

Abstract
Transcriptional regulation in mammalian cells is driven by a complex interplay of multiple transcription factors that respond to signals from either external or internal stimuli. A single transcription factor can control expression of distinct sets of target genes, dependent on its state of post-translational modifications, interacting partner proteins, and the chromatin environment of the cellular genome. Furthermore, many transcription factors can act as either transcriptional repressors or activators, depending on promoter and cellular contexts (Alvarez, et al., 2003). Even in this light, the versatility of LSF (Late SV40 Factor) is remarkable. A hallmark of LSF is its unusual DNA binding domain, as evidenced both by lack of homology to any other established DNA-binding domains and by its DNA recognition sequence. Although a dimer in solution, LSF requires additional multimerization with itself or partner proteins in order to interact with DNA. Transcriptionally, LSF can function as an activator or a repressor. It is a direct target of an increasing number of signal transduction pathways. Biologically, LSF plays roles in cell cycle progression and cell survival, as well as in cell lineage-specific functions, shown most strikingly to date in hematopoietic lineages.
This review discusses how the unique aspects of LSF DNA-binding activity may make it particularly susceptible to regulation by signal transduction pathways and may relate to its distinct biological roles. We present current progress in elucidation of both tissue-specific and more universal cellular roles of LSF. Finally, we discuss suggestive data linking LSF to signaling by the amyloid precursor protein and to Alzheimer's disease, as well as to the regulation of latency of the human immunodeficiency virus (HIV).
Keywords: GRH, DNA-binding, signal transduction, cell cycle progression, immune response, APP, HIV
1. LSF/GRH transcription factor family
LSF was first identified in HeLa cell extracts as a transcriptional activator of the late Simian Virus 40 promoter (Huang et al., 1990; Kim et al., 1987; Shirra et al., 1994). Independent identification of LSF as a DNA-binding protein and transcriptional regulator of other viral and cellular promoters (Table 1) resulted in the additional names of LBP-1 or UBP-1 (on the HIV long terminal repeat (Jones et al., 1988; Wu et al., 1988; Yoon et al., 1994)), CP2 (on the murine α-globin promoter (Barnhart et al., 1988; Kim et al., 1988; Lim et al., 1992)), and SEF1 (on the murine serum amyloid A3 promoter (Huang et al., 1994)). For each of these identified biological target promoters, independent biochemical purification of the DNA-binding activity led to sequencing of peptides (Bing et al., 1999; Lim et al., 1992; Shirra et al., 1994; Yoon et al., 1994) and cloning of cDNAs (Lim et al., 1992; Shirra et al., 1994; Yoon et al., 1994).
Table 1.1.
Multiple independent identifications of LSF.
| Name | Publication year | Reference | |
|---|---|---|---|
| LSF | Late Simian Virus 40 promoter factor | 1987 | Kim et al, 1987 |
| (α)CP2 | CCAAT binding protein-2 (on α-globin) | 1988 |
Barnhart et al, 1988
Kim et al, 1988 |
| LBP-1c | Leader binding protein-1c (on HIV LTR) | 1988 |
Jones et al, 1988
Yoon et al, 1994 |
| UBP-1 | Upstream region binding protein-1 (on HIV LTR) | 1988 | Wu et al, 1988 |
| SEF1 | Serum amyloid A3 enhancer factor-1 | 1994 |
Huang et al, 1994
Bing et al, 1999 |
With regards to nomenclature, the name of CP2, although widely used to identify LSF, has generated some confusion. CP2 originally signified “α-globin CCAAT-binding protein 2” (initially αCP2), due to the erroneous initial assumption that it recognized CCAAT motifs in the α-globin promoter (Barnhart et al., 1988). There exists an authentic CCAAT-binding protein named CP2 (Chodosh et al., 1988), which was even suggested, at first, to be identical (Kim et al., 1990). However, there is now universal acceptance that LSF/CP2 does not bind CCAAT sequences (Frith et al., 2001; Lim et al., 1993; Shirra, 1995); these two “CP2” proteins are unrelated. In order to avoid confusion, we will refer to LSF/CP2/LBP-1c solely as LSF throughout this review.
LSF is a member of a highly conserved and ancient, although surprisingly small, family of transcription factors. The family consists of two branches (Venkatesan et al., 2003; Wilanowski et al., 2002): the LSF/CP2 subfamily (Fig. 1) and the Grainyhead (GRH) subfamily. LSF/GRH-encoding genes have been identified in organisms ranging from A. gambiae (African malaria mosquito) and D. melanogaster (fruit flies) to H. sapiens (humans). In mammals, three to four members of each subfamily are represented in each genome, however most other species contain a single gene of each subfamily. The outlier species in this regard are nematodes, including C. elegans and C. briggsae, which contain one GRH gene, but none representing the LSF subfamily (Venkatesan et al., 2003). This apparent loss in nematodes is consistent with the roles for LSF in hematopoetic lineages, the liver, and the eye (Sections 5.2–5.7), as none of these tissues is present in nematodes. Finally, neither LSF nor GRH genes are found in any sequenced genomes or EST databases from plants or in any unicellular organism.
Figure 1. Identified proteins in the LSF subfamily.

Both human and mouse genomes contain three genes encoding LSF subfamily members. A splicing variant mRNA lacking one exon of the LSF gene encodes LSF-ID (LBP- 1d). A splicing variant mRNA containing an extra exon of the LBP-1a/b gene encodes the protein LBP-1b. While the LSF and LBP-1a/b genes are ubiquitously expressed, the third paralog, LBP9, exhibits a more limited expression profile; LBP9 also apparently lacks the transcriptional activation region conserved between LSF and LBP-1a/b. All LSF subfamily members oligomerize with each other.
Based on the divergence in the sequence alignment across species, the split between the LSF and GRH subfamilies is thought to have occurred ~750 million years ago, which coincides roughly with the timespan in which the first multicellular organisms are thought to have evolved. This suggests a connection of the LSF/GRH transcription factor family with development or differentiation processes. Initial studies support this viewpoint. In model organisms (e.g. D. melanogaster and C. elegans), GRH subfamily members are expressed in a tissue-specific manner, such as in the cuticle, and are essential for normal development (Bray and Kafatos 1991; Huang et al., 1995; Liaw et al., 1995; Uv et al., 1997; Venkatesan et al., 2003). In mammals, the recently identified GRH members appear to function specifically in epithelial lineages and in the neural crest (Kudryavtseva et al., 2003; Peters et al., 2002; Ting et al., 2003a; Ting et al., 2003b; Wilanowski et al., 2002). Although the LSF subfamily has not been as fully investigated in model organisms, mutation of the single orthologous gene in D. melanogaster, named gem for Gemini (http://flybase.bio.indiana.edu) or dCP2 (Wilanowski et al., 2002), results in defects in mid-stage embryos and lethality during late-stage embryonic and larval development (O'Day and Goldstein, pers. comm.). Expression of gem appears constant throughout development of the embryo and larval stages (M. O'Day and E. Goldstein, pers. comm.). In mammals, LSF subfamily members function in cell cycle and growth, as well as in differentiated cells, as discussed in Section 5.
The DNA-binding regions of GRH and LSF subfamilies exhibit a high degree of conservation, and the DNA motifs that are recognized by GRH and LSF are indeed highly related. LSF binds two directly repeated motifs (Fig. 2A) as a tetramer (discussed in more detail in Section 3) (Murata et al., 1998; Shirra and Hansen, 1998), whereas GRH binds a single, similar DNA motif (Fig. 2B (Venkatesan et al., 2003)) as a dimer (Attardi and Tjian, 1993; Shirra and Hansen, 1998). What distinguishes the subfamilies from each other is predominantly the oligomerization regions of the proteins. Consistent with this, whereas LSF or GRH can interact with members of their same subfamily, they cannot oligomerize with each other (Ting et al., 2003b; Uv et al., 1994; Wilanowski et al., 2002; Yoon et al., 1994). The distinct DNA-binding sites and inability to interact with each other suggest that GRH and LSF family members target distinct sets of genes (Venkatesan et al., 2003).
Figure 2. DNA recognition sequences for LSF and GRH subfamily members.

Known binding sites for mammalian LSF (Frith et al., 2001) (panel A) and D. melanogaster and C. elegans GRH (Venkatesan et al., 2003) (panel B) proteins were compiled. The resulting count matrices are presented as pictograms, presented according to the guidelines at http://www.genes.mit.edu/pictogram.html. The LSF and GRH pictograms are aligned in order to emphasize the similarity in sequence, with the LSF binding site containing two direct repeats of the sequence.
2. Mammalian LSF subfamily
There are three identified LSF subfamily genes in the human genome: LSF (chromosome 12q13 (Cunningham et al., 1995; Swendeman et al., 1994)), LBP-1a/b (chromosome 3), and LBP9 (chromosome 2) (Fig. 1). In the mouse genome, three orthologous genes reside on syntenic chromosomes (LBP-1a/b = NF2d9 (Sueyoshi et al., 1995) and LBP9 = CRTR-1 (Rodda et al., 2001)). Both LSF (Ramamurthy et al., 2001; Swendeman et al., 1994) and LBP-1a/b (Ramamurthy et al., 2001) mRNAs are ubiquitously expressed in the developing and adult mouse, as well as in all human cell lines examined to date. One subtlety in the two expression patterns is that LBP-1a/b mRNA is particularly abundant in the developing mouse fetal liver, the source of hematopoiesis at this developmental stage.
Mice containing a targeted disruption of the LSF/CP2 gene are viable and apparently normal (Ramamurthy et al., 2001). The lack of a mutant phenotype in the LSF knockout mice is likely to reflect functional redundancy between LSF and its paralog, LBP-1a/b (Ramamurthy et al., 2001), as LBP-1a and LSF are 72% identical in sequence, with 88% identity in the 280 N-terminal amino acids (Yoon et al., 1994), comprising the DNA-interaction and transcriptional activation regions (see Sections 4.1 and 4.3; Fig. 3). Furthermore, LBP-1a/b is ubiquitously expressed (Ramamurthy et al., 2001), binds to the identical DNA sequences as does LSF, and interacts with known LSF partner proteins (NF-E4 (Zhou et al., 2000) and RING1B (Tuckfield et al., 2002)).
Figure 3. Structural/functional regions of LSF.
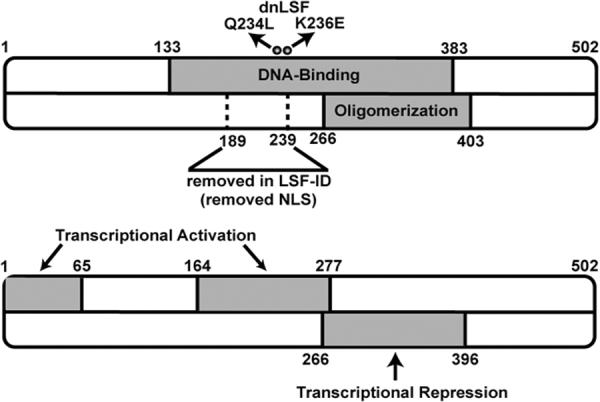
The regions containing LSF DNA-binding, oligomerization, transcriptional activation and transcriptional repression activities are indicated in gray. The two amino acid substitutions within the DNA binding domain (Q234L and K236E) comprise the dominant negative mutant (dnLSF) and render the protein incapable of binding DNA while still being able to oligomerize. The deletion in LSF-ID is also shown, which causes the protein to be localized to the cytoplasm; therefore aa 189–239 contribute to nuclear localization.
In contrast to the LSF knockout mice, mice containing a targeted disruption of the LBP-1a/b gene uncovered a unique biological function of this subfamily member. These embryos survive only to day 11.5, apparently due to defects in angiogenesis of the placenta and yolk sac (J.N. Cunningham, personal communication). No intra-embryonic deficiencies were detected. Overall, the biological functions of LSF and LBP-1a/b may overlap in most cell types, although specific functions may be found in other cell types, such as a requirement for LBP-1a/b in extra-embryonic vascularization. Based on the D. melanogaster phenotype, it is anticipated that mice deficient in both LSF and LBP-1a/b will also exhibit lethal intra-embryonic defects.
Biological roles for LSF/LBP-1a/b have been suggested in specific, differentiated lineages, most notably in hematopoietic lineages and the liver (Bing et al., 2000; Casolaro et al., 2000; Chae et al., 1999; Chae and Kim, 2003; Drouin et al., 2002; Jane et al., 1995; Kashour et al., 2003; Lim et al., 1993; Sueyoshi et al., 1995; Volker et al., 1997; Zhou et al., 2000). In addition, our laboratory has identified an apparently ubiquitous role for LSF in the regulation of cell-cycle progression and cell survival (Bruni et al., 2002; Powell et al., 2000). Further investigations are required in order to dissect the relative abilities of LSF versus LBP-1a/b to implement these roles.
Modulation of LSF and LBP-1a/b functions in some cases may result from differential RNA splicing. A number of splicing variants of LSF mRNAs have been identified, one of which encodes LSF-ID (for LSF-Internally Deleted; also named LBP-1d) (Shirra et al., 1994; Uv et al., 1994; Yoon et al., 1994). LSF-ID lacks amino acids 189 to 239, which encode part of the DNA interaction region (Fig. 3); thus LSF-ID does not bind DNA (Shirra et al., 1994; Uv et al., 1994) and inhibits the ability of full-length LSF to bind in vitro (Yoon et al., 1994; Zhong et al., 1994). Exogenously expressed LSF-ID is localized predominantly to the cytoplasm (Drouin et al., 2002; Zambrano et al., 1998). Nonetheless, it can function as a dominant-negative mutant when stably expressed in cell lines, and has therefore been a useful tool for uncovering roles of LSF (Drouin et al., 2002; Yoon et al., 1994). The biological purpose for LSF-ID is unclear, however, as its mRNA levels are much less abundant than those of LSF, and endogenous LSF-ID protein has not as yet been detected (Shirra et al., 1994). Differential RNA splicing also occurs in transcripts from the LBP-1a/b gene, leading to expression of the alternative LBP-1b isoform. LBP-1b contains an insertion of 37 amino acids relative to LBP-1a (Huang et al., 2000; Yoon et al., 1994), immediately following the DNA-interaction region. As of yet, no distinctions in activities between LBP-1a and LBP-1b have been uncovered, nor are their relative expression distributions or levels known.
Finally, although expression of the third mammalian paralog in the subfamily, LBP9/CRTR-1, was initially thought to be specific to the placenta (Huang and Miller, 2000), further studies demonstrated expression in a number of specific tissues (Rodda et al., 2001). Nonetheless, it is not as widely expressed as are LSF and LBP-1a/b. During the transition of embryonic pluripotent stem cells to primitive ectoderm-like cells, expression of LBP-9 is decreased, suggesting a biological role in pluripotent cell development (Pelton et al., 2002). LBP-9 is functionally distinct from its paralogs, in that it contains a transcriptional repression region, rather than an activation region, at its N-terminus (Rodda et al., 2001). In fact, it can reverse transcriptional activation mediated by LBP-1b (Huang and Miller, 2000). Thus, it is interesting to speculate that LBP-9 may modulate the functions of LSF and LBP-1a/b in a cell type-specific manner.
The focus of the remainder of this review will be on LSF, given the greater knowledge about this protein compared to its paralogs. The structure/function studies are likely to apply to all other subfamily members, except for transcriptional regulation by LBP9. Furthermore, the biological roles that have been shown for LSF may relate, as well, to those of LBP-1a/b given the similarities presented above in Section 2.
3. LSF binding to DNA
The interaction of LSF with DNA is unusual with regards to the target DNA recognition sequences, to the oligomerization state of LSF on the DNA, and to the lack of homology with any other known DNA-binding domains. Unfortunately, no protein structure, either from NMR or crystallography, is as yet available. The LSF recognition motif consists of directly repeated half sites on consecutive turns of the DNA helix (Huang et al., 1990). The reported consensus sequence is: CNRG N6 CNRG (Lim et al., 1993; Shirra, 1995). A more precise representation is the pictogram shown in Fig. 2A, which is a compilation of 15 known binding sequences (Frith et al., 2001); in this representation it is clear that the first (G)CTGG half site is more conserved than the second. Whereas mutational studies demonstrated that the spacing between the two CTGG motifs is important, the central specific base pairs were thought to be inconsequential (Huang et al., 1990; Lim et al., 1993; Murata et al., 1998; Yoon et al., 1994). However, the current compilation also indicates clear preferences in the sequence in between the half sites. Most notable is the predominance of three T residues following the first half site, which is highly reminiscent of the count matrix for the GRH recognition motif (Fig. 2B) (Venkatesan et al., 2003). This suggests a high degree of similarity between the LSF and GRH subfamilies in their mode of interaction with DNA.
We note that initially, there was confusion over the LSF/CP2 recognition sequence, which resulted in deposition of two incorrect matrices containing only a single motif (accession numbers M00072 or V$CP2_01, and M00644 or V$LBP1_Q6) in the TRANSFAC database of transcription factor recognition sequences (Kim et al., 1990; Wingender et al., 2000). Based on comparisons of promoter sequences against the TRANSFAC database of binding matrices, a number of promoters have been reported to contain LSF binding sites. However, our retrospective inspection of the same promoter sequences with the correct LSF recognition matrix (Fig. 2A) indicates that it is highly unlikely that LSF would bind or regulate several of these promoters (Lambertini et al., 2003; Lau et al., 1999; Xia et al., 2003; Zhao et al., 1997) (including the GSK3 promoter, Section 6.2.). The appropriate, recently added TRANSFAC matrix for LSF is M00947 (Frith et al., 2001).
LSF binds DNA as an obligate homotetramer (Murata et al., 1998; Shirra and Hansen, 1998), however sedimentation and crosslinking studies indicate that it is predominantly a dimer in solution (Shirra and Hansen, 1998; Zhong et al., 1994). Therefore, formation of a stable complex of LSF on DNA requires two steps, which may be independent or coordinate: interaction of two dimers to form an LSF tetramer and recognition of the DNA. Both steps are potentially points of regulatory control.
Not only can LSF partner with members of the same subfamily of transcription factors (e.g., LBP-1a/b) to bind LSF recognition sites (Fig. 2), but it can also partner with unrelated proteins in order to generate novel DNA-binding moieties. There is one clear example where such a partner protein leads to recognition of a novel DNA sequence - the NF-E4 plus LSF heteromeric complex is expressed specifically in a particular stage of erthyroid development, leading to a stage- and tissue-specific DNA-binding activity that regulates expression of the β-globin gene cluster (see Section 5.2. on erythroid cells) (Jane et al., 1995; Zhou et al., 2000). Another known partner protein, YY1, couples with LSF to alter transcription activity from an LSF-binding site in the HIV LTR (Coull et al., 2000; Coull et al., 2002; Romerio et al., 1997). Finally, several other DNA-binding complexes have been identified in crude nuclear extracts that contain LSF, but are clearly distinct from LSF homotetramers, and are sometimes differentially regulated. Such instances include the switch recombination site-binding complex in B cells (Drouin et al., 2002) and the IL-4 promoter-binding complex in Jurkat cells (Casolaro et al., 2000). However, in these cases, the LSF partner proteins have yet to be identified.
4. Structure/function analysis of LSF
4.1. DNA-binding
LSF is the most extensively characterized of all the mammalian paralogs in terms of its structure/function relationships. Mouse and human LSF are greater than 96% identical in amino acid sequence (Lim et al., 1992). Unlike many DNA-binding proteins, over half of the protein (amino acids 133–383) is required (Shirra and Hansen, 1998), and sufficient (Lim et al., 1992), for DNA binding activity (Fig. 3). This includes not only the DNA interaction region, but also its oligomerization region, since LSF only binds DNA efficiently as a tetramer (Shirra and Hansen, 1998). In contrast, mapping of the D. melanogaster GRH protein delineated a region of 59 aa that is sufficient to specifically, albeit weakly, recognize DNA (Uv et al., 1994). By analogy with GRH, which recognizes a DNA motif equivalent to an LSF half site (Fig. 2), this DNA-interaction region of LSF would map roughly to aa 214 to 273. Strikingly, a comparison of protein sequence profiles of the LSF and the GRH subfamilies of proteins showed alignment in the DNA-binding region up to aa 272 of LSF (Venkatesan et al., 2003), confirming the C-terminal boundary of this domain. Furthermore, six double aa substitution mutants constructed in our laboratory confirm that two short stretches of highest conservation between H. sapiens LSF and D. melanogaster GRH, which include residues 205–216 and 233–246 of LSF, are indeed critical for its DNA-binding activity (Shirra et al., 1994; Shirra, 1995). One of these mutants (Q234L/K236E; named dnLSF; Fig. 3) is not only unable to bind DNA but also inhibits the DNA-binding of wild-type LSF at equimolar concentrations, presumably due to oligomerization with LSF (Shirra et al., 1994). This mutant has therefore been a valuable dominant-negative mutant for biological studies (Drouin et al., 2002; Kashour et al., 2003; Powell et al., 2000). Since LSF and its paralogs oligomerize (Yoon et al., 1994), dnLSF inhibits DNA-binding activity not just of LSF, but of the entire LSF subfamily of proteins.
4.2. Oligomerization
Oligomerization regions of LSF have been grossly delineated through deletion mutant analyses (Shirra et al., 1994). By interaction studies with fusion proteins linked to glutathioine S-transferase (GST), plus in vitro crosslinking studies, an oligomerization region was mapped to aa 266 to 403 (Fig. 3). These assays reflect predominantly dimerization, since this is the major form of LSF in the absence of DNA, although a low level of tetramerization was apparent in solution by crosslinking studies. In yeast two-hybrid interaction experiments, oligomerization of LSF to itself was also promoted by the C-terminal region (aa 280–501) (Uv et al., 1994; Zhou et al., 2000). Finally, computational protein profiling of the LSF subfamily indicated that, outside of the DNA-binding region (Section 4.1.), evolutionarily conserved sequences were limited to aa 273–397 in LSF. This computational analysis is strikingly consistent with the experimental mapping of the oligomerization region to these sequences.
A second, distinct LSF-LSF interaction region appears to reside within the DNA-interaction region, as LSF-ID and substitution mutants in residues 211–213 or 235–237 (including dnLSF) interact less with GST-LSF than does LSF (Shirra et al., 1994; Shirra, 1995). Protein-protein interaction through the DNA-binding region, which may reflect the tetramerization function, is likely to be hydrophilic in nature, as it is inhibited by high salt. In contrast, the C-terminal oligomerization region, which remains in the DNA-binding mutants, is stable to high salt, suggesting a hydrophobic component (Shirra et al., 1994).
In summary, we hypothesize that oligomerization of LSF occurs in two steps: 1) a dimerization interaction including hydrophobic surfaces, which is stable in solution and maps to a C-terminal region of the protein, and 2) tetramerization through the DNA-binding region, which is hydrophilic in nature and is stabilized by interaction of LSF with DNA.
4.3. Transcriptional Activation and Repression
LSF both activates and represses transcription. Generally, it is a transcriptional activator, both in cell-free extracts (Kim et al., 1990; Kim et al., 1987; Lim et al., 1993; Sundseth and Hansen, 1992; Yoon et al., 1994) and in mammalian tissue culture cells (Bing et al., 1999; Bing et al., 2000; Casolaro et al., 2000; Lim et al., 1993; Murata et al., 1998; Powell et al., 2000; Yoon et al., 1994; Zheng et al., 2001), as is LBP-1a/b (Huang et al., 2000; Yoon et al., 1994). By transient expression in mammalian cells of LSF-GAL4 fusion proteins with a GAL4 reporter construct, we mapped two transcriptional activation regions (Q. Zhu, U. Hansen, in preparation): amino acids 1 to 65 and 164 to 277 (Fig. 3), each of which is sufficient independently to stimulate transcription. The Jane and Cunningham laboratories also reported unpublished data of mapping an activation region to the N-terminal 40 aa (cited in (Ramamurthy et al., 2001; Rodda et al., 2001)). LSF interacts directly with at least two components of the basal transcriptional machinery components: TBP (TATA-binding protein) and TFIIB (Zhu and Hansen, in preparation). Kinetic analyses indicate that the interaction with TFIIB is likely to be significant, as LSF increases the rate of association of TFIIB with a DNA template in vitro, as well as ultimately increasing the total number of transcription complexes (Sundseth et al., 1992). Interactions with other categories of coactivators have yet to be explored.
LSF can also act as a transcriptional repressor. In vitro, LSF represses transcription from the HIV LTR by two mechanisms: steric hindrance, preventing TBP access to the TATA (Kato et al., 1991), and inhibition of transcriptional elongation (Parada et al., 1995). In vivo, LSF also represses expression from the HIV LTR (Coull et al., 2002) as a heteromeric complex with YY1 and histone deacetylase 1 (HDAC1) (Coull et al., 2000; Romerio et al., 1997). To date, the only cellular promoter that LSF has been reported to downregulate is the IL-2 promoter, but the mechanism in this case may be indirect (Casolaro et al., 2000). We suggest that direct, negatively regulated cellular target genes still await discovery. Via transcriptional assays of LSF fusion proteins with GAL4 in mammalian cells, amino acids 266 to 396 are necessary and sufficient for transcriptional repression (Fig. 3; Q. Zhu, U. Hansen, in preparation).
The mechanisms of transcriptional repression by LSF may, in general, involve generation of repressive chromatin structure. Direct LSF-interacting proteins include not only YY1 (Romerio et al., 1997), but also RING1, a member of the polycomb repressive complex (Tuckfield et al., 2002), histone deacetylases HDAC1 and 2, and the corepressor Sin3A (Drouin et al., 2002), all of which contribute to formation of inert chromatin. In fact, LSF can mediate repression of at least one non-transcriptional process on chromatin, that of class switch recombination in B cells (Drouin et al., 2002). Such recombination between highly repetitive “switch regions” results in deletion of genomic DNA in the immunoglobulin heavy chain locus, leading to “switching” of expression from one isotype of antibody (e.g. IgM) to another (e.g. IgG), while maintaining antigen specificity. LSF specifically binds sequences in these genomic switch regions, and we hypothesize that it represses recombination by nucleation of an inert chromatin structure at the locus (Drouin et al., 2002).
4.4. Nuclear Localization
Although LSF is a nuclear protein in all cell types examined to date (Drouin et al., 2002; Zambrano et al., 1998) (Q. Zhu, U. Saxena, Ul Hansen, unpubl. observations) except for differentiated rat B103 neuroblastoma cells (Kashour et al., 2003), it lacks an obvious nuclear localization signal. However, LSF-ID, which lacks 50 internal aa, is localized almost exclusively to the cytoplasm (Drouin et al., 2002; Zambrano et al., 1998). Thus, residues 189 to 239 are likely to be important in localization to the nucleus (Fig. 3).
4.5. Regulation of LSF Activities
Given the unusual nature of the DNA-binding activity of LSF, involving not only dimerization, but also tetramerization, the potential for regulation is high – including modulation of tetramerization, as well as modulation of the DNA interaction surface or of transcriptional activation or repression (Whitmarsh and Davis, 2000). Indeed, in several cell types (details in Section 5), LSF activity is altered in response to cellular signal transduction pathways. Examples of regulation include: rapid enhancement of LSF DNA-binding activity in primary human T cells, which requires activation of Erk (Pagon et al., 2003; Volker et al., 1997); decreased LSF DNA-binding activity to sites in heavy chain immunoglobulin class switch regions, following activation of class switching in primary murine splenic B cells (Drouin et al., 2002); increased DNA-binding to the IL-4 promoter site upon induction of Jurkat cells with calcium ionophores (Casolaro et al., 2000); decreased LSF DNA-binding activity to the HIV-LTR site upon phosphorylation by Erk2 in vitro (D. Margolis, pers. comm.); and increased binding in vitro to the HIV-LTR site upon phosphorylation of LSF by p38 or in HeLa cells to an integrated HIV LTR upon induction of the p38 pathway by anisomycin (D. Margolis, pers. comm.). Finally, LSF-mediated stimulation of a serum albumin promoter in liver cells is enhanced upon induction of the inflammatory response via IL-1 signaling (Bing et al., 2000), although the mechanistic basis – DNA binding versus transcriptional activation – has not been distinguished in this case. These results indicate not only that LSF is a target of multiple signal transduction pathways, but that its DNA-binding activity is highly sensitive to modulation.
5. LSF displays both ubiquitous and cell lineage-specific biological roles
5.1. Cell Growth and Cell Cycle
The ubiquitous expression of LSF suggests global cellular functions for the transcription factor. Indeed, findings from our laboratory point to a role in regulation of cell cycle progression and possibly in cell survival (Fig. 4). In an organism, most cells are in the resting, G0, state. Entry into the cell cycle, through the G1 phase, requires activation of signal transduction cascades. A critical pathway in this process is activation of the Erk family of MAP kinases, which target a number of transcription factors (Johnson and Lapadat, 2002; Yang et al., 2003; Zhang and Liu, 2002). Subsequent commitment to proliferation and entry into S phase requires, in addition, activation of the G1 cyclin-dependent kinases and passage through the restriction point. Downstream targets of the cyclin D/cdk4 and cyclin D/cdk6 complexes include pRb, leading to activation of E2F transcription factors. Cyclin E/cdk2 also targets transcription factors, most notably to date NPAT, which stimulates histone gene expression (Ma et al., 2000; Zhao et al., 2000).
Figure 4. Role of LSF in cell cycle progression via activation of the thymidylate synthase (TS) gene.
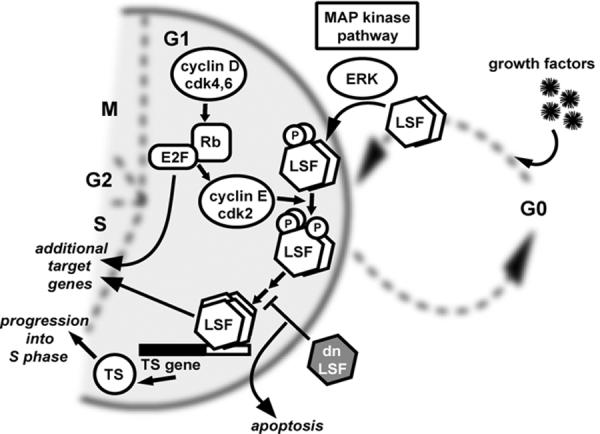
Upon entry into G1, cyclin D/cdk4 and cyclin D/cdk6 complexes phosphorylate the retinoblastoma protein (Rb), allowing release of transcription factor E2F family members. A major role for E2F in cell cycle progression is to upregulate cyclin E/cdk2. In a parallel pathway, upon stimulation of cells in the resting (G0) state with growth factors, LSF is immediately and quantitatively phosphorylated by Erk, a central kinase in the mitogen activated protein kinase (MAP kinase) pathway. LSF is subsequently phosphorylated by cdk2, as well. One major target gene of active LSF is the thymidylate synthase (TS) gene. TS is essential for DNA replication in S phase. Expression of dnLSF prevents binding of LSF to target sites, and the lack of TS causes cells to undergo apoptosis. E2F, and presumably LSF, target additional genes for regulation during cell cycle progression into S phase. If growth factors are removed from the cellular environment, cells exit the cell cycle in G1, returning to the G0 state.
A number of cellular kinases specifically phosphorylate LSF in vitro, including both Erk and cdk2 (Powell, 1999; Volker, 1998). The major site of phosphorylation by Erk in vitro, S291, is also a target in vivo. In particular, in a variety of cell types that can be obtained in a truly resting, G0, state in culture, LSF is quantitatively phosphorylated on S291 within minutes of mitogenic stimulation: e.g. upon treatment of serum-deprived fibroblasts with growth factors, or treatment of resting human peripheral blood T cells with either phorbol esters or antibodies that crosslink cell surface receptors (Pagon et al., 2003; Volker, 1998; Volker et al., 1997). That Erk is responsible for increased LSF phosphorylation was demonstrated by treatment of these cells with a specific inhibitor of MEK, which eliminates Erk activation and also abolishes phosphorylation of LSF on S291 (Pagon et al., 2003). Intriguingly, phosphorylation at S291 in cells progressing through G1 is highly reversible, depending on the presence of growth factors in the cellular medium (Z. Pagon, G.M. Cooper, U. Hansen, unpubl. observations). Thus, the phosphorylation status of LSF is an indicator of the continued presence of growth-stimulating factors, which are required until the cell passes the restriction point. Cyclin/cdk2 complexes also specifically phosphorylate LSF, both in vitro and in vivo (Fecko, 2001; Powell, 1999). These findings suggest a role for LSF in cell growth and cell-cycle progression.
Consistent with this prediction, a major cellular target gene for LSF is the thymidylate synthase (TS) gene (Powell et al., 2000), which encodes the rate limiting enzyme in production of dTTP, required for DNA synthesis. LSF binds within cell-cycle regulatory regions of the mouse TS gene (in both the promoter and intron sequences), and is required specifically for upregulation of TS mRNA levels at the G1/S transition in cells stimulated to reenter the cell cycle (Fig. 4). It is worth noting that there was initially disagreement about whether or not thymidylate synthase was up-regulated by E2F during the G1/S transition in rodent cells (DeGregori et al., 1995; Li et al., 1994b). Detailed studies with mutations in specific binding sites of the promoters from multiple species have concluded that such regulation, although sometimes repressive by E2F (Dong et al., 2000), is a minor component in normal G1/S progression (Lee and Johnson, 2000). Thus, LSF, possibly in conjunction with neighboring Ets and Sp1 sites, is the major regulator of TS cell-cycle expression.
Consistent with this notion, when LSF DNA-binding activity was inhibited by expression of dnLSF (dominant-negative LSF mutant, Section 4.1), the G1/S induction of TS expression was blocked (Fig. 4). dnLSF also caused both mouse fibroblasts and human prostate cancer cells to undergo apoptosis during S phase. Strikingly, addition of thymidine to the cell medium, which overcomes the cellular DNA replication requirement for TS, successfully protected the cells from undergoing apoptosis (Powell et al., 2000). These results indicated that not only is LSF critical for TS induction during the cell cycle, but also that TS is one of the major targets of LSF action in cell cycle progression, as overcoming its requirement allowed progression of cells through the cycle upon inhibition of LSF. Finally, given that inhibition of LSF in this case led to apoptosis, we hypothesize that LSF might promote cell survival under appropriate cellular environments.
The G1/S portion of the cell cycle was also affected when LSF activity was inhibited in a different manner, by overexpression of the interacting protein Fe65 (Section 6.1). However, in this case, cells were arrested at the G1/S transition (Bruni et al., 2002). Fe65 inhibits transactivation by LSF, but the underlying molecular mechanism is not yet defined. This arrest was also abolished by addition of thymidine, implying that the Fe65-mediated arrest was a direct consequence of inhibition of LSF and its upregulation of the thymidylate synthase gene (Powell et al., 2000).
5.2. Hematopoietic lineages: Erythroid cells
In addition to the ubiquitous cell growth functions, LSF exhibits cell type-specific roles, observed to date mainly in hematopoietic lineages. During hematopoiesis, lymphoid and myeloid cell types are differentiated from stem cells. Specific roles for LSF have been demonstrated in three of these lineages: erythrocytes, T lymphocytes, and B lymphocytes. It is intriguing to speculate that LSF may promote specific functions in other hematopoietic cell types, as well.
A major role of erythroid cells is to transport oxygen via the family of globin proteins. During development, globin gene expression is tightly regulated in a temporal and tissue-specific manner from the multigene β-globin locus. The progression from embryonic to fetal to adult globin expression is mediated by the locus control region, in conjunction with elements near specific genes. One critical element in this process, the human stage selector element in the γ-globin promoter, favors γ-globin over β-globin expression. The stage selector element binds a heteromeric protein complex, consisting of LSF (Jane et al., 1995) and the erythroid-specific factor NF-E4 (Zhou et al., 2000). Both proteins also stimulate expression of embryonic ε-globin (Chae and Kim, 2003; Zhou et al., 2000). Interestingly, the stage selector sequence contains an LSF half site (GCTGG), suggesting that a dimer of LSF interacts with this portion of the element, and NF-E4 with the remainder of the element.
LSF can also activate expression of the adult globin genes. Apparently as a homotetramer, LSF binds and activates transcription from the murine α-globin promoter (Barnhart et al., 1988; Kim et al., 1988; Lim et al., 1993), although such activation in vivo is limited to erythroid cells (Chae and Kim, 2003), suggesting the requirement for an erythroid-specific coactivator(s). Furthermore, LSF is required for maximal, inducible expression of both α-globin and β-globin genes, monitored during differentiation in vitro of mouse erythroleukemia cells (Chae et al., 1999). Binding of LSF to the β-globin promoter was observed, as well (Barnhart et al., 1988; Chae et al., 1999).
Finally, LSF is essential in human erythroid cells for activating expression of the uroporphyrinogen III synthase gene, as a mutation in the LSF-binding site in this promoter causes defects in heme biosynthesis and congenital erythropoietic porphyria in the patient (Solis et al., 2001).
5.3. Hematopoietic lineages: T lymphocytes
Primary human T cells from peripheral blood are an excellent model system to study the G0/G1 transition, as they are quiescent until stimulated with antigen/MHC complex or other specific mitogens. Upon mitogenic stimulation of naive resting peripheral T cells, LSF DNA-binding activity increases rapidly, although transiently (Pagon et al., 2003; Volker et al., 1997)(J. Volker and U. Hansen, unpubl. observations). Activation of the Erk pathway is necessary, but not sufficient, for the enhancement in DNA-binding activity of tetrameric LSF in primary T cells (Pagon et al., 2003). Furthermore, phosphorylation of LSF by Erk either in vitro or in mitogenically stimulated fibroblasts does not lead to increased DNA-binding activity. Thus, an additional, T cell-specific signaling event is required to induce LSF DNA binding. These data suggested that LSF would regulate genes rapidly expressed upon mitogenic induction of T cells.
Activation of T helper (TH) cells by the MHC-antigen complex leads to the secretion of cytokines, which qualitatively specify the type of immune response that develops. In particular, IL-4 stimulates the growth of multiple types of resting immune cells, upregulates class II MHC expression in macrophages and resting B cells, and induces class switching to IgE and IgG1 in mature B cells. Expression of IL-4 occurs only in actively dividing T cells. LSF binds the IL-4 promoter and activates IL-4 expression in the human tumor Jurkat cell line, as well as a murine TH2 cell line (Casolaro et al., 2000). As Jurkat cells cannot be made to enter a resting state, no change in tetrameric LSF DNA-binding activity was observed when the cells were stimulated with calcium ionophores. However, a novel LSF-containing complex that bound the IL-4 promoter element was induced under these circumstances. We note that unlike resting, primary T lymphocytes (Weinberg et al., 1990), Jurkat cells express a basal level of IL-4 in the absence of additional, external stimuli. Thus, not only is LSF itself regulated by Erk and additional signaling pathways, but a T cell-specific LSF partner protein is apparently induced by a calcium-dependent signaling pathway, in order to regulate IL-4 gene expression.
5.4. Hematopoietic lineages: B lymphocytes
The major function of B lymphocytes is generation of specific antibodies. During B-cell differentiation, VDJ recombination in the immunoglobulin heavy chain locus helps to confer antibody variability and specificity; subsequent class switch recombination determines the physiological properties of the antibody. The genomic switch regions in which class switch recombination occurs consist of 1–4 kb of highly repetitive sequence located upstream of constant region coding sequences. This juxtapositioning of repeated motifs generates sequences resembling LSF binding sites. Indeed, the switch regions upstream of the α (Sα) and μ (Sμ) constant region coding sequences bind LSF at multiple sites (Drouin et al., 2002). The LSF-containing protein complex in mouse splenic cell extracts binds Sα and Sμ sequences with higher affinity than does purified recombinant LSF. Furthermore, the protein/DNA complex observed in splenic cell extracts migrates more slowly upon electrophoresis than does the homotetrameric LSF/DNA complex. Thus, B cell-specific protein(s) facilitate the association of LSF with switch regions.
In a B-cell line capable of class switch recombination in vitro, overexpression of LSF reduced the amount of switching from IgM to IgA, and conversely, overexpression of either dnLSF or LSF-ID (Shirra et al., 1994) increased switching to IgA. The level of sterile transcripts was not affected by altering LSF activity; thus this is the only known non-transcriptional regulatory role for LSF (Drouin et al., 2002). It is unlikely that LSF could repress class switch recombination by steric hindrance, given the large region of the target recombination sequences (several kilobase pairs) and limiting amounts of nuclear LSF. Thus, the current model is that LSF represses class switch recombination by promoting chromatin structure that is relatively inert to recombination. One potential mechanism for achieving repressive chromatin would be by recruitment of histone deacetylases (HDAC1, HDAC2) and other corepressors (e.g. Sin3A), since LSF can interact with these proteins in vitro (Drouin et al., 2002). On the HIV LTR, YY1 is essential for the ability of LSF to repress transcription, and YY1 could potentially be involved in the complexes of LSF from B cell extracts that binds switch region sequence. Alternatively, interactions of LSF with polycomb proteins (Tuckfield et al., 2002) could lead to an inert chromatin configuration. In support of a chromatin-based model of repression of class switch recombination, VDJ recombination has been shown to be modulated by chromatin modification (Kwon et al., 2000; Schlissel, 2000).
When mouse splenic cells were stimulated to undergo class switch recombination, there was a significant decrease in the amount of LSF-containing binding activity to switch regions. This decrease was highly specific, as the amount of homotetrameric LSF DNA-binding activity to a consensus binding site was not as affected, and LSF protein levels remained constant. These data imply that LSF represses recombination by binding switch regions only in resting B cells, and with the appropriate stimulus, repression is specifically relieved to permit efficient class switch recombination. Although effects of LSF only on switch recombination to IgA have been demonstrated to date, due to the limited number of cell lines capable of switching in vitro, we speculate that LSF may generally affect switching to other immunoglobulin classes. All class switch recombination involves the switch region upstream of the IgM constant chain gene (Sμ), to which LSF binds.
Another potential role for LSF in mature B cells, not yet fully explored, and possibly other immune cells (e.g., macrophages), may be to regulate expression of class II MHC genes. LSF binds the initiator region of one of the class II MHC genes in both mice, Eα (Viville et al., 1991), and humans, Dra (Bellorini et al., 1996). Finally, LSF also binds within the Wilm's Tumor 1 (WT1) intronic hematopoietic enhancer (Bing et al., 1999). WT1, a zinc finger transcription factor, is expressed in primitive hematopoietic progenitor cells, and also at high levels in some leukemic cells (Algar, 2002; Ellisen, 2002).
5.5. Liver: regulation of immune-related genes
The systemic response to acute inflammation, involving lymphocytes and macrophages, is modulated by acute phase proteins. Transcription of genes encoding these proteins is activated by inflammatory cytokines, including IL-1 and IL-6. Especially in the liver, cytokines rapidly induce synthesis of acute phase plasma proteins, including serum amyloid A (SAA), which are secreted into the circulation and can represent up to 20% of the protein in high density lipoprotein particles during the inflammatory response.
Transcription of the SAA3 gene, as assayed in HepG2 cells, is synergistically activated by LSF and NF-κB in response to the cytokine IL-1 (Bing et al., 1999; Bing et al., 2000; Huang and Liao, 1994). In fact, NF-κB and LSF interact in a complex in vivo. Addition of IL-6 along with IL-1 dramatically enhances SAA3 promoter activity (Huang and Liao, 1999), although whether this is also related to LSF has not yet been determined. IL-1 and IL-6 stimulate the JNK and p38 pathways, respectively, in liver cells (Finch et al., 2001; Finch et al., 1997; Park et al., 1999; Zauberman et al., 1999). NF-κB is activated by these signaling pathways, but LSF may be independently targeted, as well, as it is specifically phosphorylated by both JNK and p38 in vitro (Volker, 1998). Interestingly, LSF also binds in vitro to promoters of other genes that encode acute phase proteins, but that are activated mainly by IL-6 (Baumann and Gauldie, 1994): α2-macroglobulin, Aα-fibrinogen (Bing et al., 1999), and γ-fibrinogen (Lim et al., 1993). Further experiments are required to determine whether LSF is involved in regulating expression of these genes in response to cytokines.
5.6. P450 family gene regulation
LSF family members have been implicated in the regulation of two P450 genes: the cholesterol side-chain cleavage enzyme P450scc in the placenta (Huang and Miller, 2000) and male-specific Cyp 2d–9 gene in the liver of certain strains of mice (Sueyoshi et al., 1995). In the human placental JEG-3 cells, LBP-1b and LBP9 regulate transcription of the P450scc gene in opposite directions. Whereas LBP-1b activated expression, LBP9 repressed expression, and in fact overrode activation by LBP-1b (Huang and Miller, 2000). The LSF/LBP-1b site in the Cyp 2d–9 promoter is required for maximal male-specific expression. As recombinant LBP-1b cannot bind this site by itself, an LBP-1b-partner protein was implicated in the regulation of this promoter.
5.7. Eye-specific gene expression
Finally, LSF is essential for regulation of at least two ocular genes, shown in different species, again apparently as part of tissue-specific heteromeric complexes. LSF binds the promoter of the chicken αA-crystallin gene, which is exclusively expressed in the ocular lens (Murata et al., 1998). LSF also activates expression of the human PAX6 gene (Zheng et al., 2001), which encodes a transcription factor crucial for eye development. PAX6 expression is highly restricted to the eye, nervous system and pancreas.
5.8. Summary of cell-specific functions of LSF
Overall, cell-specific functions of LSF could be said to relate to how the body responds to external influences through specific cell types (oxygen, antigens/infectious agents, and light input). But the majority of the known functions are in response to external insults through the immune system and liver functions. Most of these functions seem to involve heteromeric complexes of LSF with other proteins. One can speculate that LSF was a good candidate, during evolution, for participation in cell type-specific and developmental stage-specific DNA-binding transcription activities, due to its ubiquitous expression, to its inability to bind DNA unless in complex with a partner (including itself as a dimer), and to its ease of modulation by signal transduction pathways.
6. LSF and Alzheimer's Disease
Alzheimer's disease is a neurodegenerative disorder characterized by neuronal loss and accumulation of neurofibrillary tangles and senile plaques in the brain (Selkoe, 1996; Yankner, 1996). The extracellular plaques consist of the β-amyloid (Aβ) peptide, derived from cleavage of the amyloid precursor protein (APP). LSF has been linked to Alzheimer's disease or APP by three findings. First, LSF interacts directly with Fe65, an adaptor protein that binds the C-terminal domain of APP. Second, at least in some circumstances, the ability of APP to protect neuronal cells against apoptosis is mediated by LSF. Finally, epidemiological studies may suggest linkage between a polymorphism in the LSF gene and predisposition to developing Alzheimer's disease in human populations. These studies are discussed in the following sections, concluding with a model to potentially connect these independent reports.
6.1. Nuclear signaling by APP and potential role of LSF
APP is a member of a family of type I transmembrane proteins (including APLP1 and APLP2 (Coulson et al., 2000)) that are proteolytically processed near the membrane (Hardy and Selkoe, 2002; Steiner and Haass, 2000). The extracellular domains are shed into the medium upon cleavage by α and β secretases, while the carboxyl-terminal products of these reactions remain bound to the membrane (Fig. 5). Importantly, APP is the only family member cleaved by β secretase. The carboxyl-terminal fragments are further processed by a third protease, the membrane-associated γ secretase. One product of γ secretase activity, the APP intracellular C-terminal domain (AICD), is released into the cell, while the other product, the N-terminal peptide, is secreted. In the case of APP cleaved first by β secretase, this secreted product is the Aβ peptide. Familial Alzheimer's disease mutations, predominantly located either in APP or in presenilin, an integral component of γ secretase, lead to enhanced cleavage at the β or γ secretase sites (Hardy and Selkoe, 2002; Selkoe, 2001), generating higher levels of the Aβ peptide and plaque formation in the brain. Thus, much study of APP has focused on properties of the secreted Aβ. However, recent investigations have also turned to the function of the reciprocal product of the γ secretase reaction, AICD, which may enhance neuronal apoptosis (Kim et al., 2003; Passer et al., 2000).
Figure 5. Potential role for LSF in mediating signal transduction from APP in the nucleus.
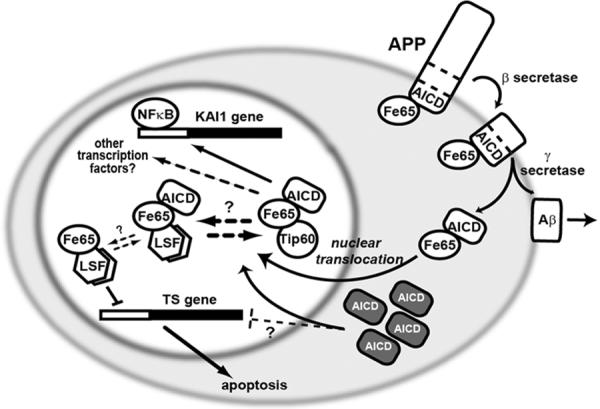
The intracellular domain of the transmembrane protein APP (amyloid precursor protein) interacts with a number of signaling proteins, including Fe65. APP cleavages produce AICD (APP intracellular C-terminal domain) and Aβ (β amyloid) peptide. Aβ is released extracellularly and is responsible for plaque formation in Alzheimer's disease. AICD is released intracellularly, where Fe65 facilitates its translocation into the nucleus. Nuclear Fe65-AICD interacts with Tip60 and LSF, perhaps in competition with each other, as indicated. Fe65 alone inhibits LSF transactivation on the TS promoter, although whether the ternary complex with AICD is also inhibitory is not known. One target of the Fe65-AICD-Tip60 complex is the tetraspanin KAI1 gene, although many other target genes are likely, including through factors other than NF-κB. Overexpression of AICD (dark boxes) increased neuronal apoptosis, presumably in part by increased nuclear translocation and misregulation of target genes, perhaps including TS.
There is striking similarity between proteolytic cleavages of APP and the well-studied Notch pathway (Selkoe and Kopan, 2003). Binding of ligand causes sequential cleavage of Notch by α and γ secretases. The Notch intracellular C-terminal fragment, NICD, then translocates to the nucleus and affects transcriptional programs. In parallel, it was hypothesized that AICD may regulate transcription through nuclear translocation and binding to cellular transcription factors. Indeed, although AICD is highly labile, its localization to both the cytoplasmic and nuclear compartments has been detected (Cupers et al., 2001; Gao and Pimplikar, 2001; Kimberly et al., 2001). AICD interacts with a number of intracellular adaptor proteins (Neve et al., 2000; Russo et al., 1998), including Fe65, which is highly expressed in brain and can shuttle between the cytoplasm and nucleus. Expression of Fe65 promotes nuclear localization of AICD (Kim et al., 2003; Kimberly et al., 2001) (Fig. 5). These observations suggest that the Fe65/AICD complex is involved in transcriptional regulation.
Current evidence supports two types of AICD/Fe65-containing nuclear complexes. (1) Fe65 interacts with the histone acetyltransferase Tip60, leading to a ternary AICD/Fe65/Tip60 complex that, upon recruitment to a promoter, can activate transcription in reporter gene assays (Cao and Südhof, 2001). The Tip60-containing ternary complex presumably normally associates with cellular promoters by Tip60 interactions directly with DNA-binding transcription factors. In particular, an NFκB p50 homodimer may recruit this complex to the tetraspanin KAI1 promoter (Baek et al., 2002) (Fig. 5). (2) The same domain of Fe65 that interacts with Tip60 can also interact with LSF (Zambrano et al., 1998), and a nuclear ternary AICD/Fe65/LSF complex has been reported (Kim et al., 2003). It is likely that such a ternary complex would be recruited to promoters through the DNA binding of LSF, although its transcriptional properties have not yet been experimentally determined. Exogenous overexpression of Fe65 alone results in its predominantly nuclear expression, coupled with repression of LSF transactivation of the thymidylate synthase promoter (Bruni et al., 2002) (Fig. 5). Whether Fe65 interferes with interaction of LSF with DNA, or affects its interaction with coactivators or corepressors is unknown. However, under normal physiological situations Fe65 may not be free to translocate to the nucleus by itself, but rather be maintained in the cytoplasm through interaction with APP (Minopoli et al., 2001). One hypothesis is that under normal circumstances, nuclear translocation of Fe65 may substantially depend on cleavage of APP to release AICD, and that nuclear Fe65 may predominantly be in association with AICD. Conceivably, association of LSF with Fe65/AICD may either promote or inhibit LSF transactivation.
The existence of these two ternary complexes raises the question of whether the two complexes compete for formation (Fig. 5; a reasonable scenario given that Tip60 and LSF interact with the same domain of Fe65 (Cao and Südhof, 2001; Zambrano et al., 1998)), or whether a quaternary AICD/Fe65/Tip60/LSF may occur. When the AICD/Fe65/Tip60 complex was artificially recruited to a synthetic promoter, LSF did not influence its transcriptional activity (Cao and Südhof, 2001). However, this would be predicted in either model, as one major role of LSF would clearly be to drive the association of the Fe65/AICD complex with a promoter, but this occurred through other mechanisms in the reporter gene experiments. Thus, additional details are required to distinguish between these alternative models.
Finally, we note that the APLP family members, upon final cleavage with γ secretase, also yield intracellular C-terminal domains that are stabilized by Fe65, translocate to the nucleus, and can activate transcription under appropriate experimental situations (Scheinfeld et al., 2002; Walsh et al., 2003). Moreover, Fe65 is also a member of a larger family, whose members are ubiquitously expressed (Russo et al., 1998). All Fe65 family members interact with AICD (Walsh et al., 2003), and all can functionally interact with LSF (Bruni et al., 2002). This large family of ternary complexes would likely regulate transcription not only in neuronal cells, but ubiquitously. Unfortunately, the presumptive biological signal(s) leading to cleavage of APP and its family members in any cell type are not yet known, making it difficult to postulate the general role that APP family members play in LSF biology.
6.2. LSF and APP-mediated cell survival
In some cell culture experiments, overexpression of APP protects neuronal cells from apoptosis. In particular, in the rat neuroblastoma B103 cell line, which lacks APP, expression of exogenous APP protects cells, when challenged with staurosporine, from undergoing apoptosis. In this system, expression of wild-type LSF enhanced protection from apoptosis, whereas expression of dnLSF augmented apoptosis, suggesting that LSF contributes to cell survival mediated by APP (Kashour et al., 2003). Strikingly, endogenous LSF was cytoplasmic in the differentiated cells, and translocated into the nucleus upon staurosporine treatment, completely dependent on the presence of wild-type APP. In sharp contrast, the familial APP disease mutant V642I (Kashour et al., 2003)(F. Amara, pers. comm.) did not promote nuclear translocation of LSF. The most parsimonious model to explain these data is that proapototic signaling in these cells causes an APP-dependent nuclear translocation of LSF, which leads to induction of expression of cell survival genes. Investigation in other cell systems of whether LSF can cooperate with APP in protecting cells from apoptosis may indicate whether this intriguing model is generally valid. [We note that although it was suggested that LSF may affect apoptosis via upregulation of GSK3β expression as a result of AICD signaling (Kim et al., 2003), this hypothesis was based on an incorrect LSF binding matrix in TRANSFAC (Section 3). There is no experimental evidence to support binding of LSF to the GSK3β promoter.]
Finally, given several previous reports that LSF is nuclear (Section 4.4), including in rat pheochromocytoma PC12 cells, it was completely unexpected that LSF was predominantly cytoplasmic in the differentiated rat neuroblastoma B103 cells. One provocative hypothesis to resolve this discrepancy is that LSF is exported from the nucleus during differentiation of (neuronal) cells; this hypothesis would be consistent with a major role for nuclear LSF in cell-cycle progression, as proposed in Section 5.1.
6.3. Reported epidemiological LSF/Alzheimer's disease linkages
One polymorphism in the 3' untranslated region of the LSF gene has been linked to a predisposition to Alzheimer's disease, although the results are not consistent throughout all human populations. In initial studies on human population cohorts in France, the United Kingdom, and the USA, the A allele (vs. G allele) correlated with protection against disease (Lambert et al., 2000) (Taylor et al., 2001) (Luedecking-Zimmer et al., 2003) (Lendon and Craddock, 2001). One study investigated LSF RNA levels, to identify a possible molecular basis for the connection, and reported that subjects with both the GG genotype and disease displayed lower LSF mRNA levels than other subjects (Lambert et al., 2000). In contrast, a recent study of a human population from southern Italy suggested that the A allele was associated with an increased risk, rather than protection, of early onset Alzheimer's disease (Panza et al., 2004). Thus, additional studies are required to determine if the A:G polymorphism in the 3' untranslated region is actually closely linked to another mutation either within LSF or in a nearby gene that uniformly influences disease outcome. This would resolve whether or not particular LSF alleles are connected with risk of disease.
6.4. Model of role of LSF in APP signaling
Overall, cleavage of APP releases AICD, which can translocate with Fe65 to the nucleus and either form a complex with LSF, Tip60, or perhaps other proteins (Scheinfeld et al., 2003), thus potentially impacting the expression of a wide variety of genes. One biological consequence of the nuclear signaling of AICD may be to enhance apoptosis. This consequence could result, in part, from repression of LSF, since inhibition of LSF by other methods promotes apoptosis by blocking thymidylate synthase expression (Fig. 5). We note that although neurons do not require DNA replication for survival, thymidylate synthase activity may nonetheless be required for DNA repair, if DNA were damaged. With regards to potential involvement of LSF in Alzheimer's disease, a highly speculative corollary would be that if an allele of the human LSF gene resulted in lower levels and activity of LSF, it may be more readily inhibited by AICD and predispose to the generation of neuronal apoptosis.
7. LSF and HIV
Evidence is more compelling that LSF regulates gene expression of the human immunodeficiency virus (HIV). Whereas NF-κB and Sp1 are potent transcriptional activators of expression from the HIV long terminal repeat (LTR) (El Kharroubi et al., 1998; Harrich et al., 1989; Li et al., 1994; Marcello et al., 2004; Perkins et al., 1993), three LSF half-site motifs around the initiation site of transcription (−10 to +27) lead to transcriptional repression (Coull et al., 2000; Coull et al., 2002; He and Margolis, 2002; Jones et al., 1988; Kato et al., 1991; Romerio et al., 1997). In vitro, LSF/LBP-1b represses transcription by two mechanisms: by preventing access to the TATA sequence (Kato et al., 1991) and by inhibiting transcriptional elongation (Parada et al., 1995). In vivo, the predominant mechanism of transcriptional repression of the LTR (Coull et al., 2002) is through a chromatin-mediated mechanism. Tetrameric LSF binds to two of the three half sites at a time (D. Margolis, pers. comm.) as a heteromeric complex with ying yang 1 (YY1), which recruits HDAC1 (Coull et al., 2000; He and Margolis, 2002; Romerio et al., 1997) (Fig. 6). Histone deacetylation then inhibits transcription (Ng and Bird, 2000; Pazin and Kadonaga, 1997).
Figure 6. LSF represses transcription from the HIV LTR: Model of a role for LSF in viral latency.
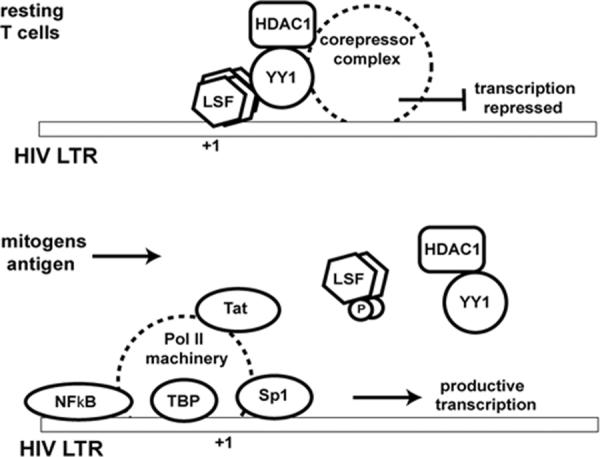
In resting T cells, transcription from the HIV LTR may be repressed by a complex including LSF, YY1 and histone deacetylase 1 (HDAC1). Upon stimulation of T cells by mitogens or by a specific antigen, the LSF-containing complex would be released, permitting assembly of the activating transcription factors, including Sp1, NF-κB, TATA-binding protein (TBP), and the rest of the RNA pol II machinery. Displacement of the repressive complex may be triggered by phosphorylation of LSF. Upon induction of HIV gene expression, Tat is produced, which further enhances productive transcription of the HIV genome.
One intriguing hypothesis that has been presented is that repression of HIV gene expression by LSF/YY1 may contribute to the reservoir of latent virus in a pool of memory CD4+ cells (He et al., 2002; Persaud et al., 2003; Pomerantz, 2003). As current HIV treatments rely on virus expression to combat the infected cells, this latent population is highly problematic for eradication of HIV from the body. Interestingly, treatment of HIV patients with IL-2, in combination with antiretroviral therapy, downregulated the DNA-binding activities of both LSF and YY1 in extracts of peripheral blood mononuclear cells (Bovolenta et al., 1999). This may aid in reactivation of virus for treatment. In line with this hypothesis, pyrrole-imidazole polyamides that inhibit the binding of LSF to the LTR both in vitro and in vivo specifically activate expression from the HIV LTR in a model tissue culture system (Coull et al., 2002). The targeting of LSF by signal transduction pathways during activation of T cells may also contribute to normal reactivation of the virus upon immunogenic stimulation of resting memory cells. In that regard, phosphorylation of LSF by Erk inhibits its binding to the HIV LTR, although phosphorylation by p38 (Volker, 1998) enhances LSF DNA binding (D. Margolis, pers. comm.); both pathways are activated in response to cytokine stimulation (Johnson and Lapadat, 2002). Although these results, taken together, suggest a role for LSF in HIV expression, and therefore, viral replication, more research is required in order to tease out the biological role for LSF in latency and reactivation of HIV.
8. LSF as a target of multiple signaling pathways
LSF is directly targeted in diverse cell types by the MEK/Erk kinase pathway, which is central to growth factor signaling (Pagon et al., 2003; Volker et al., 1997). Current data also suggest that LSF is a direct target of other MAPK signaling pathways, as LSF is phosphorylated in cells in response to UV irradiation (E. Drouin, U. Hansen, unpublished observations), its binding to an integrated HIV LTR is decreased by a p38-specific inhibitor (D. Margolis, pers. comm.), and it is specifically phosphorylated by both JNK and p38 in vitro (Volker, 1998). As suggested in Section 5.5, we hypothesize that LSF, as a downstream effector of the p38 and/or Jnk signaling pathways, is a mediator of cytokine signaling pathways in immune and liver cells.
Finally, there is evidence to suggest that LSF is a downstream effector of the PI3 kinase/Akt signaling pathway, and is involved in cell survival. The most direct data in this regard are from a unique, differentiated rat neuronal cell system (Kashour et al., 2003), in which both LSF and active Akt are required to protect the cells from apoptosis. Indeed, since LSF is directly phosphorylated by Akt in vitro (U. Saxena, Z. Pagon, G.M. Cooper, U. Hansen, unpublished observation), the hypothesis is that LSF is a direct target of Akt in this pathway. However, if it is true in this differentiated neuronal cell system, it is likely also to be true in other cells, as Akt signaling is a widespread determinant for cell survival (Datta et al., 1999).
As a direct target for multiple signaling pathways, LSF may play a general role in the cross-talk between different pathways. More specifically, LSF may integrate signals from the cellular environment to help determine decisions of cell growth versus quiescence/differentiation versus apoptosis. In addition, response to tissue-specific signaling pathways (e.g., cytokine response) may be essential for the regulatory roles of LSF in the immune response.
9. Future directions
9.1. Organismal functions of the LSF gene family
The current models for the biological functions of LSF, as described in sections 5 through 7, are all based on cell culture systems. Final elucidation of the organismal role for this transcription factor subfamily will require mating of a conditional knockout LBP-1a/b mouse with the viable LSF-deficient mouse, in order to probe functions of these proteins in different tissues. Such mice would also permit examination of the extent of the presumed functional redundancy between LSF and LBP-1a/b (see Fig. 1). In this regard, it is noteworthy that some, but not all, of the residues in LSF that are phosphorylated by different kinases appear to be conserved in LBP-1a/b. Thus, the ability to modulate LSF activity in response to certain signals may be somewhat distinct from the ability to modulate LBP-1a/b.
9.2. Structure of the LSF DNA-binding domain
One essential direction for future studies is the understanding of the structure of the DNA-binding region of the LSF/GRH family of proteins, and the molecular details of how the proteins interact with the DNA recognition sites. Armed with such a structural basis, the impact of protein modification on LSF activities would much more readily be understood.
9.3. Signaling to LSF
The molecular mechanism for how LSF modification by any signaling pathway affects its activities has not yet been elucidated. Furthermore, the relationship between signaling to LSF and its overall biological functions, either in cell-cycle progression or in tissue-specific functions is largely speculative at present. The study of LSF functions in mice deficient in particular signaling pathways, or in cell culture where signaling has been knocked down, should be highly informative to tease out distinct contributions of particular pathways for LSF overall function.
Finally, determination of whether the APP family of proteins modulate LSF activity under normal circumstances, in a variety of cell types, awaits elucidation of the presumed extracellular signals that regulate APP cleavage.
9.4. LSF target genes
One major question about LSF that remains unanswered is: what genes does LSF regulate? The current set of known targets is very limited, with some tissue-specific, some cell-cycle regulated, but clearly there are many more to be identified. To tackle this problem on a global level, a computational approach must be taken. Toward that direction, an algorithm to identify statistically overrepresented motifs determined transcription factors (NF-κB, Oct, SRF, and AP-1) whose binding sites tend to accompany known LSF binding sites in mammalian promoters (Frith et al., 2004). This information, in combination with other algorithms that identify clusters of these transcription factors sites in the human genome (Frith et al., 2003), can be utilized to predict human genes regulated by LSF. This method should prove invaluable for prediction of global LSF gene targets, although it would not predict some tissue-specific promoters that bind heteromeric protein complexes with LSF and therefore exhibit different DNA recognition motifs. As a complementary approach, global gene expression profiling, either in mice deficient of LSF/LBP-1a/b, or in knockdown tissue culture cells, should elucidate a panel of genes regulated by these proteins under particular biological conditions. Ultimately, the identification of the set(s) of LSF-regulated genes will test our current hypotheses regarding its biological role(s) and may provide clues as to additional, as yet unsuspected, biological functions.
Acknowledgements
We sincerely thank D.M. Margolis, J.M. Cunningham, S.M. Jane, and E. Goldstein for information prior to publication, and F. Amara and T. Südhof for important clarifications of their previous studies. We also thank D. Selkoe, T. Russo, G. Cooper, K. Repetny, R. Gergis, U. Saxena, M. Frith, K. Venkatesan, and H. McManus for helpful discussions, and A. van Wijnen for the opportunity to write this review. Our studies on this topic were supported by the National Institutes of Health (R01 CA81157 to U.H.). JV was supported in part by the Dragomir Nicolich Trust Scholarship from the Studenica Foundation.
Abbreviations
- Aβ
β-amyloid peptide
- AICD
APP intracellular C-terminal domain
- APP
amyloid precursor protein
- CP2
CCAAT-binding protein-2
- dnLSF
dominant negative LSF
- GRH
grainyhead
- GST
glutathioine S-transferase
- HDAC
histone deacetylase
- HIV
human immunodeficiency virus
- LBP-1
leader-binding protein-1
- LSF
late Simian Virus 40 factor
- LSF-ID
LSF-internally deleted
- LTR
long terminal repeat
- MAPK
mitogen activated protein kinase
- Rb
retinoblastoma protein
- Sα
immunoglobulin switch recombination region upstream of the coding region for IgA
- Sμ
immunoglobulin switch recombination region upstream of the coding region for IgM
- SAA
serum amyloid A
- SEF1
serum amyloid A3 enhancer factor 1
- TBP
TATA-binding protein
- TH
helper T lymphocytes
- TS
thymidylate synthase
- UBP-1
upstream region binding protein-1
- WT1
Wilm's tumor protein 1
- YY1
ying yang 1
References
- Algar E. A review of the Wilms' tumor 1 gene (WT1) and its role in hematopoiesis and leukemia. J Hematother Stem Cell Res. 2002;11:589–599. doi: 10.1089/15258160260194749. [DOI] [PubMed] [Google Scholar]
- Alvarez M, Rhodes SJ, Bidwell JP. Context-dependent transcription: all politics is local. Gene. 2003;313:43–57. doi: 10.1016/s0378-1119(03)00627-9. [DOI] [PubMed] [Google Scholar]
- Attardi LD, Tjian R. Drosophila tissue-specific transcription factor NTF-1 contains a novel isoleucine-rich activation motif. Genes Dev. 1993;7:1341–1353. doi: 10.1101/gad.7.7b.1341. [DOI] [PubMed] [Google Scholar]
- Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-κB and β-amyloid precursor protein. Cell. 2002;110:55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- Barnhart KM, Kim CG, Banerji SS, Sheffery M. Identification and characterization of multiple erythoid cell proteins that interact with the promoter of the murine α-globin gene. Mol Cell Biol. 1988;8:3215–3226. doi: 10.1128/mcb.8.8.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15:74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- Bellorini M, Dantonel JC, Yoon J-B, Roeder RG, Tora L, Mantovani R. The major histocompatibility complex class II Ea promoter requires TFIID binding to an initiator sequence. Mol Cell Biol. 1996;16:503–512. doi: 10.1128/mcb.16.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing Z, Huang JH, Liao WSL. NFκB interacts with serum amyloid A3 enhancer factor to synergistically activate mouse serum amyloid A3 gene transcription. J Biol Chem. 2000;275:31616–31623. doi: 10.1074/jbc.M005378200. [DOI] [PubMed] [Google Scholar]
- Bing Z, Reddy SAG, Ren Y, Qin J, Liao WSL. Purification and characterization of the serum amyloid A3 enhancer factor. J Biol Chem. 1999;274:24649–24656. doi: 10.1074/jbc.274.35.24649. [DOI] [PubMed] [Google Scholar]
- Bovolenta C, Camorali L, Lorini AL, Vallanti G, Ghezzi S, Tambussi G, Lazzarin A, Poli G. In vivo administration of recombinant IL-2 to individuals infected by HIV down-modulates the binding and expression of the transcription factors ying-yang-1 and leader binding protein-1/late simian virus 40 factor. J Immunol. 1999;163:6892–6897. [PubMed] [Google Scholar]
- Bray SJ, Kafatos FC. Developmental function of Elf-1: an essential transcription factor during embryogenesis in Drosophila. Genes Dev. 1991;5:1672–1683. doi: 10.1101/gad.5.9.1672. [DOI] [PubMed] [Google Scholar]
- Bruni P, Minopoli G, Brancaccio T, Napolitano M, Faraonio R, Zambrano N, Hansen U, Russo T. Fe65, a ligand of the Alzheimer's β-amyloid precursor protein, blocks cell cycle progression by down-regulating thymidylate synthase expression. J Biol Chem. 2002;277:35481–35488. doi: 10.1074/jbc.M205227200. [DOI] [PubMed] [Google Scholar]
- Cao X, Südhof TC. A transcriptionally active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Casolaro V, Keane-Myers AM, Swendeman SL, Steindler C, Zhong F, Sheffery M, Georas SN, Ono SJ. Identification and characterization of a critical CP2-binding element in the human interleukin-4 promoter. J Biol Chem. 2000;275:36605–36611. doi: 10.1074/jbc.M007086200. [DOI] [PubMed] [Google Scholar]
- Chae JH, Kim CG. CP2 binding to the promoter is essential for the enhanced transcription of globin genes in erythroid cells. Mol Cells. 2003;15:40–47. [PubMed] [Google Scholar]
- Chae JH, Lee YH, Kim CG. Transcription factor CP2 is crucial in hemoglobin synthesis during erythroid terminal differentiation in vitro. Biochem Biophys Res Commun. 1999;263:580–583. doi: 10.1006/bbrc.1999.1408. [DOI] [PubMed] [Google Scholar]
- Chodosh LA, Baldwin AS, Carthew RW, Sharp PA. Human CCAAT-binding proteins have heterologous subunits. Cell. 1988;53:11–24. doi: 10.1016/0092-8674(88)90483-7. [DOI] [PubMed] [Google Scholar]
- Coull JJ, He G, Melander C, Rucker VC, Dervan PB, Margolis DM. Targeted derepression of the human immunodeficiency virus type 1 long terminal repeat by pyrrole-imidazole polyamides. J Virol. 2002;76:12349–12354. doi: 10.1128/JVI.76.23.12349-12354.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JJ, Romerio F, Sun J-M, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM. Functional mapping of the interactions of the HIV repressors YY1 and LSF. J Virol. 2000;74:6790–6799. doi: 10.1128/jvi.74.15.6790-6799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson EJ, Paliga K, Beyreuther K, Masters CL. What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem Int. 2000;36:175–184. doi: 10.1016/s0197-0186(99)00125-4. [DOI] [PubMed] [Google Scholar]
- Cunningham JM, Vanin EF, Tran N, Valentine M, Jane SM. The human transcription factor CP2 (TFCP2), a component of the human gamma-globin stage selector protein, maps to chromosome region 12q13 and is within 250 kb of the NF-E2 gene. Genomics. 1995;30:398–399. [PubMed] [Google Scholar]
- Cupers P, Orlans I, Craessaerts K, Annaert W, De Strooper B. The amyloid precursor protein (APP)-cytoplasmic fragment generated by γ-secretase is rapidly degraded but distributes partially in a nuclear fraction of neurones in culture. J Neurochem. 2001;78:1168–1178. doi: 10.1046/j.1471-4159.2001.00516.x. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S, Lester L, Johnson LF. Transcriptional control elements and complex initiation pattern of the TATA-less bidirectional human thymidylate synthase promoter. J Cell Biochem. 2000;77:50–64. doi: 10.1002/(sici)1097-4644(20000401)77:1<50::aid-jcb6>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Drouin EE, Schrader CE, Stavnezer J, Hansen U. The ubiquitously expressed DNA-binding protein Late SV40 Factor binds Ig switch regions and represses class switching to IgA. J Immunol. 2002;168:2847–2856. doi: 10.4049/jimmunol.168.6.2847. [DOI] [PubMed] [Google Scholar]
- El Kharroubi A, Piras G, Zensen R, Martin MA. Transcriptional activation of the integrated chromatin-associated human immunodeficiency virus type 1 promoter. Mol Cell Biol. 1998;18:2535–2544. doi: 10.1128/mcb.18.5.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellisen LW. Regulation of gene expression by WT1 in development and tumorigenesis. Int J Hematol. 2002;76:110–116. doi: 10.1007/BF02982572. [DOI] [PubMed] [Google Scholar]
- Fecko JK. M. A. Thesis. Boston University; 2001. Regulation of the mammalian transcription factor LSF by cyclin E-dependent kinase. [Google Scholar]
- Finch A, Davis W, Carter WG, Saklatvala J. Analysis of mitogen-activated protein kinase pathways used by interleukin 1 in tissues in vivo: activation of hepatic c-Jun N-terminal kinases 1 and 2, and mitogen-activated protein kinase kinases 4 and 7. Biochem J. 2001;353:275–281. doi: 10.1042/0264-6021:3530275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch A, Holland P, Cooper J, Saklatvala J, Kracht M. Selective activation of JNK/SAPK by interleukin-1 in rabbit liver is mediated by MKK7. FEBS Lett. 1997;418:144–148. doi: 10.1016/s0014-5793(97)01364-1. [DOI] [PubMed] [Google Scholar]
- Frith MC, Fu Y, Yu L, Chen J-F, Hansen U, Weng Z. Detection of functional DNA motifs via statistical overrepresentation. Nucleic Acids Res. 2004;32:1372–1381. doi: 10.1093/nar/gkh299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Hansen U, Weng Z. Detection of cis-element clusters in higher eukaryotic DNA. Bioinformatics. 2001;17:878–889. doi: 10.1093/bioinformatics/17.10.878. [DOI] [PubMed] [Google Scholar]
- Frith MC, Li MC, Weng Z. Cluster-Buster: finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 2003;31:3666–3668. doi: 10.1093/nar/gkg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Pimplikar SW. The γ-secretase-cleaved C-terminal fragment of amyloid precursor protein mediates signaling to the nucleus. Proc Natl Acad Sci USA. 2001;98:14979–14984. doi: 10.1073/pnas.261463298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harrich D, Garcia J, Wu F, Mitsuyasu R, Gonazalez J, Gaynor R. Role of SP1-binding domains in in vivo transcriptional regulation of the human immunodeficiency virus type 1 long terminal repeat. J Virol. 1989;63:2585–2591. doi: 10.1128/jvi.63.6.2585-2591.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Margolis DM. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol Cell Biol. 2002;22:2965–2973. doi: 10.1128/MCB.22.9.2965-2973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Ylisastigui L, Margolis DM. The regulation of HIV-1 gene expression: the emerging role of chromatin. DNA Cell Biol. 2002;21:697–705. doi: 10.1089/104454902760599672. [DOI] [PubMed] [Google Scholar]
- Huang H-C, Sundseth R, Hansen U. Transcription factor LSF binds two variant bipartite sites within the SV40 late promoter. Genes Dev. 1990;4:287–298. doi: 10.1101/gad.4.2.287. [DOI] [PubMed] [Google Scholar]
- Huang JD, Dubnicoff T, Liaw GJ, Bai Y, Valentine SA, Shirokawa JM, Lengyel JA, Courey AJ. Binding sites for transcription factor NTF-1/Elf-1 contribute to the ventral repression of decapentaplegic. Genes Dev. 1995;9:3177–3189. doi: 10.1101/gad.9.24.3177. [DOI] [PubMed] [Google Scholar]
- Huang JH, Liao WSL. Induction of the mouse serum amyloid A3 gene by cytokines requires both C/EBP family proteins and a novel constitutive nuclear factor. Mol Cell Biol. 1994;14:4475–4484. doi: 10.1128/mcb.14.7.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JH, Liao WSL. Synergistic induction of mouse serum amyloid A3 promoter by the inflammatory mediators IL-1 and IL-6. J Interferon Cytokine Res. 1999;19:1403–1411. doi: 10.1089/107999099312867. [DOI] [PubMed] [Google Scholar]
- Huang N, Miller WL. Cloning of factors related to HIV-inducible LBP proteins that regulate steroidogenic factor-1-independent human placental transcription of the cholesterol side-chain cleavage enzyme, P450scc. J Biol Chem. 2000;275:2852–2858. doi: 10.1074/jbc.275.4.2852. [DOI] [PubMed] [Google Scholar]
- Jane SM, Nienhuis AW, Cunningham JM. Hemoglobin switching in man and chicken is mediated by a heteromeric complex between the ubiquitous transcription factor CP2 and a developmentally specific protein. EMBO J. 1995;14:97–105. doi: 10.1002/j.1460-2075.1995.tb06979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Jones KA, Luciw PA, Duchange N. Structural arrangements of transcription control domains within the 5'-untranslated leader regions of the HIV-1 and HIV-2 promoters. Genes Dev. 1988;2:1101–1114. doi: 10.1101/gad.2.9.1101. [DOI] [PubMed] [Google Scholar]
- Kashour T, Burton T, Dibrov A, Amara FM. Late Simian virus 40 transcription factor is a target of the phosphoinositide 3-kinase/Akt pathway in anti-apoptotic Alzheimer's amyloid precursor protein signalling. Biochem J. 2003;370:1063–1075. doi: 10.1042/BJ20021197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Horikoshi M, Roeder RG. Repression of HIV-1 transcription by a cellular protein. Science. 1991;251:1476–1479. doi: 10.1126/science.2006421. [DOI] [PubMed] [Google Scholar]
- Kim CG, Barnhart KM, Sheffery M. Purification of multiple erythoid cell proteins that bind the promoter of the α-globin gene. Mol Cell Biol. 1988;8:4270–4281. doi: 10.1128/mcb.8.10.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CG, Swendeman SL, Barnhart KM, Sheffery M. Promoter elements and erythroid cell nuclear factors that regulate α-globin gene transcription in vitro. Mol Cell Biol. 1990;10:5958–5966. doi: 10.1128/mcb.10.11.5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Heath C, Bertuch A, Hansen U. Specific stimulation of simian virus 40 late transcription in vitro by a cellular factor binding the simian virus 40 21-base-pair repeat promoter element. Proc Natl Acad Sci USA. 1987;84:6025–6029. doi: 10.1073/pnas.84.17.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-S, Kim E-M, Lee J-P, Park CH, Kim S, Seo J-H, Chang K-A, Yu E, Jeong S-J, Chong YH, Suh Y-H. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3β expression. FASEB J. 2003;17:1951–1953. doi: 10.1096/fj.03-0106fje. [DOI] [PubMed] [Google Scholar]
- Kimberly WT, Zheng JB, Guénette SY, Selkoe DJ. The intracellular domain of the β-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001;276:40288–40292. doi: 10.1074/jbc.C100447200. [DOI] [PubMed] [Google Scholar]
- Kudryavtseva EI, Sugihara TM, Wang N, Lasso RJ, Gudnason JF, Lipkin SM, Andersen B. Identification and characterization of Grainyhead-like epithelial transactivator (GET-1), a novel mammalian Grainyhead-like factor. Dev Dyn. 2003;226:604–617. doi: 10.1002/dvdy.10255. [DOI] [PubMed] [Google Scholar]
- Kwon J, Morshead KB, Guyon JR, Kingston RE, Oettinger MA. Histone acetylation and hSWI/SNF remodeling act in concert to stimulate V(D)J cleavage of nucleosomal DNA. Mol Cell. 2000;6:1037–1048. doi: 10.1016/s1097-2765(00)00102-7. [DOI] [PubMed] [Google Scholar]
- Lambert J-C, Goumidi L, Vrièze FWD, Frigard B, Harris JM, Cummings A, Coates J, Pasquier F, Cottel D, Gaillac M, St Clair D, Mann DMA, Hardy J, Lendon CL, Amouyel P, Chartier-Harlin M-C. The transcriptional factor LBP-1c/CP2/LSF gene on chromosome 12 is a genetic determinant of Alzheimer's disease. Hum Mol Genet. 2000;9:2275–2280. doi: 10.1093/oxfordjournals.hmg.a018918. [DOI] [PubMed] [Google Scholar]
- Lambertini E, Penolazzi L, Giordano S, Del Senno L, Piva R. Expression of the human oestrogen receptor-α gene is regulated by promoter F in MG-63 osteoblastic cells. Biochem J. 2003;372:831–839. doi: 10.1042/BJ20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau K-F, Miller CCJ, Anderton BH, Shaw P-C. Molecular cloning and characterization of the human glycogen synthase kinase-3β promoter. Genomics. 1999;60:121–128. doi: 10.1006/geno.1999.5875. [DOI] [PubMed] [Google Scholar]
- Lee Y, Johnson LF. Transcriptional control elements of the rat thymidylate synthase promoter: evolutionary conservation of regulatory features. Exp Cell Res. 2000;258:53–64. doi: 10.1006/excr.2000.4911. [DOI] [PubMed] [Google Scholar]
- Lendon C, Craddock N. Is LBP-1c/CP2/LSF a disease-modifying gene for Alzheimer's disease? Lancet. 2001;358:1029–1030. doi: 10.1016/S0140-6736(01)06228-6. [DOI] [PubMed] [Google Scholar]
- Li Y, Mak G, Franza BR., Jr. In vitro study of functional involvement of Sp1, NF-kappa B/Rel, and AP1 in phorbol 12-myristate 13-acetate-mediated HIV-1 long terminal repeat activation. J Biol Chem. 1994a;269:30616–30619. [PubMed] [Google Scholar]
- Li Y, Slansky JE, Myers DJ, Drinkwater NR, Kaelin WG, Farnham PJ. Cloning, chromosomal location, and characterization of mouse E2F1. Mol Cell Biol. 1994b;14:1861–1869. doi: 10.1128/mcb.14.3.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw GJ, Rudolph KM, Huang JD, Dubnicoff T, Courey AJ, Lengyel JA. The torso response element binds GAGA and NTF-1/Elf-1, and regulates tailless by relief of repression. Genes Dev. 1995;9:3163–3176. doi: 10.1101/gad.9.24.3163. [DOI] [PubMed] [Google Scholar]
- Lim LC, Fang L, Swendeman SL, Sheffery M. Characterization of the molecularly cloned murine α-globin transcription factor CP2. J Biol Chem. 1993;268:18008–18017. [PubMed] [Google Scholar]
- Lim LC, Swendeman SL, Sheffery M. Molecular cloning of the α-globin transcription factor CP2. Mol Cell Biol. 1992;12:828–835. doi: 10.1128/mcb.12.2.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedecking-Zimmer E, DeKosky ST, Nebes R, Kamboh MI. Association of the 3' UTR transcription factor LBP-1c/CP2/LSF polymorphism with late-onset Alzheimer's disease. Am J Med Genet. 2003;117B:114–117. doi: 10.1002/ajmg.b.10026. [DOI] [PubMed] [Google Scholar]
- Ma T, Van Tine BA, Wei Y, Garrett MD, Nelson D, Adams PD, Wang J, Qin J, Chow LT, Harper JW. Cell cycle-regulated phosphorylation of p220NPAT by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev. 2000;14:2298–2313. doi: 10.1101/gad.829500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello A, Lusic M, Pegoraro G, Pellegrini V, Beltram F, Giacca M. Nuclear organization and the control of HIV-1 transcription. Gene. 2004;326:1–11. doi: 10.1016/j.gene.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Minopoli G, De Candia P, Bonetti A, Faraonio R, Zambrano N, Russo T. The β-amyloid precursor protein functions as a cytosolic anchoring site that prevents Fe65 nuclear translocation. J Biol Chem. 2001;276:6545–6550. doi: 10.1074/jbc.M007340200. [DOI] [PubMed] [Google Scholar]
- Murata T, Nitta M, Yasuda K. Transcription factor CP2 is essential for lens-specific expression of the chicken αA-crystallin gene. Genes Cells. 1998;3:443–457. doi: 10.1046/j.1365-2443.1998.00204.x. [DOI] [PubMed] [Google Scholar]
- Neve RL, McPhie DL, Chen Y. Alzheimer's disease: a dysfunction of the amyloid precursor protein(1) Brain Res. 2000;886:54–66. doi: 10.1016/s0006-8993(00)02869-9. [DOI] [PubMed] [Google Scholar]
- Ng HH, Bird A. Histone deacetylases: silencers for hire. Trends Biochem Sci. 2000;25:121–126. doi: 10.1016/s0968-0004(00)01551-6. [DOI] [PubMed] [Google Scholar]
- Pagon Z, Volker J, Cooper GM, Hansen U. Mammalian transcription factor LSF is a target of ERK signaling. J Cell Biochem. 2003;89:733–746. doi: 10.1002/jcb.10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panza F, D'Introno A, Colacicco AM, Capurso C, Basile AM, Torres F, Capurso A, Solfrizzi V. LBP-1c/CP2/LSF gene polymorphism and risk of sporadic Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2004;75:166–168. [PMC free article] [PubMed] [Google Scholar]
- Parada CA, Yoon J-B, Roeder RG. A novel LBP-1-mediated restriction of HIV-1 transcription at the level of elongation in vitro. J Biol Chem. 1995;270:2274–2283. doi: 10.1074/jbc.270.5.2274. [DOI] [PubMed] [Google Scholar]
- Park J, Tadlock L, Gores GJ, Patel T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology. 1999;30:1128–1133. doi: 10.1002/hep.510300522. [DOI] [PubMed] [Google Scholar]
- Passer B, Pellegrini L, Russo C, Siegel RM, Lenardo MJ, Schettini G, Bachmann M, Tabaton M, D'Adamio L. Generation of an apoptotic intracellular peptide by γ-secretase cleavage of Alzheimer's amyloid β protein precursor. J Alzheimers Dis. 2000;2:289–301. doi: 10.3233/jad-2000-23-408. [DOI] [PubMed] [Google Scholar]
- Pazin MJ, Kadonaga JT. What's up and down with histone deacetylation and transcription? Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- Pelton TA, Sharma S, Schulz TC, Rathjen J, Rathjen PD. Transient pluripotent cell populations during primitive ectoderm formation: correlation of in vivo and in vitro pluripotent cell development. J Cell Sci. 2002;115:329–339. doi: 10.1242/jcs.115.2.329. [DOI] [PubMed] [Google Scholar]
- Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J. 1993;12:3551–3558. doi: 10.1002/j.1460-2075.1993.tb06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persaud D, Zhou Y, Siliciano JM, Siliciano RF. Latency in human immunodeficiency virus type 1 infections: no easy answers. J. Virol. 2003;77:1659–1665. doi: 10.1128/JVI.77.3.1659-1665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters LM, Anderson DW, Griffith AJ, Grundfast KM, San Agustin TB, Madeo AC, Friedman TB, Morell RJ. Mutation of a transcription factor, TFCP2L3, causes progressive autosomal dominant hearing loss, DFNA28. Hum Mol Genet. 2002;11:2877–2885. doi: 10.1093/hmg/11.23.2877. [DOI] [PubMed] [Google Scholar]
- Pomerantz RJ. Reservoirs, sanctuaries, and residual disease: the hiding spots of HIV-1. HIV Clin. Trials. 2003;4:137–143. doi: 10.1310/80jh-148k-nadq-u927. [DOI] [PubMed] [Google Scholar]
- Powell CMH. Ph. D. Thesis. Harvard University; 1999. Role of the mammalian transcription factor LSF at the G1/S transition of the cell cycle. [Google Scholar]
- Powell CMH, Rudge TL, Zhu Q, Johnson LF, Hansen U. Inhibition of the mammalian transcription factor LSF induces S-phase-dependent apoptosis by downregulating thymidylate synthase expression. EMBO J. 2000;19:4665–4675. doi: 10.1093/emboj/19.17.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy L, Barbour V, Tuckfield A, Clouston DR, Topham D, Cunningham JM, Jane SM. Targeted disruption of the CP2 gene, a member of the NTF family of transcription factors. J Biol Chem. 2001;276:7836–7842. doi: 10.1074/jbc.M004351200. [DOI] [PubMed] [Google Scholar]
- Rodda S, Sharma S, Scherer M, Chapman G, Rathjen P. CRTR-1, a developmentally regulated transcriptional repressor related to the CP2 family of transcription factors. J Biol Chem. 2001;276:3324–3332. doi: 10.1074/jbc.M008167200. [DOI] [PubMed] [Google Scholar]
- Romerio F, Gabriel MN, Margolis DM. Repression of Human Immunodeficiency Virus Type 1 through the novel cooperation of human factors YY1 and LSF. J Virol. 1997;71:9375–9382. doi: 10.1128/jvi.71.12.9375-9382.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo T, Faraonio R, Minopoli G, De Candia P, De Renzis S, Zambrano N. Fe65 and the protein network centered around the cytosolic domain of the Alzheimer's β-amyloid precursor protein. FEBS Lett. 1998;434:1–7. doi: 10.1016/s0014-5793(98)00941-7. [DOI] [PubMed] [Google Scholar]
- Scheinfeld MH, Ghersi E, Laky K, Fowlkes BJ, D'Adamio L. Processing of β-amyloid precursor-like protein-1 and -2 by γ-secretase regulates transcription. J Biol Chem. 2002;277:44195–44201. doi: 10.1074/jbc.M208110200. [DOI] [PubMed] [Google Scholar]
- Scheinfeld MH, Matsuda S, D'Adamio L. JNK-interacting protein-1 promotes transcription of Aβ protein precursor but not Aβ precursor-like proteins, mechanistically different than Fe65. Proc Natl Acad Sci USA. 2003;100:1729–1734. doi: 10.1073/pnas.0437908100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlissel MS. Perspectives: transcription. A tail of histone acetylation and DNA recombination. Science. 2000;287:438–440. doi: 10.1126/science.287.5452.438. [DOI] [PubMed] [Google Scholar]
- Selkoe D, Kopan R. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci. 2003;26:565–597. doi: 10.1146/annurev.neuro.26.041002.131334. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Amyloid beta-protein and the genetics of Alzheimer's disease. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shirra MK. Ph. D. Thesis. Harvard University; 1995. Characterization of DNA-binding and oligomerization properties of the mammalian transcription factor LSF. [Google Scholar]
- Shirra MK, Hansen U. LSF and NTF-1 share a conserved DNA-recognition motif yet require different oligomerization states to form a stable protein-DNA complex. J Biol Chem. 1998;273:19260–19268. doi: 10.1074/jbc.273.30.19260. [DOI] [PubMed] [Google Scholar]
- Shirra MK, Zhu Q, Huang H-C, Pallas D, Hansen U. One exon of the human LSF gene includes conserved regions involved in novel DNA-binding and dimerization motifs. Mol Cell Biol. 1994;14:5076–5087. doi: 10.1128/mcb.14.8.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis C, Aizencang GI, Astrin KH, Bishop DF, Desnick RJ. Uroporphyrinogen III synthase erythroid promoter mutations in adjacent GATA1 and CP2 elements cause congenital erythropoietic porphyria. J Clin Invest. 2001;107:753–762. doi: 10.1172/JCI10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Haass C. Intramembrane proteolysis by presenilins. Nat Rev Mol Cell Biol. 2000;1:217–224. doi: 10.1038/35043065. [DOI] [PubMed] [Google Scholar]
- Sueyoshi T, Kobayashi R, Nishio K, Aida K, Moore R, Wada T, Handa H, Negishi M. A nuclear factor (NF2d9) that binds to the male-specific P450 (Cyp 2d-9) gene in mouse liver. Mol Cell Biol. 1995;15:4158–4166. doi: 10.1128/mcb.15.8.4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundseth R, Hansen U. Activation of RNA polymerase II transcription by the specific DNA-binding protein LSF: Increased rate of binding of the basal promoter factor TFIIB. J Biol Chem. 1992;267:7845–7855. [PubMed] [Google Scholar]
- Swendeman SL, Spielholz C, Jenkins NA, Gilbert DJ, Copeland NG, Sheffery M. Characterization of the genomic structure, chromosomal location, promoter, and developmental expression of the α-globin transcription factor CP2. J Biol Chem. 1994;269:11663–11671. [PubMed] [Google Scholar]
- Taylor AE, Yip A, Brayne C, Easton D, Evans JG, Xuereb J, Cairns N, Esiri MM, Rubinsztein DC. Genetic association of an LBP-1c/CP2/LSF gene polymorphism with late onset Alzheimer's disease. J Med Genet. 2001;38:232–233. doi: 10.1136/jmg.38.4.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting SB, Wilanowski T, Auden A, Hall M, Voss AK, Thomas T, Parekh V, Cunningham JM, Jane SM. Inositol- and folate-resistant neural tube defects in mice lacking the epithelial-specific factor Grhl-3. Nat Med. 2003a;9:1513–1519. doi: 10.1038/nm961. [DOI] [PubMed] [Google Scholar]
- Ting SB, Wilanowski T, Cerruti L, Zhao L-L, Cunningham JM, Jane SM. The identification and characterization of human Sister-of-Mammalian Grainyhead (SOM) expands the grainyhead-like family of developmental transcription factors. Biochem J. 2003b;370:953–962. doi: 10.1042/BJ20021476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuckfield A, Clouston DR, Wilanowski TM, Zhao L-L, Cunningham JM, Jane SM. Binding of the RING polycomb proteins to specific target genes in complex with the grainyhead-like family of developmental transcription factors. Mol Cell Biol. 2002;22:1936–1946. doi: 10.1128/MCB.22.6.1936-1946.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uv AE, Harrison EJ, Bray SJ. Tissue-specific splicing and functions of the Drosophila transcription factor Grainyhead. Mol Cell Biol. 1997;17:6727–6735. doi: 10.1128/mcb.17.11.6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uv AE, Thompson CRL, Bray SJ. The Drosophila tissue-specific factor grainyhead contains novel DNA-binding and dimerization domains which are conserved in the human protein CP2. Mol Cell Biol. 1994;14:4020–4031. doi: 10.1128/mcb.14.6.4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan K, McManus HR, Mello CR, Smith TF, Hansen U. Functional conservation between members of an ancient duplicated transcription factor family, LSF/Grainyhead. Nucleic Acids Res. 2003;31:4304–4316. doi: 10.1093/nar/gkg644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viville S, Jongeneel V, Koch W, Mantovani R, Benoist C, Mathis D. The E alpha promoter: a linker-scanning analysis. J Immunol. 1991;146:3211–3217. [PubMed] [Google Scholar]
- Volker JL. Ph. D. Thesis. Harvard University; 1998. Regulation of the mammalian transcription factor LSF at the G0/G1 boundary. [Google Scholar]
- Volker JL, Rameh LE, Zhu Q, DeCaprio J, Hansen U. Mitogenic stimulation of resting T cells causes rapid phosphorylation of the transcription factor LSF and increased DNA-binding activity. Genes Dev. 1997;11:1435–1446. doi: 10.1101/gad.11.11.1435. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Fadeeva JV, LaVoie MJ, Paliga K, Eggert S, Kimberly WT, Wasco W, Selkoe DJ. γ-Secretase cleavage and binding to FE65 regulate the nuclear translocation of the intracellular C-terminal domain (ICD) of the APP family of proteins. Biochem. 2003;42:6664–6673. doi: 10.1021/bi027375c. [DOI] [PubMed] [Google Scholar]
- Weinberg AD, English M, Swain SL. Distinct regulation of lymphokine production is found in fresh versus in vitro primed murine helper T cells. J Immunol. 1990;144:1800–1807. [PubMed] [Google Scholar]
- Whitmarsh AJ, Davis RJ. Regulation of transcription factor function by phosphorylation. Cell Mol Life Sci. 2000;57:1172–1183. doi: 10.1007/PL00000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilanowski T, Tuckfield A, Cerruti L, O'Connell S, Saint R, Parekh V, Tao J, Cunningham JM, Jane SM. A highly conserved novel family of mammalian developmental transcription factors related to Drosophila grainyhead. Mech Dev. 2002;114:37–50. doi: 10.1016/s0925-4773(02)00046-1. [DOI] [PubMed] [Google Scholar]
- Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, Meinhardt T, Pruss M, Reuter I, Schacherer F. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–319. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FK, Garcia JA, Harrich D, Gaynor RB. Purification of the human immunodeficiency virus type 1 enhancer and TAR binding proteins EBP-1 and UBP-1. EMBO J. 1988;7:2117–2129. doi: 10.1002/j.1460-2075.1988.tb03051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Zhou Z-H, Bubien JK, Fuller CM, Markert JM, Mapstone TB, Gillespie GY, Benos DJ. Molecular cloning and characterization of human acid sensing ion channel (ASIC)2 gene promoter. Gene. 2003;313:91–101. doi: 10.1016/s0378-1119(03)00633-4. [DOI] [PubMed] [Google Scholar]
- Yang S-H, Sharrocks AD, Whitmarsh AJ. Transcriptional regulation by the MAP kinase signaling cascades. Gene. 2003;320:3–21. doi: 10.1016/s0378-1119(03)00816-3. [DOI] [PubMed] [Google Scholar]
- Yankner BA. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- Yoon J-B, Li G, Roeder RG. Characterization of a family of related cellular transcription factors which can modulate human immunodeficiency virus type 1 transcription in vitro. Mol Cell Biol. 1994;14:1776–1785. doi: 10.1128/mcb.14.3.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano N, Minopoli G, De Candia P, Russo T. The Fe65 adaptor protein interacts through its PID1 domain with the transcription factor CP2/LSF/LBP1. J Biol Chem. 1998;273:20128–20133. doi: 10.1074/jbc.273.32.20128. [DOI] [PubMed] [Google Scholar]
- Zauberman A, Zipori D, Krupsky M, Ben-Levy R. Stress activated protein kinase p38 is involved in IL-6 induced transcriptional activation of STAT3. Oncogene. 1999;18:3886–3893. doi: 10.1038/sj.onc.1202738. [DOI] [PubMed] [Google Scholar]
- Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- Zhao J, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA, Harlow E. NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes Dev. 2000;14:2283–2297. [PMC free article] [PubMed] [Google Scholar]
- Zhao X-F, Underhill DA, Hammond GL. Hepatic nuclear proteins that bind cis-regulatory elements in the proximal promoter of the rat corticosteroid-binding globulin gene. Mol Cell Endocrinol. 1997;126:203–212. doi: 10.1016/s0303-7207(96)03992-5. [DOI] [PubMed] [Google Scholar]
- Zheng JB, Zhou Y-H, Maity T, Liao WSL, Saunders GF. Activation of the human PAX6 gene through the exon 1 enhancer by transcription factors SEF and Sp1. Nucleic Acids Res. 2001;29:4070–4078. doi: 10.1093/nar/29.19.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong F, Swendeman SL, Popik W, Pitha PM, Sheffery M. Evidence that levels of the dimeric cellular transcription factor CP2 play little role in the activation of the HIV-1 long terminal repeat in vivo or following superinfection with herpes simplex virus type 1. J Biol Chem. 1994;269:21269–21276. [PubMed] [Google Scholar]
- Zhou W, Clouston DR, Wang X, Cerruti L, Cunningham JM, Jane SM. Induction of human fetal globin gene expression by a novel erythroid factor, NF-E4. Mol Cell Biol. 2000;20:7662–7672. doi: 10.1128/mcb.20.20.7662-7672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]