

Programmed cell death culminating in apoptosis is responsible for normal tissue homeostasis and has increasingly been implicated in mediating pathological cell loss (1–3). Apoptosis is accompanied by characteristic morphological changes, including cell body and nuclear condensation, chromatin margination, and the formation of membrane-bound remnants (apoptotic bodies) (4). In recent years much has been learned about the molecular underpinnings of apoptosis, including the release of cytochrome c from mitochondria, modulation by bcl-2 and related genes, and a central role for caspase activation in triggering final events (5). However, despite the universal prominence of cell volume loss in cells undergoing apoptosis, the relationship between this event and ensuing cell death remains uncertain. As posed by Bortner and Cidlowski (6), a key question is whether cell volume loss is “a passive, secondary feature of the cell death process” or a driver of the process.
In this issue of PNAS, Maeno et al. (7) present intriguing data supporting the latter possibility. Examining multiple cell types (HeLa, lymphoid U937, NG108–15, and PC12 cells) and methods of inducing apoptosis (treatment with staurosporine, or tumor necrosis factor α plus cycloheximide), they make three major observations: (i) apoptotic volume decrease (AVD) occurred early, before characteristic ultrastructural or biochemical events (DNA fragmentation, cytochrome c release, and caspase-3 activation); (ii) AVD was inhibited by pharmacological blockers of K+ or Cl− channels implicated in cellular volume regulation, but not by a caspase inhibitor; and (iii) pharmacological block of K+ or Cl− channels (and AVD) inhibited subsequent ultrastructural and biochemical events including cell death. In addition, they noted that the regulatory volume decrease triggered by transient exposure to hypotonic medium occurred more quickly in apoptotic cells than in control cells. These observations lead Maeno et al. to favor the hypothesis that K+ and Cl− channel-mediated AVD is an early prerequisite for apoptosis.
A visible decrease in cell body volume was recognized as a hallmark of apoptosis in the pioneering studies of Kerr et al. (4)—indeed, those workers originally used the term “shrinkage necrosis” to refer to the process. These and other groundbreaking morphological studies suggested that cell shrinkage developed relatively quickly in the course of apoptosis (4). Although events occurring before cellular fragmentation were not distinctly resolved in these early studies, more recent studies with greater temporal resolution generally have reached the same conclusion. Rat thymocytes subjected to irradiation (8) or renal tubule epithelial cells microinjected with cytochrome c (9) exhibited AVD 30–60 min later, before most other morphological changes. Studies with CD4+ T cells (10) or S49 Neo cells (11) demonstrated that AVD could precede DNA fragmentation (although see Nardi et al., ref. 12). Sympathetic ganglion neurons undergoing apoptosis after nerve growth factor (NGF) deprivation developed AVD more slowly—19 h after NGF deprivation—but still before widespread neurite fragmentation, failure of NGF rescue (commitment point), or caspase activation (13, 14).
Maeno et al. (7) observed that U937 cells exhibited AVD 1 h after exposure to staurosporine, whereas cytochrome c release, caspase-3 activation, DNA laddering, and loss of viability all required more than 2 h to appear. Administration of the broad-spectrum caspase inhibitor, benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (Z-VAD-FMK) blocked cell death but not AVD, a finding consistent with AVD occurring upstream of caspase activation. Similarly, boc-aspartyl(OMe)-fluoromethylketone or Z-VAD-FMK blocked sympathetic neuronal death triggered by nerve growth factor removal, but the volume of surviving cells fell to about half normal (15). Maeno et al. also cite a demonstration that Z-VAD-FMK failed to block AVD in lymphoma cells exposed to a Ca2+ ionophore or thapsigargin (16), but because ensuing cell death itself was largely caspase-independent, implications for apoptotic event sequencing may be limited.
In contrast with the findings of Maeno et al., other studies have reported that Z-VAD-FMK blocked both AVD and cell death, for example in ML-1 cells exposed to etoposide (17), B lymphocytes exposed to transforming growth factor β (18), or thymocytes exposed to glucocorticoids (19). How can these observations be reconciled with evidence that AVD is an early event? Although early, AVD is not the first event in the apoptotic cascade. Some biochemical events including reductions in protein synthesis, glucose uptake, mitochondrial transmembrane potential (Δψm), and the activity of certain kinases (mitogen-activated protein and phosphoinositide 3-kinase kinases), as well as increased c-jun expression, typically precede AVD (13, 14, 20). Different pathways and sequences of events may mediate apoptosis in different cell types, or after different insults. Moreover, the caspase(s) mediating AVD may differ from those involved in late apoptotic execution, e.g., caspase-1 for the former and caspase-3 for the latter (18, 21).
The suggestion that K+ channel activation mediates AVD fits with previous work. K+ is the predominant intracellular cation (≈140 mM), so its efflux seems a likely requirement for serious volume reduction (Fig. 1). Linkage between K+ efflux and apoptosis was raised by experiments showing that the K+ ionophore valinomycin could induce apoptosis (22–24) and gained strength with demonstrations that AVD in eosinophils could be attenuated by K+ channel blockers 4-aminopyridine, sparteine, and quinidine (25). We observed that serum deprivation- or staurosporine-induced apoptosis of cortical neurons was associated with an early enhancement of the delayed rectifier current (IK), leading to net loss of cellular K+ and cell shrinkage; attenuating this outward K+ current and AVD by adding a K+ channel blocker or elevating extracellular K+ inhibited apoptosis, even if secondary increases in intracellular Ca2+ concentration were prevented (26, 27). Furthermore, Bortner et al. (28) found in lymphoma cells, increasing cellular K+ loss (by triggering regulatory volume decrease) enhanced apoptosis, and that reducing cellular K+ loss (by elevating extracellular K+) inhibited apoptosis. Other studies have implicated N-methyl-d-aspartate receptor-mediated K+ efflux in neuronal apoptosis (29) and demonstrated that reducing K+ efflux with blockers or elevated extracellular K+ attenuated apoptosis in cholinergic septal SN56 cells, thymocytes, or hepatoma cells (30–32). The idea that K+ efflux promotes apoptosis is clouded by reports that the K+ channel blockers 4-aminopyridine or clofilium can be intrinsically cytotoxic (33, 34), although clofilium reduces ceramide-induced apoptosis (35). These toxicities may reflect deleterious actions at sites other than K+ channels. In addition to blocking K+ channels, clofilium blocks other cation currents (unpublished data) and induces cellular acid release (36).
Figure 1.
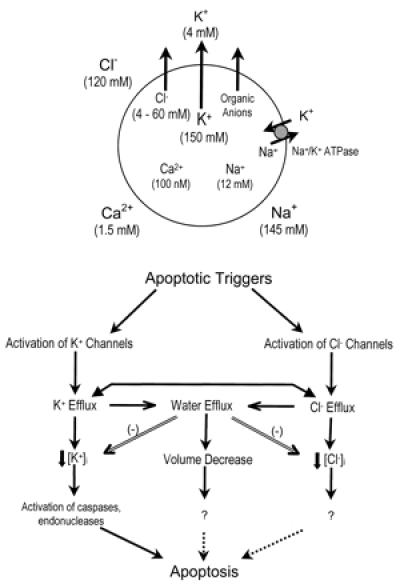
Apoptotic volume decrease. (Upper) K+ efflux, accompanied by the efflux of Cl−, organic anions, and water, is a plausible prerequisite for cellular volume reduction and has been specifically implicated in regulatory volume decrease. The Na+,K+-ATPase and other ion transporters and exchangers likely also participate in cell volume control and ion homeostasis. Typical ion concentrations are depicted; these vary somewhat in different cell types (54–56). (Lower) Speculative model outlining the possible participation of three highly interlinked events in AVD. Double lines depict inhibitory events, and dotted lines depict undefined relationships. Apoptotic triggers may activate K+ or Cl− channels, leading to K+ and Cl− efflux, water efflux, and AVD. If K+ or Cl− efflux to greater extent than water efflux, [K+]i or [Cl−]i will fall. Decreases in [K+]i, [Cl−]i, or volume each may independently promote apoptosis; alternatively one event may critically promote apoptosis, and the effects of other events may be primarily mediated through that critical event. For example, Cl− efflux might promote apoptosis primarily by facilitating K+ efflux and hence reduction in [K+]i, permitting the K+-inhibited activation of caspases and endonucleases.
Besides facilitating AVD through osmolyte reduction, the efflux of intracellular K+ may specifically promote two key apoptotic events, the activation of caspases and endonucleases (37–40). Several enzymes and cellular processes, including cell growth, protein synthesis, and mitosis, are sensitive to the concentration of intracellular free K+ ([K+]i) (41, 42), and hence might be affected if K+ efflux occurred to a greater extent than water efflux. In murine peritoneal macrophages, K+ efflux appeared necessary for posttranslational maturation of IL-1β (later named caspase-1) (37), likely reflecting relief of the ability of physiological (>100 mM) concentrations of [K+]i to inhibit the enzyme responsible for its proteolytic activation (39). In a similar fashion, intracellular K+ may inhibit the proteolytic activation of pro-caspase-3 (Ki for K+ about 40 mM in cell-free extracts) and endonucleases (Ki for K+ about 65 mM) (39). Isolated nuclei exposed to supernatants of mitochondrial that have undergone permeability transition exhibited DNA fragmentation if the solution [K+] was reduced below physiological levels (40).
Although the specificity of fluorescent dye tools currently available for measuring [K+]i is limited, several studies have suggested that [K+]i falls in cells undergoing apoptosis. Barbiero et al. (43) estimated that [K+]i dropped below 50 mM in apoptotic mouse L cells; Hughes et al. (39) reached a similar estimate of 56 mM in apoptotic thymocytes. Also examining thymocytes, Dallaporta et al. (40) found that the drop in [K+]i occurred after loss of Δψm and exhibited sensitivity to Z-VAD-FMK (characteristics of AVD, see discussion above).
The suggestion by Maeno et al. (7) that Cl− channel activation also participates in AVD and apoptosis is consistent with three other recent studies. An outwardly rectifying chloride channel was activated in leukemia cells by CD95 receptor stimulation, and resultant apoptosis was inhibited by blockers of this channel (44). In leukemia cells exposed to apoptotic triggers, internucleosomal DNA fragmentation, but not loss of Δψm, was inhibited by reducing Cl− efflux (45). Finally, hepatoma cells exposed to tumor necrosis factor α exhibited increases in both K+ and Cl− currents, and apoptosis was delayed by either K+ or Cl− channel blockers (32). Maeno et al. mention that their preliminary studies using the Cl−-sensitive dye, N-ethoxycarbonylmethyl-6-methoxyquinolinium bromide (MQAE) have suggested decreased [Cl−]i in HeLa cells exposed to staurosporine, so it seems highly worthwhile to test steps in the apoptotic cascade for sensitivity to ambient Cl− concentration. It has been proposed that Cl− channel opening might permit enhanced HCO3− efflux and intracellular acidification, leading to enhanced endonuclease activity (44), although evidence for endonuclease sensitivity to Cl− or H+ concentrations was not seen in cell lysates (45).
Additional studies are needed to define the relationships linking K+ efflux, Cl− efflux, volume loss, and subsequent apoptosis. Each of these first three events may promote apoptosis independently; alternatively, because the three events are themselves highly interrelated, one event's influence on apoptosis might be mediated strictly through another event's actions (Fig. 1). K+ efflux can be expected to induce an accompanying Cl− efflux to maintain electroneutrality, and vice versa; water can be expected to follow KCl efflux. Thus either K+ or Cl− channel blockers will likely attenuate the efflux of both ions (46). As outlined above, reduction in [K+]i below normal physiological levels has been specifically proposed to facilitate the activation of caspases and endonucleases involved in apoptosis, so an extreme, albeit parsimonious, perspective, alternative to that proposed by Maeno et al., might be that: (i) [K+]i directly modulates the apoptotic cascade; (ii) Cl− efflux promotes downstream apoptotic events only by facilitating K+ efflux; and (iii) volume loss passively reflects K+ (and Cl−) efflux, without independent proapoptotic effects (Fig. 1).
This is not to argue against the idea that Cl− channels may be activated early during apoptosis, or that this activation facilitates at least AVD. And, as Maeno et al. note, there is evidence that cell volume loss per se might promote the execution of downstream apoptotic events. Exposure to hyperosmolar solutions can trigger apoptosis (47, 48), especially in cell types that do not recover normal volume (lack a regulatory volume increase) (49). Although the alternative possibility that hyperosmolar solutions trigger apoptosis nonspecifically by inducing cellular injury is difficult to exclude, it is easy to envisage mechanisms by which cell volume loss could be identified by intracellular sensors and transduced into proapoptotic signals, such as an increased concentrations of intracellular signals such as cytochrome c, alterations in the behavior of membrane channels (50), or the activation of key proapoptotic kinases such as p38 mitogen-activated protein kinase (51, 52).
Regardless of how specifically K+ efflux, Cl− efflux, and volume loss might be linked to the apoptotic cascade, the suggestion by Maeno et al. that apoptosis co-opts normal volume regulatory systems to achieve these events is attractive and heuristically useful. This co-opting may not always occur, as alterations in regulatory volume homeostasis were not observed in CEM-C7A cells treated with dexamethasone (53), but the importance of apoptosis in biology justifies vigorous exploration of the idea.
Acknowledgments
This work was supported in part by National Institutes of Health Grant NS30337 and an unrestricted grant from Bristol-Myers Squibb to D.W.C.
Footnotes
See companion article on page 9487.
References
- 1.Raff M C, Barres B A, Burne J F, Coles H S, Ishizaki Y, Jacobson M D. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 2.Thompson C B. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 3.Choi D W. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- 4.Kerr J F R, Wyllie A H, Currie A R. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H, Yuan J. Curr Opin Cell Biol. 1999;11:261–266. doi: 10.1016/s0955-0674(99)80035-0. [DOI] [PubMed] [Google Scholar]
- 6.Bortner C D, Cidlowski J A. Biochem Pharmacol. 1998;56:1549–1559. doi: 10.1016/s0006-2952(98)00225-1. [DOI] [PubMed] [Google Scholar]
- 7.Maeno E, Ishizaki Y, Kanaseki T, Hazama A, Okada Y. Proc Natl Acd Sci USA. 2000;97:9487–9492. doi: 10.1073/pnas.140216197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klassen N V, Walker P R, Ross C K, Cygler J, Lach B. Int J Radiat Biol. 1993;64:571–581. doi: 10.1080/09553009314551791. [DOI] [PubMed] [Google Scholar]
- 9.Chang S H, Phelps P C, Berezesky I K, Ebersberger M L, Jr, Trump B F. Am J Pathol. 2000;156:637–649. doi: 10.1016/S0002-9440(10)64768-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wesselborg S, Kabelitz D. Cell Immunol. 1993;148:234–241. doi: 10.1006/cimm.1993.1106. [DOI] [PubMed] [Google Scholar]
- 11.Bortner C D, Hughes F M, Jr, Cidlowski J A. J Biol Chem. 1997;272:32436–32442. doi: 10.1074/jbc.272.51.32436. [DOI] [PubMed] [Google Scholar]
- 12.Nardi N, Avidan G, Daily D, Zilkha-Falb R, Barzilai A. J Neurochem. 1997;68:750–759. doi: 10.1046/j.1471-4159.1997.68020750.x. [DOI] [PubMed] [Google Scholar]
- 13.Deckwerth T L, Johnson E M., Jr J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deshmukn M, Johnson E M., Jr Mol Pharmacol. 1997;51:897–906. doi: 10.1124/mol.51.6.897. [DOI] [PubMed] [Google Scholar]
- 15.Deshmukh M, Vasilakos J, Deckwerth T L, Lampe P A, Shivers B D, Johnson E M., Jr J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bortner C D, Cidlowski J A. J Biol Chem. 1999;274:21953–21962. doi: 10.1074/jbc.274.31.21953. [DOI] [PubMed] [Google Scholar]
- 17.Wolf C M, Reynolds J E, Morana S J, Eastman A. Exp Cell Res. 1997;230:22–27. doi: 10.1006/excr.1996.3401. [DOI] [PubMed] [Google Scholar]
- 18.Schrantz N, Blanchard D A, Auffredou M T, Sharma S, Leca G, Vazquez A. Oncogene. 1999;18:3511–3519. doi: 10.1038/sj.onc.1202718. [DOI] [PubMed] [Google Scholar]
- 19.Hughes F M, Jr, Cidlowski J A. J Steroid Biochem Mol Biol. 1998;65:207–217. doi: 10.1016/s0960-0760(97)00188-x. [DOI] [PubMed] [Google Scholar]
- 20.Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin S A, Petit P X, Mignotte B, Kroemer G. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhivotovsky B, Gahm A, Ankarcrona M, Nicotera P, Orrenius S. Exp Cell Res. 1995;221:404–412. doi: 10.1006/excr.1995.1391. [DOI] [PubMed] [Google Scholar]
- 22.Allbritton N L, Verret C R, Wolley R C, Eisen H N. J Exp Med. 1988;167:514–527. doi: 10.1084/jem.167.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ojcius D M, Zychlinsky A, Zheng L M, Young J D. Exp Cell Res. 1991;197:43–49. doi: 10.1016/0014-4827(91)90477-c. [DOI] [PubMed] [Google Scholar]
- 24.Inai Y, Yabuki M, Kanno T, Akiyama J, Yasuda T, Utsumi K. Cell Struct Funct. 1997;22:555–563. doi: 10.1247/csf.22.555. [DOI] [PubMed] [Google Scholar]
- 25.Beauvais F, Michel L, Dubertret L. J Leukocyte Biol. 1995;57:851–855. doi: 10.1002/jlb.57.6.851. [DOI] [PubMed] [Google Scholar]
- 26.Yu S P, Yeh C-H, Sensi S L, Gwag B J, Canzoniero L M T, Farhangrazi Z S, Ying H S, Tian M, Dugan L L, Choi D W. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- 27.Wang, X., Xiao, A. Y., Ichinose, T. & Yu, S. P. (2000) J. Pharmacol. Exp. Ther., in press. [PubMed]
- 28.Bortner C D, Hughes F M, Jr, Cidlowski J A. J Biol Chem. 1997;272:32436–32442. doi: 10.1074/jbc.272.51.32436. [DOI] [PubMed] [Google Scholar]
- 29.Yu S P, Yeh C-H, Strasser U, Tian M, Choi D W. Science. 1999;284:336–339. doi: 10.1126/science.284.5412.336. [DOI] [PubMed] [Google Scholar]
- 30.Colom L V, Diaz M E, Beers D R, Neely A, Xie W J, Appel S H. J Neurochem. 1997;70:1925–1934. doi: 10.1046/j.1471-4159.1998.70051925.x. [DOI] [PubMed] [Google Scholar]
- 31.Dallaporta B, Marchetti P, de Pablo M A, Maisse C, Duc H T, Metivier D, Zamzami N, Geuskens M, Kroemer G. J Immunol. 1999;162:6534–6542. [PubMed] [Google Scholar]
- 32.Nietsch H H, Roe M W, Fiekers J F, Moore A L, Lidofsky S D. J Biol Chem. 2000;275:20556–20561. doi: 10.1074/jbc.M002535200. [DOI] [PubMed] [Google Scholar]
- 33.Chin L S, Park C C, Zitnay K M, Sinha M, DiPatri A J, Jr, Perillan P, Simard J M. J Neurosci Res. 1997;48:122–127. [PubMed] [Google Scholar]
- 34.Choi B Y, Kim H Y, Lee K H, Cho Y H, Kong G. Cancer Lett. 1999;147:85–93. doi: 10.1016/s0304-3835(99)00280-3. [DOI] [PubMed] [Google Scholar]
- 35.Yu S P, Yeh C-H, Gottron F, Choi D W. J Neurochem. 1999;73:933–941. doi: 10.1046/j.1471-4159.1999.0730933.x. [DOI] [PubMed] [Google Scholar]
- 36.Rabinowitz J D, Rigler P, Carswell-Crumpton C, Beeson C, McConnell H M. Life Sci. 1997;61:87–94. doi: 10.1016/s0024-3205(97)00543-2. [DOI] [PubMed] [Google Scholar]
- 37.Perregaux D, Gabel C A. J Biol Chem. 1994;269:15195–15203. [PubMed] [Google Scholar]
- 38.Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. EMBO J. 1995;14:1607–1614. doi: 10.1002/j.1460-2075.1995.tb07149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hughes F M, Jr, Bortner C D, Purdy G D, Cidlowski J A. J Biol Chem. 1997;272:30567–30576. doi: 10.1074/jbc.272.48.30567. [DOI] [PubMed] [Google Scholar]
- 40.Dallaporta B, Hirsch T, Susin S A, Zamzami N, Larochette N, Brenner C, Marzo I, Kroemer G. J Immunol. 1998;160:5605–5615. [PubMed] [Google Scholar]
- 41.Ledbetter M L, Lubin M. Exp Cell Res. 1977;105:223–236. doi: 10.1016/0014-4827(77)90120-3. [DOI] [PubMed] [Google Scholar]
- 42.Albano J, Bhoola K D, Kingsley G. J Physiol. 1977;267:35P–36P. [PubMed] [Google Scholar]
- 43.Barbiero G, Duranti F, Bonelli G, Amenta J S, Baccino F M. Exp Cell Res. 1995;217:410–418. doi: 10.1006/excr.1995.1104. [DOI] [PubMed] [Google Scholar]
- 44.Szabo I, Lepple-Wienhues A, Kaba K N, Zoratti M, Gulbins E, Lang F. Proc Natl Acad Sci USA. 1998;95:6169–6174. doi: 10.1073/pnas.95.11.6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rasola A, Farahi Far D, Hofman P, Rossi B. FASEB J. 1999;13:1711–1723. doi: 10.1096/fasebj.13.13.1711. [DOI] [PubMed] [Google Scholar]
- 46.Nilius B, Sehrer J, De Smet P, Van Driessche W, Droogmans G. J Physiol. 1995;487:367–378. doi: 10.1113/jphysiol.1995.sp020886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matthews C C, Feldman E L. J Cell Physiol. 1996;166:323–331. doi: 10.1002/(SICI)1097-4652(199602)166:2<323::AID-JCP10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 48.Rosette C, Karin M. Science. 1996;274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- 49.Bortner C D, Cidlowski J A. Am J Physiol. 1996;271:C950–C961. doi: 10.1152/ajpcell.1996.271.3.C950. [DOI] [PubMed] [Google Scholar]
- 50.Koch J, Korbmacher C. J Membr Biol. 1999;168:131–139. doi: 10.1007/s002329900503. [DOI] [PubMed] [Google Scholar]
- 51.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 52.Roger F, Martin P Y, Rousselot M, Favre H, Feraille E. J Biol Chem. 1999;274:34103–34110. doi: 10.1074/jbc.274.48.34103. [DOI] [PubMed] [Google Scholar]
- 53.Benson R S, Heer S, Dive C, Watson A J. Am J Physiol. 1996;270:C1190–C1203. doi: 10.1152/ajpcell.1996.270.4.C1190. [DOI] [PubMed] [Google Scholar]
- 54.Inoue M, Hara M, Zeng X T, Hirose T, Ohnishi S, Yasukura T, Uriu T, Omori K, Minato A, Inagaki C. Neurosci Lett. 1991;134:75–78. doi: 10.1016/0304-3940(91)90512-r. [DOI] [PubMed] [Google Scholar]
- 55.Hill B, editor. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer; 1992. p. 15. [Google Scholar]
- 56.Garcia L, Rigoulet M, Georgescauld D, Dufy B, Sartor P. FEBS Lett. 1997;400:113–118. doi: 10.1016/s0014-5793(96)01365-8. [DOI] [PubMed] [Google Scholar]