infiltrating cells was washed for use in subsequent experiments.
In vitro isolation of tumour macrophages
Tumour macrophages were isolated from suspensions of tumour-infiltrating cells by plastic adherence or FACS. For isolation by plastic adherence, cells were resuspended in tissue culture media and seeded at a concentration of 1 × 106 cells/cm2 surface area of cell-culture-treated multiwell plates. Cultures were incubated for 30 min at room temperature, then washed three times with media to leave an adherent macrophage population that was > 90% CD11b+. For FACS, tumour-infiltrating cells were resuspended in PBS containing 5% fetal bovine serum and stained with CD11b-FITC, Gr1-phycoerythrin and IA-phycoerythrin-Cy5 (all Ebioscience, San Diego, CA). Stained cells were washed, re-filtered over 100-μm nylon mesh and the CD11b+ Gr1lo IAhi cell population was sorted on a BD FACSAria to > 98% purity.
Stimulation of interferon-γ release by tumour macrophages
Macrophages from tumour cell suspensions were isolated by plastic adherence as above or sorted by FACS to > 98% pure CD11b+ Gr1lo IAhi cells and seeded to flat-bottomed 96-well plates. Replicate wells were variably treated with 1 ng/ml IL-12 (R&D Systems, Minneapolis, MN) and/or 1 or 10 ng/ml IL-18 (R&D Systems) for 48 hr. Supernatants of treated cells were tested for interferon-γ (IFN-γ) secretion by ELISA using matched antibodies from BD Biosciences (San Jose, CA) and compared to a standard curve of recombinant IFN-γ. For in vivo studies, mice bearing MCA205 tumours were treated with 250 μg αOX40 or control antibody on days 10 and 17 following tumour challenge and divided into groups receiving daily doses of 150 ng IL-18 along with 100 ng IL-12 intraperitoneally over days 12–19 or control treatment. Toxicity studies were first performed to confirm the tolerability of the IL-18/IL-12 doses used. For isolation of tumour infiltrates, tumours were harvested at day 17 before administration of the second antibody dose.
Microarray analysis
Total RNA was prepared from FACS sorted CD11b+ Gr1lo IAhi cells using a Qiagen RNA extraction kit (Valencia, CA). The Affymetrix Microarray Core Facility at Oregon Health and Sciences University (Portland, OR) prepared DNA probes and performed microarray analysis. Relative levels of gene expression were determined using GeneSifter (Geospiza Inc., Seattle, WA).
Western blotting
Macrophages isolated by plastic adherence or sorting by FACS were lysed in RIPA buffer and the protein content was quantified using a bicinchoninic acid assay kit (Sigma). Between 10 and 20 μg protein was denatured in SDS loading buffer containing β2-mercaptoethanol, electrophoresed on 12% denaturing gels and transferred to nitrocellulose. Blots were blocked and probed with antibodies to Arginase I (BD Biosciences), Ym1, GAPdH, or Actin (all R&D Systems) followed by anti-mouse-horseradish peroxidase (HRP), anti-rat-HRP or streptavidin-HRP (all Jackson Immunoresearch, West Grove, PA). Quantification of band intensity was performed on scanned blots using the public domain ImageJ analysis software.
Arginase enzyme activity
Arginase enzyme activity was quantified as previously described.26 Briefly, 10 or 20 μg protein from macrophage lysates was treated with 1 mm MnCl2 at 56° for 10 min and incubated with an equal volume of 0·5 m l-arginine (Sigma) for 30 min at 37°. The reaction was stopped by addition of 10× volume of H2SO4/H3PO4/H2O (in a ratio of 1/3/7). The conversion of l-arginine to urea was quantified by incubation at 95° for 30 min with α-isonitrosopropiophenone, alongside a standard curve of known urea concentration, and measurement of the absorption at 540 nm.
In vivo arginase inhibition
For in vivo studies of arginase enzyme inhibition, mice bearing MCA205 tumours were treated with 250 μg αOX40 or control antibody on days 10 and 17 following tumour challenge and divided into groups receiving daily doses of 10 mg/kg Nω-hydroxy-nor-l-arginine (NOHA) (EMD Biosciences, San Diego, CA) intraperitoneally over days 12–19 or control treatment. For analysis of infiltrate, tumours were isolated from treated and control groups on the day 17 time-point before administration of the second antibody dose.
Statistics
Statistical analysis between individual groups was performed using Student’s t-test in Graphpad Prism (Graphpad Software Inc., LaJolla, CA). Differences in survival of mice between treatment groups were investigated using the Log-rank test in Graphpad Prism. Where shown, bar charts show the mean ± standard error and NS indicates that differences were not significant; *P < 0·05; **P < 0·01; ***P < 0·001.
Results
Therapy of tumour-bearing mice with agonistic antibodies to OX40 results in tumour regression and long-term protection against rechallenge in a wide variety of tumour models.20 However, treatment of tumours that have been established for more than 10 days with a single dose of 250 μg αOX40 results in a transient decrease in tumour volume, but not a significant increase in survival.21 Established MCA205 tumours in C57BL/6 mice treated with a single dose of αOX40 show a decrease in size 7 days after αOX40 therapy (Fig. 1a). Flow cytometry of the tumour-infiltrating cells identified an αOX40-mediated increase in infiltrating CD8 T cells that was accompanied by a decrease in CD11b+ macrophages. This decrease was particularly noticeable in the CD11b+ Gr1hi population (Fig. 1b) that represents both immature macrophages and granulocytes and has been broadly defined as myeloid-derived suppressor cells.27 Despite these presumably positive effects on the tumour environment, a single dose of αOX40 provides only transient control of tumour growth to mice bearing established tumours, and only a minimal survival benefit (not shown).
Figure 1.
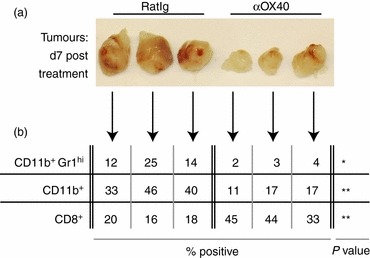
MCA205 cells were injected subcutaneousl in C57BL/6 mice and allowed to grow to 5–7 mm in diameter (approximately day 10). Mice were then injected intraperitoneally with 250 μg αOX40 or control RatIg and the tumour was removed 7 days later. (a) Photographs of individual isolated tumours are shown. (b) The percentage CD11b+Gr1hi IAlo immature macrophages, the overall CD11b+ macrophage population, and the percentage infiltrating CD8+ T cells from those same tumours are provided. NS: not significant; *P < 0·05; **P < 0·01; ***P < 0·001.
We previously demonstrated that the influx of effector CD8 T cells into the tumour initiated by OX40 therapy was associated with a decrease in a range of suppressive mechanisms in the tumour.21 These data suggest that remodelling of the tumour environment is a T-cell-mediated event following OX40 therapy. Although OX40 is predominantly described as a T-cell activation molecule, Baumann et al.28 demonstrated that neutrophils express functional OX40, and Pardee et al.29demonstrated that CD11b+ MHCII+ cells in the tumour express OX40. To determine the possibility of direct action of the αOX40 antibody on the macrophages or other non-T-cell populations, we incorporated a novel genetic model to identify OX40-expressing cells.24 This model avoids the possible artefacts associated with immunostaining of phagocytic and Fc-expressing cell populations. Genetically modified mice engineered to express Cre recombinase under control of the OX40 locus were bred with reporter mice that have floxed LacZ blocking the expression of GFP. In F1 mice, OX40-expressing cells excise LacZ and become irreversibly GFP+. Distinct GFP+ populations are detectable in the peripheral blood and in the tumour-infiltrating cells of OX40Cre × GFP mice (Fig. 2a). Gating on GFP+ cells in the peripheral blood of naive mice or in the tumour exclusively identified CD4+ or CD8+ cells (Fig. 2b), suggesting that no other cell populations significantly express OX40. The major population of GFP+ cells was CD4+ (Fig. 2b), and may include antigen-experienced memory CD4 T cells that had transiently expressed OX40, along with regulatory T cells that are known to constitutively express OX40.30 By first gating on CD11b+ cells infiltrating the tumour, we confirmed that macrophages showed no evidence of OX40 expression as opposed to the large proportion of tumour-infiltrating CD4 T cells that were GFP+ (Fig. 2c). A greater percentage of CD8 T cells were GFP+ in the tumour than in the tumour-draining lymph node or non-draining lymph node (Fig. 2d). In each case, expression of GFP corresponded with expression of CD44, confirming that GFP+ cells represent antigen-experienced cells. This genetic model: 1) shows that only T cells express OX40 within the tumour; 2) shows that CD8 T cells that have expressed OX40 can accumulate within the tumour environment; and 3) confirms previous studies in OX40 knockout mice showing that changes in macrophage and other myeloid populations within the tumour following αOX40 therapy are indirect, occurring through stimulation of OX40 on T cells.21
Figure 2.
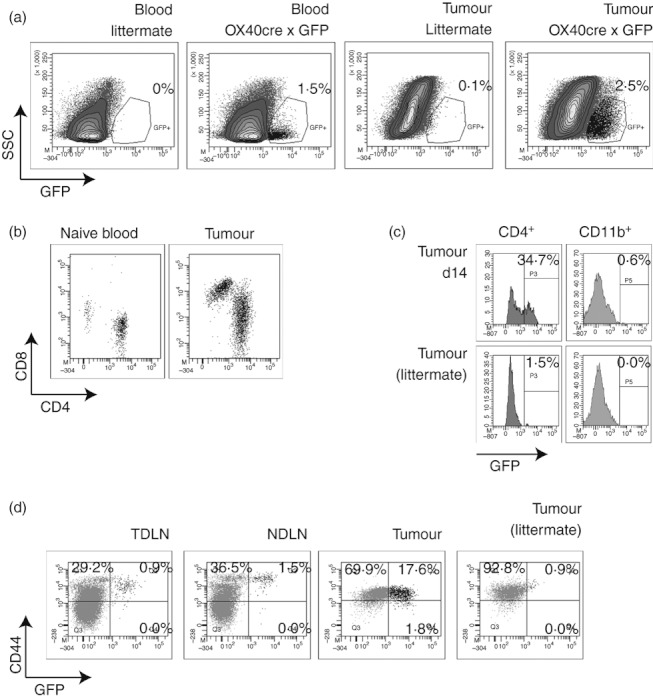
Transgenic mice with Cre recombinase under the control of the endogenous OX40 promoter were crossed with GFP reporter mice to generate F1 mice that show stable expression of GFP in cells that have expressed OX40. OX40Cre+ GFP+ or littermates were injected subcutaneously with MCA205 tumours and the tumour-infiltrating cells were harvested at day 14. (a) Dot plots show GFP expression in peripheral blood mononuclear cells (PBMC) or tumour-infiltrating cells from tumour-bearing OX40Cre+ GFP+ F1 mice or littermates. (b) CD4 and CD8 expression on gated GFP+ cells in the peripheral blood of a naive mouse or in a tumour. (c) GFP expression in gated d14 tumour-infiltrating CD4+ or CD11b+ cells from OX40Cre+ GFP+ or littermates. (d) Analysis of CD44 expression on gated CD8+ cells in the tumour-draining lymph node, non-draining lymph node and tumour of day 17 tumour-bearing OX40Cre+ GFP+ mice or littermates.
To explain the transient control of tumour growth achieved by αOX40 therapy, we proposed that tumour recurrence is a result of local tumour suppression of adaptive immunity. Acute responses to infectious agents are terminated by inflammatory resolution in the target environment.31–34 Such an occurrence in the tumour could suppress the pro-inflammatory changes induced by the influx of CD8 T cells and re-establish an anti-inflammatory and suppressive environment. Macrophages are a major determinant of acute versus resolving inflammation and are regulated by αOX40 therapy (Fig. 1). To examine the macrophage contribution to the failure of OX40 therapy, we isolated tumour macrophages 7 days after antibody administration, which represents not only the peak of T-cell infiltration but also the time beyond which tumours begin to regrow. We have previously established that the CD11b+ tumour macrophages can be subdivided into MHC class II (IA) negative Gr1hi granulocytic and myeloid-derived suppressor cells, and IA+ Gr1lo macrophages.21 We performed microarray gene expression analysis to examine the response of tumour macrophages to αOX40 therapy. Tumour-bearing mice received a single dose of αOX40 or control antibody. Seven days later, CD11b+ IA+ Gr1lo mature macrophages were sorted from the tumours of control or αOX40-treated mice (Fig. 3a and b), macrophage RNA was purified and Gene Expression Microarray analysis was performed. The gene expression pattern confirmed the isolated macrophage phenotype (Fig. 3c); there was abundant expression of CD14, F4/80, and low or absent expression of B-cell, T-cell and endothelial markers. Interestingly, Chi3l3 (Ym1), Retnla (Fizz1) and Chi3l4 (Ym2) are the three most up-regulated genes in tumour macrophages from αOX40-treated mice (see Supplementary material, Fig. S1). Macrophages can be polarized into distinct phenotypes with very different consequences on immune responses; Ym1 and Fizz1 are associated with alternative macrophage activation.35 As opposed to classically activated (M1) macrophages, alternatively activated (M2) macrophages can inhibit T-cell function,36 in part through expression of arginase I.37,38 To determine the differentiation of the tumour macrophages, we examined the expression of characteristic M1 and M2 markers as described by Mantovani et al.39 Interestingly, the tumour macrophages showed a mixed M1 and M2 pattern of gene expression. Whereas the macrophages expressed M2-associated phagocytic and scavenger receptors, the cells expressed M1-associated chemokines and cytokines.39 This chemokine expression pattern would be more able to attract T helper type 1 cells, type 1 cytotoxic T cells and natural killer cells via CCL5, CXCL16 and CXCL9, and less likely to attract T helper type 2 and T regulatory cells in view of the low or absent CCL1, CCL17, CCL22, CCL24 and CXCL13. Nevertheless, the tumour macrophages express abundant arginase I, and like Fizz1 and Ym1, expression of arginase I increases following αOX40 therapy. Hence, the mixture of macrophages in the tumour actively recruit adaptive immune effectors to the tumour, but macrophages in the tumour simultaneously express the immunosuppressive M2 marker arginase I.
Figure 3.
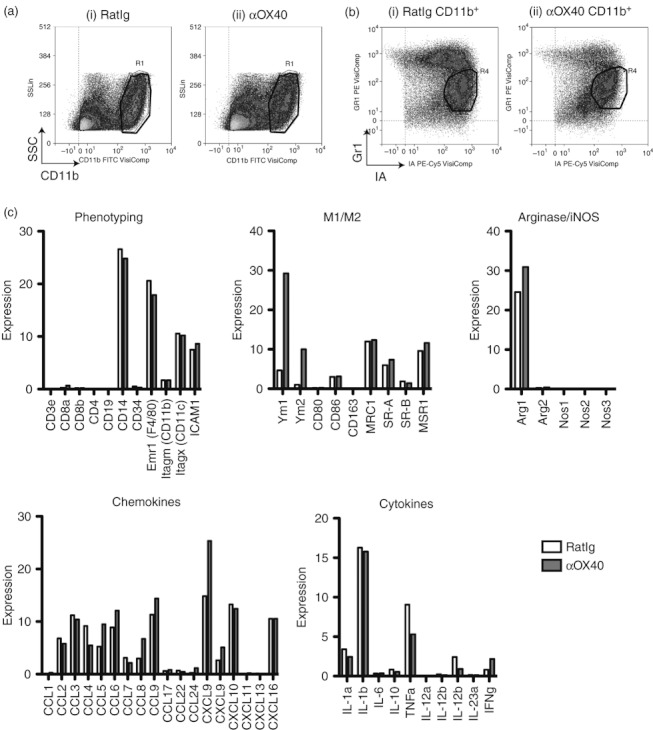
MCA205 cells were injected subcutaneously in C57BL/6 mice and allowed to grow to 5–7 mm in diameter (approximately day 10), whereupon mice were injected intraperitoneally with 250 μg αOX40 or control RatIg and 7 days later the tumour was removed. (a) CD11b+ tumour macrophages from (i) RatIg and (ii) αOX40-treated mice were (b) sub-gated to select only the Gr1lo IAhi cells to > 99% purity. Total RNA was isolated from FACSorted CD11b+ Gr1lo IAhi macrophages and microarray assays were performed in the Affymetrix Microarray Core of the OHSU Gene Microarray Shared Resource. (c) Relative levels of gene expression were determined using GeneSifter. Where multiple probes were matched to an individual gene, representative expression results are shown.
To validate the microarray differences we confirmed the gene expression patterns by performing Western blots. C57BL/6 mice bearing MCA205 tumours were treated with a single dose of αOX40 or control antibody and tumours were harvested 7 days later. CD11b+ Gr1lo mature macrophages were sorted from pooled tumours from control or αOX40-treated mice and lysed for Western blot analysis. Consistent with the microarray data, an increased arginase I protein expression was observed in the tumour macrophages from αOX40-treated animals (Fig. 4a). We also observed increased expression of the M2-associated marker Ym1 in the tumour macrophages from αOX40-treated animals, though the detected signal was much weaker than the gene expression would suggest, potentially because of secretion of the Ym1 protein.35 To examine arginase expression in tumours from individual mice treated with αOX40 or control antibodies, tumour macrophages were enriched from tumour-infiltrating cell populations by adherence to plastic. Tumour macrophages from αOX40-treated mice demonstrated a significant up-regulation in arginase expression when compared with macrophages isolated from control tumours (P < 0·05) (Fig. 4b). To confirm that the protein levels correspond to functional enzyme activity, macrophage lysates were tested for their ability to convert l-arginine into urea. We demonstrate a correlation between protein expression detected by Western blot and arginase enzyme activity, confirming that αOX40 treatment increased arginase enzyme in macrophages in the tumour (Fig. 4c). These data strongly suggest that αOX40 therapy increases arginase within tumour-infiltrating macrophages. Such an up-regulation of arginase is highly consistent with T-cell immunosuppression.37,38 We hypothesize that these high levels of arginase will limit the efficacy of αOX40 therapy.
Figure 4.
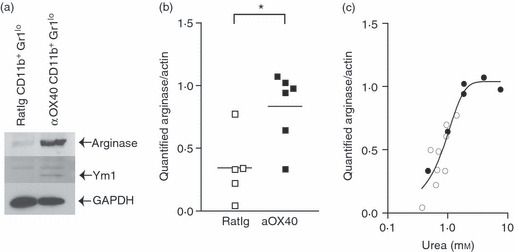
(a) MCA205 cells were injected subcutaneously in C57BL/6 mice and allowed to grow to 5–7 mm in diameter (approximately day 10). Mice were injected intraperitoneally with 250 μg αOX40 or control RatIg and after 7 days the tumour was removed, tumour-infiltrating cells were sorted by FACS to purify CD11b+ Gr1lo IAhi populations, and these cells were lysed and Western blotted for arginase, Ym1 and GAPDH expression. (b) Tumour-infiltrating cells were harvested as in (a), then the adherent macrophage population from individual tumours (> 90% CD11b+ by FACS analysis) was lysed and Western blotted using antibodies specific for arginase, then stripped and reprobed with antibodies specific for actin. Graph shows quantification of arginase bands standardized to actin expression. Each symbol represents the data from one tumour-bearing animal. (c) Lysates of tumour macrophages from (b) were tested for arginase enzyme activity by conversion of l-arginine to urea, and compared to a standard curve of known urea concentration by colorimetric assay. The graph correlates quantified arginase expression levels by Western blot to arginase enzyme activity in macrophages from mice treated with RatIg (open circles) or αOX40 (closed circles). Each symbol represents the data from one tumour-bearing animal. *P < 0·05.
To test whether the increase in arginase expression limited the efficacy of αOX40 therapy, arginase activity was blocked in vivo. C57BL/6 mice bearing MCA205 tumours were treated with αOX40 or control immunoglobulin. Arginase activity was blocked with daily injections of the specific inhibitor NOHA beginning 2 days after antibody treatment. Tumour-infiltrating cells were analysed at day 17 following tumour injection (day 7 following αOX40 therapy). Although αOX40 treatment significantly increased CD8 infiltration (P < 0·01) and decreased the proportion of immature CD11b+ Gr1hi macrophages (P < 0·001), arginase inhibition did not significantly alter the percentage of CD8 or immature macrophages infiltrating the tumour (Fig. 5a). To determine the effect of arginase inhibition on the efficacy of αOX40 therapy, MCA205 tumour-bearing mice were treated at day 10 with αOX40 or control antibody and again 7 days later. Arginase enzyme activity was blocked with daily injections of the specific inhibitor NOHA over days 12–19. αOX40 alone significantly delayed tumour growth (P < 0·001), but few animals were tumour-free at the end of the experiment (Fig. 5b). While treatment with NOHA alone also significantly increased survival (P < 0·001), the change in median survival was minimal and no mice survived long-term. Importantly, combined treatment with αOX40 and NOHA significantly increased survival compared with either agent alone (P < 0·001 versus NOHA; P < 0·05 versus αOX40), and resulted in approximately 40% of animals completely rejecting their established tumours. These data support the hypothesis that expression of arginase as a result of inflammatory resolution in the tumour limits the efficacy of αOX40 therapy.
Figure 5.
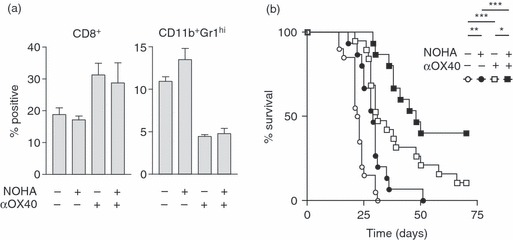
(a) C57BL/6 mice were challenged with MCA205 and treated at day 10 with 250 μg αOX40 or control RatIg. Half of these mice received additional treatment with daily intraperitoneal injections of the arginase inhibitor NOHA (10 mg/kg) from day 12. Tumours were harvested at day 17 for analysis of tumour-infiltrating cells. Graphs show the mean and standard deviation of tumour-infiltrating CD8 T cells and CD11b+ Gr1hi IAlow macrophages. (b) Survival of C57BL/6 mice challenged with MCA205 and treated at day 10 and day 17 with 250 μg αOX40 or control RatIg. Half of these mice received additional treatment with daily intraperitoneal injections of the arginase inhibitor NOHA (10 mg/kg) from day 12 to day 19. NS: not significant; *P < 0·05; **P < 0·01; ***P < 0·001.
If inflammatory resolution is responsible for the termination of OX40-mediated control of tumour growth, we hypothesized that prolonging acute inflammation would extend the effect of OX40 therapy. Interestingly, of the genes associated with antigen processing and presentation, IFNg is one of the few genes showing evidence of up-regulation in tumour macrophages following OX40 therapy (Fig. S2). We considered strategies to prolong the presence of IFN-γ in the tumour environment. The effects of αOX40 have been improved by combination with IL-12 40 or IL-18,41 and while each is a potent IFN-γ-inducing pro-inflammatory cytokine, they are known to synergize strongly.42 Macrophage populations have been shown to produce IFN-γ in response to dual stimulation with IL-18 and IL-12,43 and we considered that this might be a method to prolong the presence of the M1-driving cytokine IFN-γ in the tumour environment. Interestingly, IL18R1 was up-regulated in tumour macrophages from OX40-treated mice (Fig. 6a), suggesting that these cells may be responsive to cytokine stimulation. To test the functional response of tumour-isolated macrophages to IL-18 and IL-12, mice bearing MCA205 tumours were treated with αOX40 or control antibody and 7 days later macrophages were enriched from tumour-infiltrating cells by adherence to plastic. Macrophage IFN-γ secretion was assayed following treatment with IL-18, IL-12, or both. Adherent cells secreted significant quantities of IFN-γ only in the presence of both IL-12 and IL-18 (Fig. 6bi). Importantly, tumour macrophages from mice treated with αOX40 secreted significantly more IFN-γ than those from control treated animals, suggesting that these macrophages may be functionally different following αOX40 therapy. Both IL-12 and IL-18 have been shown to have biological activity on non-macrophage populations that may have contaminated the adherent culture,43 therefore we used FACS to sort to high purity CD11b+ Gr1lo mature macrophages from pooled αOX40 or control treated tumours. Again, only the combination of IL-18 and IL-12 resulted in IFN-γ secretion from tumour macrophages and the tumour macrophages from mice treated with αOX40 secreted significantly more IFN-γ than those from control treated animals (Fig. 6bii).
Figure 6.
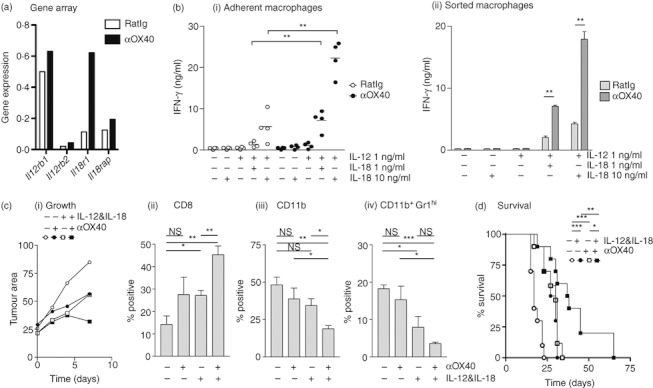
(a) Affymetrix gene expression analysis of interleukin-12 (IL-12) and IL-18 receptor expression in FACS-analysed CD11b+ Gr1lo IAhi macrophages. (b) MCA205 cells were injected subcutaneously in C57BL/6 mice and allowed to grow to 5–7 mm in diameter (approximately day 10). Mice were injected intraperitoneally with 250 μg αOX40 or control RatIg and after 7 days tumours were removed, tumour-infiltrating cells were isolated. (i) The adherent macrophage population from individual tumours (> 90% CD11b+ by FACS analysis) was treated in vitro with IL-12, IL-18 or both. Supernatants were harvested at 48 hr and tested for interferon-γ (IFN-γ) secretion by ELISA. Each symbol represents the data from one tumour-bearing animal. (ii) Repeat of the experiment shown in (i) but using CD11b+ Gr1lo IAhi cells FACS sorted from the tumour. (c) C57BL/6 mice were challenged with MCA205 and treated at day 10 with 250 μg αOX40 or control RatIg. Half of these mice received additional treatment with daily intraperitoneal injections of IL-12 and IL-18 from day 12. Tumours were harvested at day 17 for analysis of tumour-infiltrating cells. Graphs show (i) the mean tumour area following initiation of therapy, and the mean and standard error of (ii) CD8 T cells (iii) CD11b+ cells and (iv) CD11b+ Gr1hi IAlow cells infiltrating the tumour. (d) Survival of C57BL/6 mice challenged with MCA205 and treated at day 10 and day 17 with 250 μg αOX40 or control RatIg. Half of these mice received additional treatment with daily intraperitoneal injections of IL-12 and IL-18 from day 12 to day 19. NS: not significant; *P < 0·05; **P < 0·01; ***P < 0·001.
In view of these data, we hypothesized that treatment with IL-12 and IL-18 would synergize with αOX40 therapy to improve control of tumours in vivo. To test this, MCA205 tumour-bearing mice were treated at day 10 with αOX40 or control antibody with or without daily injection of IL-12 and IL-18 beginning on day 14. While tumour growth was slowed by either αOX40 or IL-12 + IL-18, only the combination of all of these led to tumour regression by day 17 (Fig. 6ci). Tumours were harvested at day 17 and tumour-infiltrating cells were analysed. αOX40 combined with IL-12 + IL-18 increased infiltration by CD8 T cells and decreased the proportion of macrophages, particularly Gr1hi immature macrophages in the tumour environment (Fig. 6cii–iv). To determine the effect of combination treatment on survival, MCA205 tumour-bearing mice were treated at day 10 and day 17 with αOX40 or control antibody with or without daily IL-12 and IL-18 over days 12–19. Whereas treatment with either αOX40 alone or combined therapy with IL-12 and IL-18 enhanced survival (αOX40 P < 0·001, IL-12 + IL-18 P < 0·001), the combination of αOX40 with IL-12 and IL-18 significantly enhanced survival compared with either strategy alone (αOX40 + IL-12 + IL-18 P < 0·05 versus αOX40, P < 0·01 versus IL-12 + IL-18) (Fig. 6d). These data demonstrate that IL-12 and IL-18 in combination with αOX40 therapy heightens changes in the tumour environment, and results in increased survival of mice bearing large tumours in vivo. Interestingly, administration of IL-12 and IL-18 alone had little effect beyond the final dose; we hypothesize that inflammatory resolution is rapidly initiated following cytokine withdrawal. These data support the hypothesis that manipulation of inflammation in the tumour environment can enhance the effect of OX40 therapy.
Discussion
The experiments shown here demonstrate that therapy with OX40 agonistic antibodies acts on T cells to alter the tumour environment (Figs 1, 2). These alterations include a favourable increase in CD4 and CD8 T cells, but depending on the ‘age’ of the tumour may be followed by potentially unfavourable negative feedback mechanisms that include up-regulation of arginase by macrophages. (Figs 3, 4). We demonstrate that blocking macrophage-mediated immune suppression by inhibiting arginase I with the specific inhibitor NOHA significantly increased αOX40-mediated survival in tumour-bearing animals (Fig. 5). We show that the combination of IL-12 and IL-18 results in enhanced IFN-γ release by tumour macrophages from αOX40-treated mice. Hence, therapy with αOX40 along with IL-12 and IL-18 results in increased CD8 infiltrates and decreased macrophage proportions in the tumour, and enhanced therapeutic efficacy (Fig. 6).
These data have significant implications for the translation of T-cell therapies to the clinic. Whereas many T-cell-based therapies are highly effective in animal models, their efficacy depends either on transfer very early in tumour development, or the transfer of extremely large numbers of tumour-specific T cells. In view of these and other data, one explanation for these observations is that the tumour takes time to establish an immune suppressive environment. In transplantable tumour models it takes 9–10 days to establish this suppressive environment,44,45 which includes both suppressive macrophages and regulatory T cells.3 These data also imply that in cancer patients, the existence of a suppressive immune environment in the tumour will be a significant impediment to the use of adjuvant antibodies or T-cell therapies as single agents. Though we see significant influxes of effector CD8 T cells, the presence or restoration of suppression resists long-term control of tumour growth. For this reason we hypothesize that αOX40 therapy for established tumours would be most effective as part of a combination therapy that includes strategies to prevent inflammatory resolution at the tumour.
The array of suppressive mechanisms demonstrated to be operative in the tumour continues to grow. One of the more recent discoveries is the role of the enzyme arginase I. The relative expression of arginase versus iNOS has been used to discriminate between M2 and M1 macrophages, respectively.11 The products of arginase activity have been demonstrated to suppress T-cell activation, whereas specific inhibition of arginase has been shown to restore T-cell activity in vitro and in vivo.37,38 Administration of the arginase inhibitor NOHA as a single agent has been previously shown to delay tumour growth.37 Our data demonstrate that arginase inhibition can be an effective addition to a T-cell-targeted therapy, which both enhances effector function and reduces tumour-induced suppressive activity.
Our data showed that the increase in tumour-infiltrating CD8 T cells following αOX40 therapy was associated with changes in macrophages in the tumour environment. These data are consistent with the results of Zhang et al., 46who demonstrated that effector T cells caused tumour destruction by targeting the non-malignant cells in the tumour stroma. We show that αOX40 therapy led to a decrease in the proportion of macrophages in the tumour, and further experiments will be necessary to distinguish whether this process occurs through selective macrophage destruction by effector T cells, macrophage differentiation within the tumour, or recruitment of new macrophages to the altered tumour site. Appropriately activated macrophages can become tumoricidal 47 via TNF-α and nitric oxide; our gene array shows down-regulation of TNF-α and absent iNOS in macrophages from αOX40-treated mice (see Supplementary material, Fig. S2 and Fig. 3). However, whether T cells can activate macrophages to become tumoricidal is influenced by the tumour environment.48 This, together with our previous data demonstrating decreased T-cell inhibition by tumour macrophages following OX40 therapy,21 suggests a complicated immune environment in the tumour. Surprisingly, despite the influx of effectors expressing IFN-γ,21 the mixture of tumour macrophages exhibited M2 markers alongside M1 markers. These data suggest that continued presence of other differentiation factors in the tumour limit M1 differentiation and initiate inflammatory resolution. The ability of IL-12 and IL-18 to cause IFN-γ secretion by macrophages has previously been demonstrated in bone marrow macrophage and peritoneal macrophage populations.49 We demonstrate for the first time that tumour macrophages also respond to this cytokine combination ex vivo. However, IL-12 and IL-18 have each been shown to synergize with αOX40 therapy in enhancing the differentiation and effector function of T cells in vivo40,41,50 and potently synergize to induce IFN-γ release from T cells.51 The contribution of IFN-γ secretion by tumour-resident macrophages in our model remains to be determined; nevertheless the potential to extend in situ differentiation of suppressive tumour macrophages may be of great advantage to prolong effective T-cell-mediated control of tumours.
These data are consistent with a model where αOX40 therapy increases the activity of tumour antigen-specific effector T cells, resulting in increased trafficking to the tumour and acute inflammation. The consequence to the tumour is growth suppression, but the influence of the tumour immune environment ultimately suppresses the T-cell response, in part by arginase up-regulation in tumour-associated macrophages. We demonstrated that blocking this suppression via arginase inhibition, or by extending acute inflammation with T helper type 1/M1 cytokines can overcome the suppressive influence of the tumour. The data presented in this manuscript have implications for the application of T-cell-targeted co-stimulatory antibodies for cancer immunotherapy, and suggest that addressing tumour immune suppression will be a necessary addition for effective therapy of established tumours with suppressive environments.
Acknowledgments
These experiments would not have been possible without the assistance of Daniel Haley for flow cytometry analysis and sorting. Acknowledgements are due to Dr Walter Urba for excellent comments and advice on the manuscript. This work was supported by National Institutes of Health grants CA122701 and CA102577.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Figure S1. Analysis of the 50 most (a)up-regulated and (b) down-regulated genes in tumor macrophages 7 days after systemic OX40 stimulation. To exclude outliers, genes designated as present in both categories, and genes expressed in αOX40-treated tumor macrophages with an intensity < 2 (dotted line) are not shown.
Figure S2. Analysis of expression of genes within the KEGG Antigen Procession and Presentation pathway. Genes are sorted in order of expression, and higher and lower expression genes are shown on different axes to improve visibility. Graphs only show genes designated as present, and expressed at an intensity > 0·5.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than about missing material) should be directed to the corresponding author for the article.
References
- 1.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–12. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 2.Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315–22. doi: 10.1007/s10555-006-9001-7. [DOI] [PubMed] [Google Scholar]
- 3.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–66. doi: 10.1172/JCI31422. PMCID: 1857267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86:1159–66. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 5.Linehan WM, Walther MM, Alexander RB, Rosenberg S. Adoptive immunotherapy of renal cell carcinoma: studies from the Surgery Branch, National Cancer Institute. Semin Urol. 1993;11:41–3. [PubMed] [Google Scholar]
- 6.Gough M, Crittenden M, Thanarajasingam U, et al. Gene therapy to manipulate effector T cell trafficking to tumors for immunotherapy. J Immunol. 2005;174:5766–73. doi: 10.4049/jimmunol.174.9.5766. [DOI] [PubMed] [Google Scholar]
- 7.Huang B, Pan PY, Li Q, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 8.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 9.Filippi CM, von Herrath MG. IL-10 and the resolution of infections. J Pathol. 2008;214:224–30. doi: 10.1002/path.2272. [DOI] [PubMed] [Google Scholar]
- 10.Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–9. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruby CE, Redmond WL, Haley D, Weinberg AD. Anti-OX40 stimulation in vivo enhances CD8+ memory T cell survival and significantly increases recall responses. Eur J Immunol. 2007;37:157–66. doi: 10.1002/eji.200636428. [DOI] [PubMed] [Google Scholar]
- 13.Redmond WL, Gough MJ, Charbonneau B, Ratliff TL, Weinberg AD. Defects in the acquisition of CD8 T cell effector function after priming with tumor or soluble antigen can be overcome by the addition of an OX40 agonist. J Immunol. 2007;179:7244–53. doi: 10.4049/jimmunol.179.11.7244. [DOI] [PubMed] [Google Scholar]
- 14.Evans DE, Prell RA, Thalhofer CJ, Hurwitz AA, Weinberg AD. Engagement of OX40 enhances antigen-specific CD4+ T cell mobilization/memory development and humoral immunity: comparison of αOX-40 with αCTLA-4. J Immunol. 2001;167:6804–11. doi: 10.4049/jimmunol.167.12.6804. [DOI] [PubMed] [Google Scholar]
- 15.Gramaglia I, Weinberg AD, Lemon M, Croft M. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol. 1998;161:6510–7. [PubMed] [Google Scholar]
- 16.Song A, Tang X, Harms KM, Croft M. OX40 and Bcl-xL promote the persistence of CD8 T cells to recall tumor-associated antigen. J Immunol. 2005;175:3534–41. doi: 10.4049/jimmunol.175.6.3534. [DOI] [PubMed] [Google Scholar]
- 17.Matsumura Y, Hori T, Nishigori C, et al. Expression of CD134 and CD134 ligand in lesional and nonlesional psoriatic skin. Arch Dermatol Res. 2003;294:563–6. doi: 10.1007/s00403-002-0363-6. [DOI] [PubMed] [Google Scholar]
- 18.Yoshioka T, Nakajima A, Akiba H, et al. Contribution of OX40/OX40 ligand interaction to the pathogenesis of rheumatoid arthritis. Eur J Immunol. 2000;30:2815–23. doi: 10.1002/1521-4141(200010)30:10<2815::AID-IMMU2815>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 19.Weinberg AD, Wegmann KW, Funatake C, Whitham RH. Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J Immunol. 1999;162:1818–26. [PubMed] [Google Scholar]
- 20.Weinberg AD, Rivera MM, Prell R, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol. 2000;164:2160–9. doi: 10.4049/jimmunol.164.4.2160. [DOI] [PubMed] [Google Scholar]
- 21.Gough MJ, Ruby CE, Redmond WL, Dhungel B, Brown A, Weinberg AD. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res. 2008;68:5206–15. doi: 10.1158/0008-5472.CAN-07-6484. [DOI] [PubMed] [Google Scholar]
- 22.Ko K, Yamazaki S, Nakamura K, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med. 2005;202:885–91. doi: 10.1084/jem.20050940. PMCID: 2213162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang YF, Zou JP, Mu J, et al. Enhanced induction of antitumor T-cell responses by cytotoxic T lymphocyte-associated molecule-4 blockade: the effect is manifested only at the restricted tumor-bearing stages. Cancer Res. 1997;57:4036–41. [PubMed] [Google Scholar]
- 24.Klinger M, Kim JK, Chmura SA, Barczak A, Erle DJ, Killeen N. Thymic OX40 expression discriminates cells undergoing strong responses to selection ligands. J Immunol. 2009;182:4581–9. doi: 10.4049/jimmunol.0900010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JK, Klinger M, Benjamin J, et al. Impact of the TCR signal on regulatory T cell homeostasis, function, and trafficking. PLoS ONE. 2009;4:e6580. doi: 10.1371/journal.pone.0006580. PMCID: 2719063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corraliza IM, Campo ML, Soler G, Modolell M. Determination of arginase activity in macrophages: a micromethod. J Immunol Methods. 1994;174:231–5. doi: 10.1016/0022-1759(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 27.Gabrilovich DI, Bronte V, Chen SH, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. author reply 6. PMCID: 1941787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baumann R, Yousefi S, Simon D, Russmann S, Mueller C, Simon HU. Functional expression of CD134 by neutrophils. Eur J Immunol. 2004;34:2268–75. doi: 10.1002/eji.200424863. [DOI] [PubMed] [Google Scholar]
- 29.Pardee AD, McCurry D, Alber S, Hu P, Epstein AL, Storkus WJ. A therapeutic OX40 agonist dynamically alters dendritic, endothelial, and T cell subsets within the established tumor microenvironment. Cancer Res. 2010;70:9041–52. doi: 10.1158/0008-5472.CAN-10-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeda I, Ine S, Killeen N, et al. Distinct roles for the OX40–OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol. 2004;172:3580–9. doi: 10.4049/jimmunol.172.6.3580. [DOI] [PubMed] [Google Scholar]
- 31.Serhan CN, Brain SD, Buckley CD, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–32. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–7. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 33.Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discovery. 2004;3:401–16. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- 34.Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol. 2002;2:787–95. doi: 10.1038/nri915. [DOI] [PubMed] [Google Scholar]
- 35.Nair MG, Cochrane DW, Allen JE. Macrophages in chronic type 2 inflammation have a novel phenotype characterized by the abundant expression of Ym1 and Fizz1 that can be partly replicated in vitro. Immunol Lett. 2003;85:173–80. doi: 10.1016/s0165-2478(02)00225-0. [DOI] [PubMed] [Google Scholar]
- 36.Liu Y, Van Ginderachter JA, Brys L, De Baetselier P, Raes G, Geldhof AB. Nitric oxide-independent CTL suppression during tumor progression: association with arginase-producing (M2) myeloid cells. J Immunol. 2003;170:5064–74. doi: 10.4049/jimmunol.170.10.5064. [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez PC, Quiceno DG, Zabaleta J, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez PC, Zea AH, DeSalvo J, et al. l-Arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol. 2003;171:1232–9. doi: 10.4049/jimmunol.171.3.1232. [DOI] [PubMed] [Google Scholar]
- 39.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 40.Kuriyama H, Watanabe S, Kjaergaard, et al. Mechanism of third signals provided by IL-12 and OX-40R ligation in eliciting therapeutic immunity following dendritic–tumor fusion vaccination. Cell Immunol. 2006;243:30–40. doi: 10.1016/j.cellimm.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 41.Maxwell JR, Yadav R, Rossi RJ, et al. IL-18 bridges innate and adaptive immunity through IFN-γ and the CD134 pathway. J Immunol. 2006;177:234–45. doi: 10.4049/jimmunol.177.1.234. [DOI] [PubMed] [Google Scholar]
- 42.Carson WE, Dierksheide JE, Jabbour S, et al. Coadministration of interleukin-18 and interleukin-12 induces a fatal inflammatory response in mice: critical role of natural killer cell interferon-γ production and STAT-mediated signal transduction. Blood. 2000;96:1465–73. [PubMed] [Google Scholar]
- 43.Darwich L, Coma G, Pena R, et al. Secretion of interferon-γ by human macrophages demonstrated at the single-cell level after costimulation with interleukin (IL)-12 plus IL-18. Immunology. 2009;126:386–93. doi: 10.1111/j.1365-2567.2008.02905.x. PMCID: 2669819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bursuker I, North RJ. Generation and decay of the immune response to a progressive fibrosarcoma II. Failure to demonstrate postexcision immunity after the onset of T cell-mediated suppression of immunity. J Exp Med. 1984;159:1312–21. doi: 10.1084/jem.159.5.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.North RJ, Bursuker I. Generation and decay of the immune response to a progressive fibrosarcoma I. Ly-1+2– suppressor T cells down-regulate the generation of Ly-1–2+ effector T cells. J Exp Med. 1984;159:1295–311. doi: 10.1084/jem.159.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang B, Karrison T, Rowley DA, Schreiber H. IFN-γ- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J Clin Invest. 2008;118:1398–404. doi: 10.1172/JCI33522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cleveland RP, Meltzer MS, Zbar B. Tumor cytotoxicity in vitro by macrophages from mice infected with Mycobacterium bovis strain BCG. J Natl Cancer Inst. 1974;52:1887–95. doi: 10.1093/jnci/52.6.1887. [DOI] [PubMed] [Google Scholar]
- 48.Vicetti Miguel RD, Cherpes TL, Watson LJ, McKenna KC. CTL induction of tumoricidal nitric oxide production by intratumoral macrophages is critical for tumor elimination. J Immunol. 2010;185:6706–18. doi: 10.4049/jimmunol.0903411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Munder M, Mallo M, Eichmann K, Modolell M. Murine macrophages secrete interferon γ upon combined stimulation with interleukin (IL)-12 and IL-18: a novel pathway of autocrine macrophage activation. J Exp Med. 1998;187:2103–8. doi: 10.1084/jem.187.12.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruby CE, Montler R, Zheng R, Shu S, Weinberg AD. IL-12 is required for anti-OX40-mediated CD4 T cell survival. J Immunol. 2008;180:2140–8. doi: 10.4049/jimmunol.180.4.2140. [DOI] [PubMed] [Google Scholar]
- 51.Tomura M, Maruo S, Mu J, et al. Differential capacities of CD4+, CD8+, and CD4–CD8– T cell subsets to express IL-18 receptor and produce IFN-γ in response to IL-18. J Immunol. 1998;160:3759–65. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.