

Abstract
Activation of the p53 signaling pathway by DNA-damaging agents was originally proposed to result either in cell cycle checkpoint activation to promote survival or in apoptotic cell death. This model provided the impetus for numerous studies focusing on the development of p53-based cancer therapies. According to recent evidence, however, most p53 wild-type human cell types respond to ionizing radiation by undergoing stress-induced premature senescence (SIPS) and not apoptosis. SIPS is a sustained growth-arrested state in which cells remain viable and secrete factors that may promote cancer growth and progression. The p21WAF1 (hereafter p21) protein has emerged as a key player in the p53 pathway. In addition to its well-studied role in cell cycle checkpoints, p21 regulates p53 and its upstream kinase (ATM), controls gene expression, suppresses apoptosis, and induces SIPS. Herein, we review these and related findings with human solid tumor-derived cell lines, report new data demonstrating dynamic behaviors of p53 and p21 in the DNA damage response, and examine the gain-of-function properties of cancer-associated p53 mutations. We point out obstacles in cancer-therapeutic strategies that are aimed at reactivating the wild-type p53 function and highlight some alternative approaches that target the apoptotic threshold in cancer cells with differing p53 status.
1. Introduction
Extensive research has been directed towards targeting the p53 tumor suppressor and other key players in the DNA damage surveillance network in an attempt to improve the outcome of conventional cancer therapies [1]. This approach has met with limited success [2]. The impetus behind most of these studies has been the model, proposed in the mid 1990s, suggesting that the principal role of the p53 pathway in determining cell fate following genotoxic stress is to either promote survival by activating cell cycle checkpoints and facilitating repair or to induce apoptotic cell death [3, 4]. However, several laboratories [5–13], ours included [14–18], have demonstrated that the primary response triggered by moderate doses of DNA-damaging agents in most human cell types is a sustained proliferation block, and not apoptosis. The proliferation block triggered by ionizing radiation predominantly reflects stress-induced premature senescence (SIPS) in p53-proficient cultures [6, 11], and the development of multinucleated and polyploid giant cells in p53-deficient cultures [5, 8]. Accumulating evidence has revealed that such responses may represent cell survival mechanisms consequent to therapeutic exposures.
Below, we will first review the current knowledge on the p53 signaling pathway that is activated in human solid tumor-derived cell lines after exposure to ionizing radiation, with a particular focus on the multiple functions of p21WAF1 (hereafter p21), a key downstream effector of p53. Next, we will discuss the properties of mutant p53 commonly found in human cancers and will examine some promising pharmacological approaches for potentiating the radiosensitivity of cancer cells with differing p53 status.
2. Wild-type p53 Signaling
The human p53 protein is a 393 amino acid transcription regulator consisting of five structural and functional domains [19, 20] (also see Figure 1). The N-terminal acidic transcriptional transactivation domain is required for activating p53-inducible genes. The central DNA-binding domain facilitates sequence-specific binding of p53 to p53-response elements in DNA. The tetramerization domain facilitates the interaction of p53 monomers to form dimers, and the interactions of dimers to form tetramers. Tetramerization is essential for the ability of p53 to positively regulate gene expression. These three domains contribute to transcriptional activation of a large number of p53 targets, including the multifunctional protein p21 (see below) and the proapoptotic proteins BAX, PUMA, and NOXA.
Figure 1.
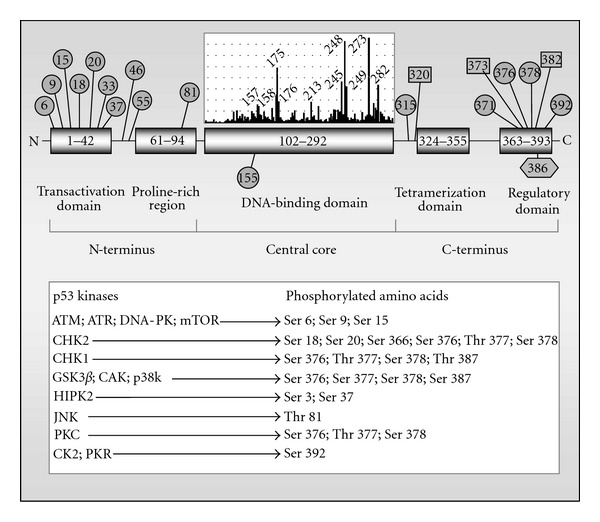
Structure and posttranscriptional modification of the human p53 protein. Circles, Ser/Thr phosphorylation sites; squares, acetylation sites; hexagon, SUMOylation site. The insert vertical bars above the DNA-binding domain illustrate the distribution and the prevalence of point mutations found in p53 in human cancers [19, 20]. The most frequently mutated codons (“hotspot” codons) are identified. Several proteins that phosphorylate p53 and the target amino acids of p53 are also shown in the box [21]. ATM, ataxia-telangiectasia mutated; ATR, ataxia-telangiectasia and RAD3 related; DNA-PK, DNA-dependent protein kinase; mTOR; mammalian target of rapamycin; CHK1 and 2, checkpoint kinases 1 and 2; p38k, p38 kinase; GSK3β, glycogen synthase kinase-3 beta; HIPK2, homeodomain interacting protein kinase 2; JNK, c-Jun-NH(2)-terminal kinase; PKC, protein kinase C; CK2, casein kinase 2; PKR, protein kinase R.
In addition to inducing gene expression, p53 negatively regulates the transcription of a large number of genes, including those that encode BCL-2 [22], MCL-1 [23], and survivin [24], all of which suppress apoptosis, and MDR-1 [25–27] that confers the multidrug resistance phenotype. This property of p53 is associated with the proline-rich domain which is located between the DNA binding and transactivation regions. Repression by p53 occurs indirectly through the transcriptional activation of a repressor protein(s) [27].
In normal unstressed cells, the p53 protein is maintained at low steady-state levels that restrict its impact on cell fate [28]. Turnover of p53 is predominantly regulated by MDM2-mediated ubiquitination and degradation. Genotoxic stress triggers a series of posttranslational modifications on p53 (Figure 1) that contribute to its stabilization, nuclear accumulation and biochemical activation [29, 30]. The rapid activation of p53 by ionizing radiation is mediated by the kinase activity of the ataxia-telangiectasia-mutated (ATM) protein. Upon activation, p53 and its downstream effectors (e.g., p21) regulate different responses, including cell cycle checkpoints, apoptosis, and SIPS.
An assessment of p53 in terms of its regulation, function, and transcriptional targets in response to different stimuli is beyond the scope of the current paper, and the reader is referred to previous reviews (e.g., [20, 31, 32]). In what follows, we discuss the multiple functions of p21 (Figure 2) in the p53 signaling network that is activated by ionizing radiation, a key component in cancer therapy.
Figure 2.
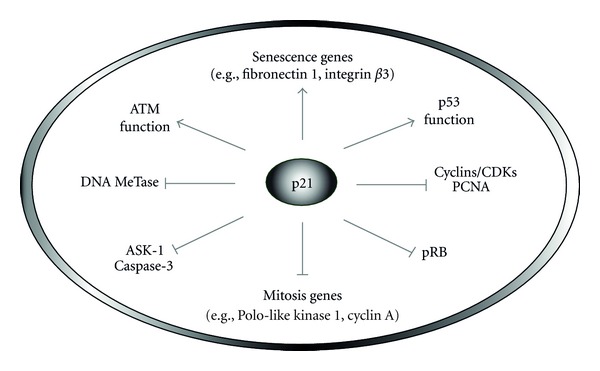
Multiple functions of the p21 tumor suppressor. This protein plays a crucial role in the p53 signaling pathway owing to its ability to inhibit cell cycle progression (e.g., by interacting with PCNA and cyclin/CDK complexes), influence gene expression indirectly, through interfering with DNA methyltransferase (MeTase) activity, and directly, by controlling the transcription of genes involved in senescence and mitosis, down-regulating apoptosis (e.g., through interaction with ASK-1), and positively regulating ATM and p53.
2.1. p21 and Cell Cycle
The p21 protein is classified as a member of the CIP/KIP family of the cyclin-dependent kinase (CDK) inhibitors and is considered to be a universal inhibitor of cell cycle progression [35, 36]. p21 exerts its effect on the cell cycle not only by inhibiting cyclin/CDK complexes, but also by inhibiting proliferating cell nuclear antigen (PCNA), degrading retinoblastoma protein (pRB), and inhibiting cyclin B1. The N-terminal region of p21 binds to CDKs and inhibits their ability to phosphorylate pRB, an event that is required for the release of the E2F factor from pRB/E2F complexes and progression of cells from G1 to S phase [37]. p21 also inactivates pRB through proteasome-mediated degradation, independent of its effect on cyclin/CDK complexes [38]. The C-terminal region of p21 suppresses DNA synthesis by interacting with PCNA, an auxiliary factor for DNA polymerase δ [37]. Collectively, p21-mediated suppression of cyclin/CDK functions, degradation of pRB, and inhibition of PCNA result in activation of G1/S and S-phase checkpoints. In addition, p21 contributes to the G2/M checkpoint in response to DNA damage [39, 40] and this effect has been attributed to p21-mediated degradation of the G2-associated cyclin, cyclin B1 [40].
2.2. p21 and Gene Expression
In 1997, Chuang and associates demonstrated that p21 negatively regulates DNA-methyltransferase activity [41]. This finding raised the possibility that p21 might indirectly influence the expression of the many human genes that contain CpG islands within their transcription start sites [14, 42]. In 1998, we reported studies with non-cancerous human fibroblasts that supported the model in which DNA damage-induced sustained growth arrest (now commonly called SIPS) is a delayed response, predominantly reflecting p21-mediated regulation of gene expression rather than activation of early cell cycle checkpoints [14]. More recently, p21 was established to actively participate in the regulation of genes involved in growth arrest, senescence, and aging [43–46]. Roninson and coworkers showed that p21 positively regulates the expression of a battery of genes associated with senescence, and suppresses the transcription of numerous genes that control mitosis [6, 43].
More recently, Löhr et al. [27] made a striking discovery when testing their hypothesis that p53-mediated repression of genes might involve a repressor. The negative regulation of all p53-repressed genes that were analyzed by these authors proved to be entirely dependent on the presence of p21, and overexpression of p21 was shown to repress similar sets of genes. Thus, p53-mediated gene repression occurs mainly (if not solely) through p53-mediated transcriptional activation of p21.
2.3. p21 and Senescence
The p21 protein was identified in three different laboratories in the early 1990's, and was variously called CIP1 for CDK-interacting protein 1 [35], WAF1 for wild-type p53-activated fragment 1 [47], and SDI1 for senescent cell-derived inhibitor 1 [48]. Its pivotal role in regulating different types of senescence, including replicative senescence (seen in aging human fibroblasts) and SIPS, is universally accepted. Prominent biomarkers of senescence include the acquisition of enlarged and flattened cell morphology, presence of β-galactosidase activity at sub-optimal conditions (i.e., pH 6), and absence of cell division in metabolically-active cells. Replicative senescence results primarily from impaired telomerase activity and shortened telomeres [49], whereas SIPS is triggered by nonspecific genome-wide DNA damage, independent of telomerase function and telomere status [50, 51].
Most authors attribute the DNA damage-induced SIPS response to p21-mediated control of the G1/S checkpoint. Specifically, it has been proposed that p21 might switch on the SIPS program mainly through activating this checkpoint, whereas another G1 checkpoint activator, the CDK inhibitor p16Ink4a (hereafter p16), might serve to maintain the growth-arrested state associated with SIPS. Several reports, however, did not support a role for p16 in SIPS (e.g., [52, 53]). Our recent studies with human normal, ataxia telangiectasia (AT) and Li-Fraumeni syndrome fibroblasts led us to propose the model in which p16 serves as a backup regulator of SIPS, triggering this response only in the absence of wild-type p53 function [18]. In addition, ATM-deficient (AT) fibroblasts, which are known to fail to activate early cell cycle checkpoints in response to ionizing radiation, exhibited hypersensitivity to undergo SIPS after irradiation when compared to normal fibroblasts. AT fibroblasts undergoing SIPS did not express p16, but exhibit nuclear accumulation of p21, which was observed several days (but not ≤24 h) after irradiation. These results clearly dissociate early cell cycle checkpoints from SIPS, at least in the fibroblast background, and are consistent with the premise that p21-mediated SIPS may largely reflect its ability to regulate gene expression.
According to Crescenzi et al. [54], p53 wild-type cancer cell lines (e.g., A549, MCF7, and HCT116) that do not express p16 exhibit a high degree of SIPS in response to DNA damage. SIPS cancer cells that were maintained in culture for several months showed neither proliferation nor loss of viability and were mainly arrested with a G2-M DNA content. The apoptosis resistance of such cells was shown to be a direct consequence of the sustained upregulation of p21.
2.4. p21 and Apoptosis
It is becoming appreciated that under certain conditions p21 functions as a potent antiapoptotic factor, acting at different levels of the death cascade. Phosphorylation of p21 at the Thr 145 residue in some cell types triggers the exit of p21 from the nucleus and its accumulation in the cytoplasm, where it interacts with apoptosis signal-regulating kinase 1 (ASK-1), downregulates the stress-induced mitogen-activated protein kinase (MAPK) cascade, and results in resistance of cells to apoptosis induced by different stimuli [55–57]. Thr 146 phosphorylation of p21 and its cytoplasmic retention occurs in Her2/neu-expressing cancers and is mediated by the phosphatidylinositol-3 kinase (PI3K)/AKT pathway [55]. In addition, p21 has been reported to downregulate apoptosis by forming a complex with procaspase-3 and suppressing its activation by masking the protease cleavage site [58], inhibiting activation of caspase-9 [59], and inhibiting cytochrome c release from mitochondria [60]. Recent studies with p53 wild-type A549 human lung carcinoma cells treated with the histone deacetylase inhibitor trichostatin A [61] or the CDK inhibitor roscovitine [62] have provided new insight into the relationship between p21 and caspase-3 in the regulation of apoptosis. Treatment of A549 cells with high concentrations of each of these chemicals triggered apoptosis, and this response was accompanied by caspase-3-dependent cleavage of p21. Surprisingly, a 15-kDa fragment of p21 was essential in caspase-3-mediated apoptosis. In short, it appears that intact p21 protein suppresses apoptosis through inhibition of caspase-3 activity, whereas a 15-kDa cleavage product of p21 facilitates caspase-3-directed apoptosis.
As alluded to earlier, the negative regulation of transcription by p53 is mediated in part by p21. This observation is puzzling because p53-repressed proteins (e.g., BCL-2, MCL-1, survivin) are anti-apoptotic, and the factor (p21) that mediates this repression is also intrinsically anti-apoptotic. The aforementioned discoveries with trichostatin A and roscovitine raise the intriguing possibility that the 15-kDa cleavage product of p21 might mediate the p53-directed repression rather than the full-length p21 protein. Further studies are warranted to test this model.
2.5. p21, p53, and ATM
In response to DNA damage, the p21 protein regulates the abundance, subcellular localization, and transcriptional function of p53. Loss of p21 in p53 wild-type human colon carcinoma cells is associated with increased p53 protein levels without exposure to genotoxic agents [63]. In addition, p21 deficiency results in elevated expression of p14ARF (hereafter ARF) which is known to stabilize p53 by promoting MDM2 degradation [64, 65]. These findings led Javelaud and Besançon [63] to propose that increased ARF levels in p21-null cells may increase the steady-state levels of p53 by interrupting the MDM2-p53 feedback loop.
Although p21 negatively regulates p53 protein abundance, recent work of Pang et al. [66] paradoxically demonstrated the requirement of p21 in the maintenance of the p53 transcriptional program. In human colon carcinoma cells exposed to ionizing radiation, knocking out the p21 gene resulted in a p53 protein shift from chromatin into the cytoplasm and attenuation of the specific activity of p53 as a transcription factor. Complementation of p21 knockout cells with the p21 gene restored nuclear localization of p53 and its transcriptional activity after irradiation. Moreover, ATM inhibition in p21 wild-type cells also resulted in nuclear exclusion of p53, suggesting a positive interaction between ATM and p21. The authors concluded that the p53 transcription program has evolved a negative and a positive feedback loop, the balance of which controls the transcriptional function of p53 in response to DNA damage. The well-known negative loop is maintained by MDM2, whereas the novel positive loop is maintained by the ATM-p21 axis.
3. Sequential Waves of p53 and p21 in the DNA-Damage Response
Zhang et al. [67, 68] constructed an integrative computational model of the p53 network for ionizing radiation which was composed of four modules: DNA repair, ATM sensor, p53-centered feedback control, and cell fate decision. The cell fate decision module was limited to G1 arrest and apoptosis. The model predicted multiple waves for p53 and p21 after irradiation and two forms of p53: the “arrester” p53 that was phosphorylated at Ser 15 and mediated early checkpoints through p21, and the “killer” p53 that was additionally phosphorylated at Ser 46 and mediated apoptosis through caspase-3. Under normal conditions (no gene manipulations), the level of p53 killer (Ser 46-phosphorylated) was ~6 times higher than that of the p53 arrester (Ser 15-phosphorylated), and the former was fully manifested at around 24 h after exposure to 7.5 Gy of ionizing radiation and was predicted to induce 100% apoptosis [68]. This model remains to be experimentally tested in different biological systems.
Unfortunately, the model did not incorporate SIPS which is a major response triggered by ionizing radiation in most p53-proficient cell types. With respect to p21, induction of apoptosis after irradiation was predicated to be invariably associated with a decline of p21 to the basal levels. This prediction is consistent with the well-known anti-apoptotic property of p21 (see above). As discussed below, some other key aspects of the model are not supported experimentally for human solid tumor-derived cell lines and noncancerous fibroblast strains.
3.1. Sequential Waves of p53 and p21 in Cancer Cells
Batchelor et al. [69] reported the dynamics of the global p53 protein levels in MCF7 breast cancer cells (p53 wild-type) consequent to ionizing radiation exposure (10 Gy). Western blot analysis revealed two p53 peaks over a period of 10 h after irradiation, the first at ~2 h and the second at ~8 h. The authors suggested a model in which the initial p53 pulse would allow the cells to activate repair/checkpoints and the late pulse to trigger apoptosis. Unfortunately, no repair/checkpoint/apoptosis data were reported to support the model. These measurements would be particularly critical for MCF7 cells because these cells lack caspase-3 [70] which is a key mediator of radiation-induced apoptosis [68, 71]. These authors also did not incorporate SIPS in their model. Furthermore, the influence of radiation exposure on p21 dynamics was not reported.
Radiation-induced SIPS is a late response, being fully manifested several days after irradiation [72]. In our studies with several p53 wild-type human cancer cell lines (e.g., A172 malignant glioma), senescence-associated β-galactosidase staining (a marker of SIPS) was observed at 3 days and beyond after irradiation (e.g., 5 Gy), but not at earlier times (unpublished data). These observations, together with the “digital” response of p53 to DNA damage and the role of p21 in regulating p53, prompted us to determine the relationship between p53 and p21 protein levels at early (2–12 h) and late times (1–7 days) after exposure to ionizing radiation. We used the A172 cell line and a 5 Gy dose of γ radiation. This dose causes ~95% “cell kill” in the clonogenic assay which largely reflects SIPS [15], but induces only marginal (<5%) apoptosis when evaluated at various times between 1 and 6 days after irradiation (Figure 3). The caspase cascade is functional in A172 cells (e.g., [73]). Representative Western blots and the p53/p21 protein measurements from multiple experiments are presented in Figure 4. Three observations should be noted:
over a period of 12 h after irradiation (5 Gy), there were two peaks for p53, one at ~2 h and one at ~6 h. These data are in general agreement with those reported by Batchelor et al. [69] who used MCF7 cells exposed to 10 Gy of γ radiation. In their experiments, the p53 levels peaked between 2 and 3 h and between 6 and 9 h after irradiation;
in irradiated cultures, p53 levels decreased to nearly background levels after day 1 and remained low on day 2. Three days after irradiation, however, p53 levels increased markedly and remained elevated over the duration of the experiments (7 days);
the p21 response involved two major waves over the time window that we examined. The levels of p21 increased at early times and peaked at ~6 h. The levels subsequently declined at later times and reached the background levels on day 2. This was followed by a second wave of p21 upregulation on day 3, and p21 levels remained high for the duration of the experiments. This late (≥3 days) p21 wave coincides with the onset of SIPS noted above, suggesting a relationship between these responses.
Figure 3.
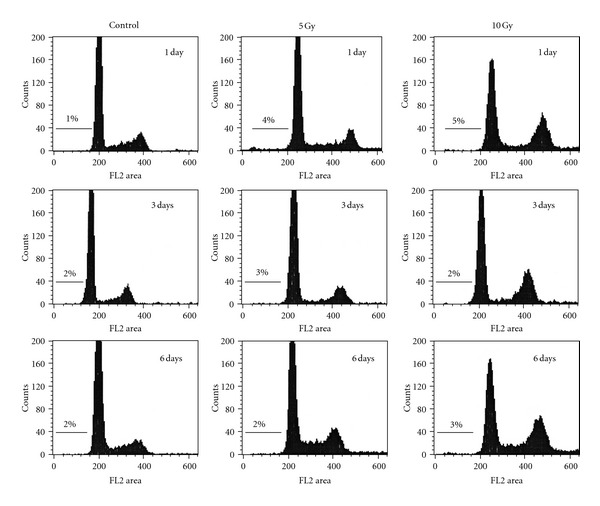
Flow cytometric profiles of A172 cell populations before (control) and after exposure to 5 Gy or 10 Gy doses of γ radiation and incubation for the indicated times. For each sample, the adherent and floating cells were combined and analyzed as described [33]. Relative cell number is plotted as a function of fluorescence intensity. The percentage of cells with sub-G1 (apoptotic) nuclei is indicated above the marked area in each diagram.
Figure 4.
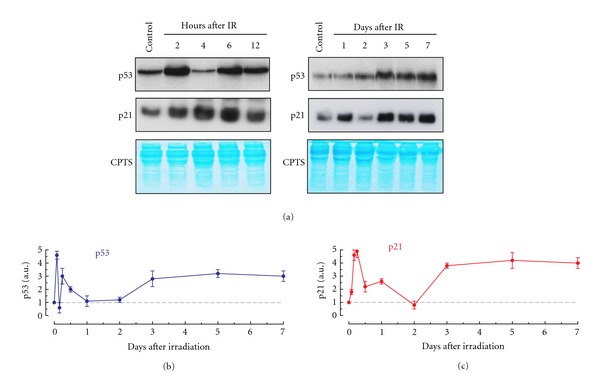
Dynamics of p53 and p21 protein levels in A172 (p53 wild-type) cells after exposure to ionizing radiation (IR; 5 Gy). (a) Representative Western blots showing p53 and p21 levels before (control) and at indicated times post-irradiation. CPTS staining (blue) was used to confirm equal amount of protein loading in each well. (b and c) Relative amounts of p53 and p21 protein levels at indicated times after irradiation. The data are expressed as normalized p21 and p53 values for irradiated samples relative to the values for the corresponding controls (set at 1). Bars, SE. Western blot analysis of p53 and p21 protein levels was performed using our published techniques [15].
Immunofluorescence microscopy generated results consistent with the Western blotting, with radiation exposure triggering three and two waves of p53 and p21, respectively (Figure 5). The former assay also revealed that the p53 and p21 upregulation post-irradiation reflects nuclear accumulation of these proteins (Figure 5(a)).
Figure 5.
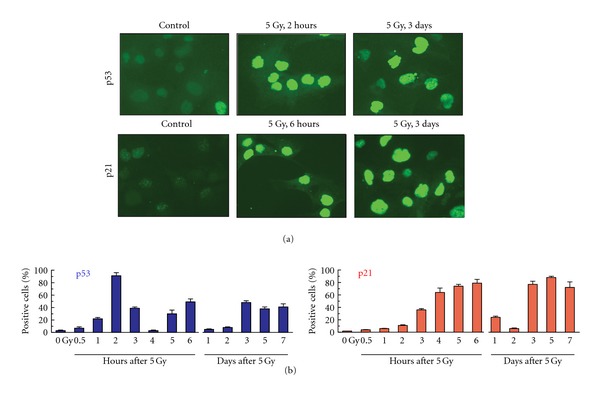
Immunofluorescence analysis of p53 and p21 protein expression in A172 cells after exposure to ionizing radiation (IR; 5 Gy). (a) Representative images showing p53 and p21 protein levels in control (non-irradiated) and irradiated cells. [b] Percentages of cells exhibiting nuclear accumulation of p53 or p21 before (control) and at indicated times post-irradiation. Each datum point represents the mean (±SD) values of duplicate samples. At least 200 cells were evaluated for each sample. Immunofluorescence analysis of p53 and p21 protein levels was performed using our published techniques [18].
These results suggest that p53 in irradiated cultures might be phosphorylated at Ser 15 because this phosphorylation is essential for the nuclear accumulation of the protein, and p21 contributes to this response [66]. Immunofluorescence experiments performed at selected times (2 h and 3 days) after irradiation (5 Gy) confirmed this prediction. Under these conditions, we observed strong nuclear fluorescence staining using a Ser 15-phosphospecific antibody (Figure 6(a)). Similar results were obtained when cultures were exposed to 10 Gy (data not shown). We also used a Ser 46-phosphospecific antibody in these experiments. In response to 5 Gy (Figure 6(b)) or 10 Gy (data not shown) doses of γ radiation, we observed a strong nuclear staining at 2 h after irradiation, but low (background) staining at the late time point (Figure 6(b)). The importance of Ser 46 phosphorylation of p53 at early times after exposure to subapoptotic doses of ionizing radiation remains to be elucidated.
Figure 6.
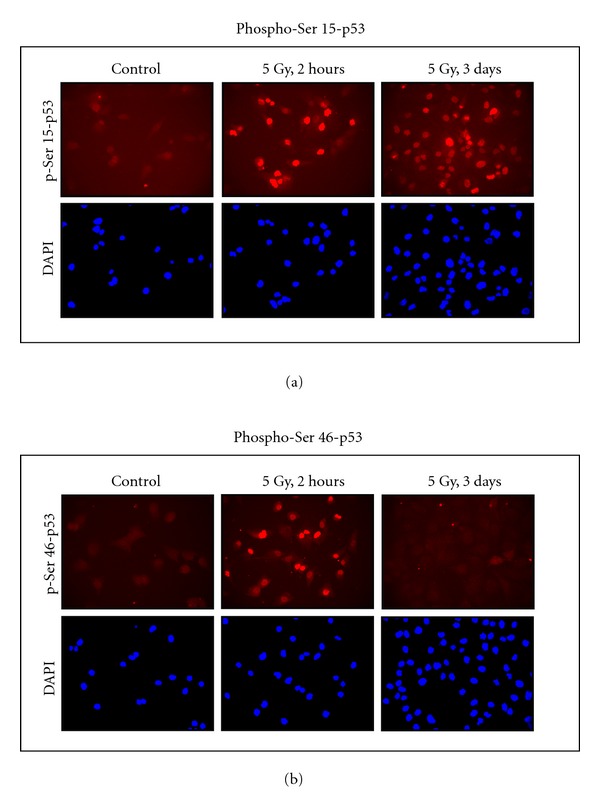
Immunofluorescence analysis of p53 phosphorylation (red) in A172 cells before and at indicated times after exposure to ionizing radiation (5 Gy). These experiments were performed using our published techniques [18], except for the use of phosphospecific antibodies to Ser 15 (a) or Ser 46 (b) of p53 (Cell Signaling, Beverly, MA, USA). Nuclei were counterstained with DAPI (blue).
Figure 7 illustrates the multiple functions of p53 and p21 discussed above, together with their dynamic behaviors shown in Figures 4 and 5, for human solid tumor-derived cells exposed to moderate (sub-apoptotic) doses of ionizing radiation. The early response involves ATM-mediated activation of p53 which accelerates repair and induces p21, which in turn downregulates apoptosis and activates cell cycle checkpoints. At late times (several days) after irradiation, persistent genomic injury in a proportion of cells triggers the delayed wave of ATM-p53-p21 induction, leading to p21-dependent activation of the SIPS program accompanied by suppression of apoptosis. The sustained proliferation block and apoptosis resistance is maintained through positive feedback interactions between ATM and p21, and between p53 and p21.
Figure 7.
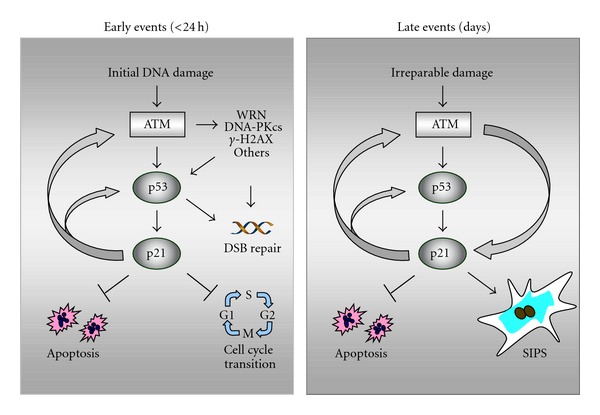
ATM-p53-p21 signaling in p53 wild-type human solid tumor-derived cells exposed to ionizing radiation. (a) Radiation exposure results in ATM-dependent activation of several proteins (e.g., p53, WRN, DNA-PKcs) that play important roles in DSB repair, as well as p53-mediated activation of p21 that suppresses apoptosis and activates cell cycle checkpoints. Proper activation of these early events provides time for the repair of potentially cytotoxic and mutagenic lesions. (b) Persistence of DNA damage (i.e., irreparable damage) at late times (several days) after irradiation leads to sustained induction of p21 which suppresses apoptosis and triggers SIPS. Positive feedback loops between ATM and p53 and between p53 and p21 ensure the maintenance of the SIPS response for extended times (several months in culture). WRN, Werner's syndrome protein; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; γ-H2AX, H2A variant histone H2AX phosphorylated on Ser 139.
That p21 is responsible for the apoptosis resistance of cancer cells undergoing SIPS has also been documented with different cancer cell lines treated with the chemotherapeutic drug doxorubicin [54]. Inhibition of either ATM or p21 after full manifestation of the SIPS response readily induced apoptosis in the absence of additional stimuli [54].
3.2. Sequential Waves of p53 Signaling in Fibroblasts
The above observations with solid tumor-derived cells are compatible with the two-wave model of DNA damage response that we proposed in 1998 for human fibroblasts [14]. According to our model, while the early activation of the p53-p21 axis causes transient activation of cell cycle checkpoints to facilitate repair, the persistence of genomic injury (e.g., chromosome aberrations) provides the critical signal for the late activation of the p53-p21 axis, leading to p21-mediated SIPS or p53-mediated apoptosis, depending on the type and extent of DNA damage. Recently, we have shown that human fibroblast strains expressing wild-type (normal, AT) or mutant (Li-Fraumeni syndrome) p53 lose their clonogenic potential following exposure to ionizing radiation (2–8 Gy) through undergoing SIPS and not apoptosis [18]. The radiation-induced SIPS correlated with expression of p21 but not of p16 in p53-proficient fibroblasts, and with expression of p16 but not of p21 in p53-deficient fibroblasts.
4. Mutant p53 Signaling
Mutation of the p53 gene is one of the most frequent genetic changes in human cancer. Most of the p53 mutations are located within the central DNA-binding domain (residues 102–292) and a few hot spots are found in the conserved regions of the gene (Figure 1). Although loss of p53 expression has been observed in various cancers, the majority of p53 mutations result in expression of mutant p53 that exhibits gain-of-function properties. Such mutations actively promote pro-survival signals and tumorigenesis, independent of the loss of wild-type p53 function [76–78].
Sub-cellular localization of mutant p53 is critical for the biological outcome. Cytoplasmic sequestration of mutant p53 is observed in some cancers and has been associated with its ability to interact with mitochondrial caspase-3, thereby conferring the apoptosis-resistant phenotype [79]. These mutations are not frequent. In fact, we did not observe cytoplasmic sequestration of p53 in any of the p53-mutated breast cancer cell lines commonly used in our laboratory (Table 1). The majority of cancer-specific p53 mutations result in nuclear accumulation of mutant p53 protein that exhibits properties such as long half-life and the ability to transcriptionally activate genes that are not activated by wild-type p53. Of the panel of mutant p53 breast cancer cell lines examined by us, ten showed high levels of nuclear p53 (Table 1), and two (SUM159PT and MDA-MB-436) expressed low levels or no p53.
Table 1.
p53 mutations in commonly used human breast cancer cell lines.
| Cell line | Exon | Codon | Type | Nucleotide change | Residue change |
|---|---|---|---|---|---|
| MDA-MB-231 | 8 | 280 | Missense | AGA → AAA | Arg → Lys |
| CAMA-1 | 8 | 280 | Missense | AGA → ACA | Arg → Thr |
| MDA-MB-468 | 8 | 273 | Missense | CGT → CAT | Arg → His |
| BT-474 | 8 | 285 | Missense | GAG → AAG | Glu → Lys |
| MDA-MB453s | 8 | 266 | Missense | GGA → GAA | Glu → Lys |
| BT-549 | 7 | 249 | Missense | AGG → AGC | Arg → Ser |
| T-47D | 6 | 194 | Missense | CTT → TTT | Leu → Phe |
| Hs578T | 5 | 157 | Missense | GTC → TTC | Val → Phe |
| SK-BR-3 | 5 | 175 | Missense | CGC → CAC | Arg → His |
| BT-20 | 5 | 132 | Missense | AAG → CAG | Lys → Gln |
A large number of genes are induced by gain-of-function p53 mutants [76–78]. These include the epidermal growth factor (EGF) receptor [80], c-Myc [81], Fos [82], and MDR-1 [83]. The finding that MDR-1 transcription is induced by some p53 mutants but is repressed by wild-type p53 is intriguing. The MDR-1 gene encodes P-glycoprotein, an energy-dependent anti-cancer drug efflux pump [84]. Over-expression of MDR-1could result in the selective resistance of cancer cells to drugs such as vinblastine, etoposide, and taxol, which are transported by this membrane pump [85].
The gain-of-function effects are not limited to regulation of gene expression by mutant p53. In a number of in vitro and in vivo models, for example, mutant p53 was shown to augment the promigratory, proinvasive, and prometastatic properties of TGF-β [86]. Furthermore, many gain-of-function effects of mutant p53 rely on its ability to bind and inactivate the p63 and p73 tumor suppressors [77]. In p53-deficient cells, these p53-related proteins play important roles in maintaining genome stability by suppressing aneuploidy and polyploidy [87, 88].
Although the gain-of-function effects are complex [77], the remarkable apoptosis-resistance feature appears to be directly linked to high expression of mutant p53, rather than reflecting some secondary effect such as changes in gene expression patterns associated with genomic instability. The basis for this conclusion is that suppressing mutant p53 by the transient siRNA approach induced death through apoptosis in the MDA-MB-231 breast cancer cell line [89]. Using a similar approach, in a preliminary study we confirmed this observation with MDA-MB-231 cells, and further observed that transient silencing of mutant p53 induced a high degree of apoptosis (>50%) in several breast cancer cell lines that carry missense p53 mutations, including the widely-used cell lines MDA-MB-435s, MDA-MB-468, BT-474 (which express exon 8-mutant p53) and BT-549 (which expresses exon 7-mutant p53) (unpublished observations).
The finding that suppression of at least certain types of mutant p53 renders cancer cells highly susceptible to undergo apoptosis provides the opportunity to design novel cancer therapy strategies, some of which are discussed below.
5. p53-Based Cancer Therapies: Reality or Empty Promises?
Normal cells possess tight regulatory mechanisms that enable constitutive p53 to maintain cellular homeostasis without impeding normal growth and function. These regulatory mechanisms are subverted in cells within most cancers that lack wild-type p53 activity. These observations led to the notion that sudden reactivation of p53 may trigger lethality or permanent growth arrest in p53-deficient cancer cells and might lead to the development of successful anticancer treatments. Accordingly, modulating p53 in the context of cancer therapy has been a very active area of research for many years. Approaches to restoring p53 function included the use of virus-mediated p53 gene replacement, pharmacological agents capable of activating endogenous wild-type p53 (e.g., RITA, Nutlins), and pharmacological agents that reactivate some of the wild-type function of endogenous mutant p53 (e.g., CP31398, MIRA, PRIMA, Ellipticine). Despite intensive research and new drug discoveries, the conventional p53-based therapeutic strategies have met with limited success [2]. Numerous obstacles have been identified, including unwanted side effects in normal tissues and the development of p53-resistant tumors [2].
Furthermore, in view of the current knowledge on p53-mediated responses reviewed above, activation of p53 in some cancer cells would be expected to result in p21-mediated induction of SIPS and down-regulation of apoptosis. Triggering SIPS in some cancers (e.g., sarcomas) has been associated with a positive therapeutic outcome [90, 91]. With most other cancers, however, the long-term consequences of SIPS remain unknown. As mentioned above, cells undergoing SIPS maintain viability and secrete factors that, among other detrimental effects, can promote tumor growth and mediate the transformation process of pre-neoplastic cells [6, 12, 92–95]. In addition, although cells undergoing SIPS do not form macroscopic colonies in clonogenic survival assays, it is becoming increasingly appreciated that under some conditions SIPS might not be a “permanent” growth-arrested state. Thus, a proportion of cells exhibiting features of SIPS can escape the proliferation block and give rise to aneuploid progeny that can re-enter the mitotic cycle [12, 94, 95]. The crosstalk between SIPS and autophagy is considered to influence the capacity of the tumor cells to maintain a prolonged state of growth inhibition that unfortunately can be succeeded by tumor regrowth and disease recurrence [95].
Despite their limitations when used singly, the usefulness of small-molecule modulators of p53 are being explored in other contexts, such as in combination therapy with conventional anti-cancer agents (e.g., doxorubicin) [2]. As pointed out by Desilet et al. [2], whether p53 will fulfill its promise of playing a leading role in cancer therapy remains to be seen. The potential and limitations of p53-based cancer therapies have been extensively reviewed by us [96] and others [2] and will not be considered further. Below, we will outline some alternative pharmacological approaches that modulate the apoptosis-resistance phenotype of cancer cells with differing p53 status.
5.1. Targeting Phospholipase D in Cancer Therapy
Phospholipase D (PLD) promotes tumor progression by generating “survival signals” that suppress default apoptotic programs [97–99]. PLD is known to be responsible for activation of the mammalian target of rapamycin (mTOR) [100–103], stabilization of mutant p53 [89], and regulation of aerobic glycolysis (the “Warburg effect”) [104], all of which suppress apoptosis and promote cancer cell proliferation and migration. Consistent with these properties, elevated PLD activity has been observed in several human cancers including breast [99], and in several human cancer cell lines including those from breast [98, 105, 106]. In view of the emerging paradigm of PLD survival signals in the context of therapeutic intervention, Hui et al. [89] determined the influence of PLD on the mutant p53 in the MDA-MB-231 breast cancer cell line. PLD was shown to mediate the stabilization of mutant p53 in these cells, and targeting either PLD or mutant p53 by the siRNA approach enhanced their sensitivity to undergo apoptosis in response to serum withdrawal. The impact of targeting PLD on the apoptotic response of mutant p53-expressing cancers after exposure to ionizing radiation and chemotherapeutic drugs remains to be, and should be, elucidated.
5.2. Targeting mTOR in Cancer Therapy
mTOR is a key regulator of cellular metabolism and apoptosis [107]. Accordingly, there is a great deal of interest in exploring mTOR as a target for cancer therapy [75, 107–109]. Its expression is regulated by PLD [100] and the PI3K/AKT pathway [75]. Ionizing radiation and other stress stimuli are known to activate mTOR [75]. Targeting mTOR using the small-molecule inhibitor RAD0001 (Everolimus) resulted in marked radiosensitization of MDA-MB-231 breast cancer cells through induction of caspase-3-dependent apoptosis [75].
5.3. Targeting Heat Shock Protein 90 in Cancer Therapy
In cancer cells, the molecular chaperone heat shock protein 90 (HSP90) is used to facilitate the function of numerous oncoproteins, and contributes to resistance of cancers to therapy [110, 111]. HSP90 is known to mediate the maturation of mTOR, facilitating this kinase to form functional complexes with its specific interacting partners [112]. BIIB021, a novel small-molecule inhibitor of HSP90, was recently reported to potentiate the apoptotic response of several human head and neck squamous cell carcinoma (HNSCC) cell lines after exposure to ionizing radiation, and to elicit a remarkable impact on the outcome of radiotherapy of HNSCC xenografts [113].
5.4. Targeting Aurora B in Cancer Therapy
As mentioned earlier, loss of wild-type p53 function is permissive for the development of giant cells with extensive genetic abnormalities (e.g., multinucleation, polyploidy) in response to genotoxic stress [114]. It is becoming apparent that multinucleation/polyploidy may provide a radiation-survival mechanism in p53-deficient cancer cells. Giant cells with such genetic abnormalities may give rise to rapidly proliferating offspring by different mechanisms, including neosis, an ill-defined parasexual somatic reduction division which resembles division of the budding yeast [12, 13, 94], as well as depolyploidization through meiotic or pseudo-meiotic pathways [115, 116] (also see Figure 8). The Aurora B kinase, a key regulator of chromosome segregation and cytokinesis [117], has been identified as the gatekeeper between the life and death of such multinucleated/polyploid giant cells; expression of catalytically-active Aurora-B kinase promoted the survival of giant cells, whereas its absence was associated with giant-cell apoptosis [118]. Moreover, the Aurora B inhibitor AZD1152 markedly potentiated the radiosensitivity of p53-null human colon carcinoma cells when evaluated in vitro and in vivo [119]. The influence of targeting Aurora B and other mitotic kinases (e.g., PLK-1) on the radiosensitivity of mutant p53-expressing cancers is unknown.
Figure 8.
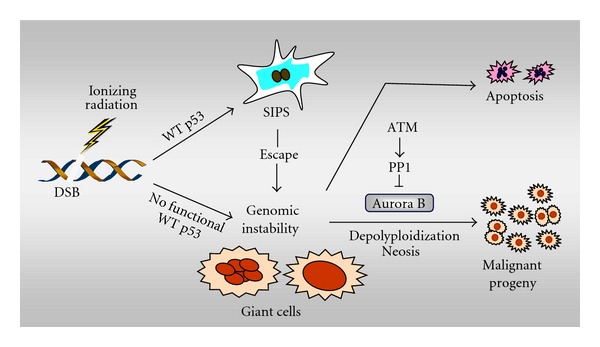
Formation and fate of multinucleated/polyploid giant cancer cells. Ionizing radiation triggers the development of giant cells within cultures of cancer cells lacking wild-type (WT) p53 function. In addition, although p53 wild-type cancer cells respond to ionizing radiation by undergoing SIPS, a small proportion of cells “escape” from the SIPS response and give rise to multinucleated/polyploid giants. While some giant cells may die through apoptosis, others may undergo complex genome-reduction processes (e.g., depolyploidization; neosis), ultimately giving rise to rapidly-proliferating progeny. The mitotic kinase Aurora B plays an important role in regulating the survival of giant cells. Recent evidence suggests that ATM may prevent the propagation of giant cells and their descendants by activating protein phosphatase 1 (PP1) and inhibiting Aurora B kinase activity (for details, consult [34]).
5.5. Targeting Cell-Metabolic Pathways in Cancer Therapy
In normal cells, adenosine triphosphate (ATP), the principal molecule that drives all energy-dependent cellular processes (including synthesis of proteins, nucleic acids and lipids), is mainly generated by two metabolic pathways: oxidative phosphorylation when oxygen is plentiful, and anaerobic glycolysis when oxygen levels are low. Cancer cells, on the other hand, metabolize glucose to generate ATP by glycolysis even when oxygen is abundant (the “Warburg” effect). This and other metabolic differences between normal and cancer cells have led to growing interest in metabolic inhibitors in cancer therapy [120–125]. Promising results have been reported with 2-deoxyglucose (2DG) and the antidiabetic agent metformin [121]. Treatment with these drugs depleted ATP and induced apoptosis in p53 wild-type prostate cancer cells but not in normal prostate cells. Dichloroacetate (DCA), an inhibitor of the enzyme pyruvate dehydrogenase that is a key mediator of the Warburg effect, is also known to promote apoptosis in human cancers [126–128]. Whether targeting the tumor-specific metabolic pathways by small-molecule inhibitors would improve the outcome of radio/chemotherapy of cancers expressing wild-type, mutant, or no p53 remains to be determined.
6. Conclusion
In this article, we have discussed the growing complexity of p53 signaling, pointed out some challenges associated with conventional p53-based cancer therapeutic strategies, and highlighted novel targets that might be exploited for improved therapeutic outcome. These targets include PLD, mTOR, HSP90, Aurora B, and the Warburg effect (Figure 9). Many small-molecule inhibitors of these targets are commercially available and are known to have suitable half-lives and bioavailability for clinical applications (see, e.g., [119, 126–130]). The mTOR inhibitor RAD0001 (Everolimus), for example, has been in clinical development since 1996 and is currently in clinical trials for various cancers, including triple-negative metastatic breast cancer. Although the studies that have identified these targets were performed with different solid tumors and cell lines, the results are nonetheless encouraging and warrant further in-depth evaluation with a specific cancer type.
Figure 9.
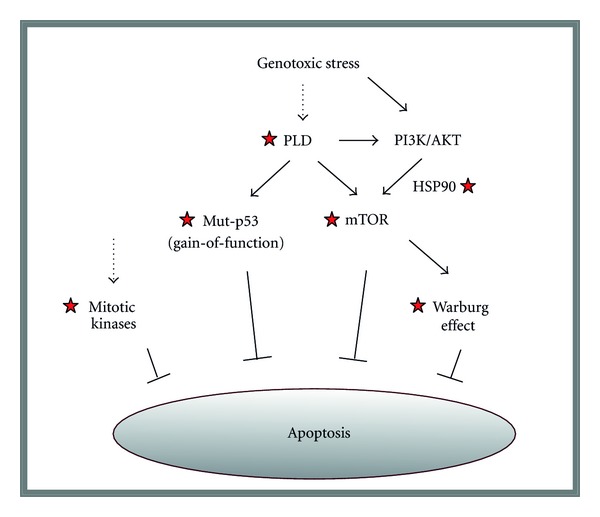
Highly simplified model showing responses triggered by genotoxic stress in human cancer cells. Broken arrows indicate that the mechanism by which cancer-therapeutic agents (e.g., ionizing radiation) might trigger activation of phospholipase D (PLD) and mitotic kinases remain to be determined. However, it is known that PLD contributes to activation of PI3K/AKT signalling [74], and that ionizing radiation triggers mTOR activation through PI3K/AKT [75]. Asterisks mark molecules/pathways that might be targeted, for example, using small-molecule inhibitors, for improving the outcome of cancer radiotherapy (see text for details).
In this article, we have further demonstrated for the first time that ionizing radiation triggers sequential waves of p21 in p53 wild-type human cancer cells. The waves of p21 induction peaked at early times (~6 h) and late times (several days) post-irradiation, which presumably contribute to activation of cell cycle checkpoints and SIPS, respectively. That p21-dependent SIPS is a prominent response of p53 wild-type cancer cells exposed to clinically-relevant doses of ionizing radiation is well established. Interestingly, consistent with our observations with Li-Fraumeni syndrome fibroblasts [18], studies with non-small cell lung carcinoma cell lines have revealed that ionizing radiation also triggers p16-dependent (but p21-independent) SIPS in some cell lines with mutant p53 [131]. However, for most cancers, whether the treatment-induced SIPS may lead to cancer cure or may be associated with tumor recurrence, ultimately leading to the emergence of highly metastatic and therapy-resistant disease, remains largely unknown. Unraveling the long-term effects of SIPS in a given cancer type will lead to novel therapeutic strategies for solid tumors with differing p53 status.
Acknowledgments
The authors wish to thank Ying W. Wang for technical assistance. This work was supported by the Canadian Breast Cancer Foundation-Prairies/NWT region.
References
- 1.Zhang T, Brazhnik P, Tyson JJ. Exploring mechanisms of the DNA-damage response: p53 pulses and their possible relevance to apoptosis. Cell Cycle. 2007;6(1):85–94. doi: 10.4161/cc.6.1.3705. [DOI] [PubMed] [Google Scholar]
- 2.Desilet N, Campbell TN, Choy FYM. p53-based anti-cancer therapies: an empty promise? Current Issues in Molecular Biology. 2010;12(3):143–146. [PubMed] [Google Scholar]
- 3.Enoch T, Norbury C. Cellular responses to DNA damage: cell-cycle checkpoints, apoptosis and the roles of p53 and ATM. Trends in Biochemical Sciences. 1995;20(10):426–430. doi: 10.1016/s0968-0004(00)89093-3. [DOI] [PubMed] [Google Scholar]
- 4.Meyn MS. Ataxia-telangiectasia and cellular responses to DNA damage. Cancer Research. 1995;55(24):5991–6001. [PubMed] [Google Scholar]
- 5.Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resistance Updates. 2001;4(5):303–313. doi: 10.1054/drup.2001.0213. [DOI] [PubMed] [Google Scholar]
- 6.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Research. 2003;63(11):2705–2715. [PubMed] [Google Scholar]
- 7.Han Z, Wei W, Dunaway S, et al. Role of p21 in apoptosis and senescence of human colon cancer cells treated with camptothecin. Journal of Biological Chemistry. 2002;277(19):17154–17160. doi: 10.1074/jbc.M112401200. [DOI] [PubMed] [Google Scholar]
- 8.Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23(16):2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 9.Sohn D, Essmann F, Schulze-Osthoff K, Jänicke RU. p21 blocks irradiation-induced apoptosis downstream of mitochondria by inhibition of cyclin-dependent kinase-mediated caspase-9 activation. Cancer Research. 2006;66(23):11254–11262. doi: 10.1158/0008-5472.CAN-06-1569. [DOI] [PubMed] [Google Scholar]
- 10.Afshar G, Jelluma N, Yang X, et al. Radiation-induced caspase-8 mediates p53-independent apoptosis in glioma cells. Cancer Research. 2006;66(8):4223–4232. doi: 10.1158/0008-5472.CAN-05-1283. [DOI] [PubMed] [Google Scholar]
- 11.Gewirtz DA, Holt SE, Elmore LW. Accelerated senescence: an emerging role in tumor cell response to chemotherapy and radiation. Biochemical Pharmacology. 2008;76(8):947–957. doi: 10.1016/j.bcp.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 12.Sliwinska MA, Mosieniak G, Wolanin K, et al. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mechanisms of Ageing and Development. 2009;130(1-2):24–32. doi: 10.1016/j.mad.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 13.Mansilla S, Bataller M, Portugal J. A nuclear budding mechanism in transiently arrested cells generates drug-sensitive and drug-resistant cells. Biochemical Pharmacology. 2009;78(2):123–132. doi: 10.1016/j.bcp.2009.03.027. [DOI] [PubMed] [Google Scholar]
- 14.Barley RDC, Enns L, Paterson MC, Mirzayans R. Aberrant p21WAF1-dependent growth arrest as the possible mechanism of abnormal resistance to ultraviolet light cytotoxicity in Li-Fraumeni syndrome fibroblast strains heterozygous for TP53 mutations. Oncogene. 1998;17(5):533–543. doi: 10.1038/sj.onc.1202271. [DOI] [PubMed] [Google Scholar]
- 15.Mirzayans R, Scott A, Cameron M, Murray D. Induction of accelerated senescence by γ radiation in human solid tumor-derived cell lines expressing wild-type TP53. Radiation Research. 2005;163(1):53–62. doi: 10.1667/rr3280. [DOI] [PubMed] [Google Scholar]
- 16.Mirzayans R, Murray D. Cellular senescence: implications for cancer therapy. In: Garvey RB, editor. Monograph. Hauppauge, NY, USA: Nova Science Publishers; 2009. pp. 1–130. [Google Scholar]
- 17.Mirzayans R, Scott A, Andrais B, Pollock S, Murray D. Ultraviolet light exposure triggers nuclear accumulation of p21WAF1 and accelerated senescence in human normal and nucleotide excision repair-deficient fibroblast strains. Journal of Cellular Physiology. 2008;215(1):55–67. doi: 10.1002/jcp.21284. [DOI] [PubMed] [Google Scholar]
- 18.Mirzayans R, Andrais B, Scott A, Paterson MC, Murray D. Single-cell analysis of p16INK4a and p21WAF1 expression suggests distinct mechanisms of senescence in normal human and Li-Fraumeni syndrome fibroblasts. Journal of Cellular Physiology. 2010;223(1):57–67. doi: 10.1002/jcp.22002. [DOI] [PubMed] [Google Scholar]
- 19.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265(5170):346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 20.Bouchet BP, de Fromentel CC, Puisieux A, Galmarini CM. p53 as a target for anti-cancer drug development. Critical Reviews in Oncology/Hematology. 2006;58(3):190–207. doi: 10.1016/j.critrevonc.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 21.Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death and Differentiation. 2006;13(6):941–950. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- 22.Akhtar RS, Geng Y, Klocke BJ, et al. BH3-only proapoptotic Bcl-2 family members Noxa and Puma mediate neural precursor cell death. The Journal of Neuroscience. 2006;26(27):7257–7264. doi: 10.1523/JNEUROSCI.0196-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pietrzak M, Puzianowska-Kuznicka M. p53-dependent repression of the human MCL-1 gene encoding an anti-apoptotic member of the BCL-2 family: the role of Sp1 and of basic transcription factor binding sites in the MCL-1 promoter. Biological Chemistry. 2008;389(4):383–393. doi: 10.1515/BC.2008.039. [DOI] [PubMed] [Google Scholar]
- 24.Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. Journal of Biological Chemistry. 2002;277(5):3247–3257. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 25.Chin KV, Ueda K, Pastan I, Gottesman MM. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science. 1992;255(5043):459–462. doi: 10.1126/science.1346476. [DOI] [PubMed] [Google Scholar]
- 26.Johnson RA, Shepard EM, Scotto KW. Differential regulation of MDR1 transcription by the p53 family members: role of the DNA binding domain. Journal of Biological Chemistry. 2005;280(14):13213–13219. doi: 10.1074/jbc.M414646200. [DOI] [PubMed] [Google Scholar]
- 27.Löhr K, Möritz C, Contente A, Dobbelstein M. p21/CDKN1A mediates negative regulation of transcription by p53. Journal of Biological Chemistry. 2003;278(35):32507–32516. doi: 10.1074/jbc.M212517200. [DOI] [PubMed] [Google Scholar]
- 28.Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Seminars in Cancer Biology. 2003;13(1):49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- 29.Ljungman M. Dial 9-1-1 for p53: mechanisms of p53 activation by cellular stress. Neoplasia. 2000;2(3):208–225. doi: 10.1038/sj.neo.7900073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meek DW. The p53 response to DNA damage. DNA Repair. 2004;3(8-9):1049–1056. doi: 10.1016/j.dnarep.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 31.Ling B, Zuo Zhu WG. p53: structure, function and therapeutic applications. Journal of Cancer Molecules. 2006;2:141–153. [Google Scholar]
- 32.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137(3):413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 33.Enns L, Barley RDC, Paterson MC, Mirzayans R. Radiosensitivity in ataxia telangiectasia fibroblasts is not associated with deregulated apoptosis. Radiation Research. 1998;150(1):11–16. [PubMed] [Google Scholar]
- 34.Mirzayans R, Murray D. Role of the ataxia telangiectasia mutated protein in stress-induced premature senescence. In: Hong SH, editor. Ataxia: Causes, Symptoms and Treatment. Hauppauge, NY, USA: Nova Science; 2012. pp. 1–34. [Google Scholar]
- 35.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin- dependent kinases. Cell. 1993;75(4):805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 36.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366(6456):701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 37.Rousseau D, Cannella D, Boulaire J, Fitzgerald P, Fotedar A, Fotedar R. Growth inhibition by CDK-cyclin and PCNA binding domains of p21 occurs by distinct mechanisms and is regulated by ubiquitin-proteasome pathway. Oncogene. 1999;18(30):4313–4325. doi: 10.1038/sj.onc.1202686. [DOI] [PubMed] [Google Scholar]
- 38.Broude EV, Swift ME, Vivo C, et al. p21Waf1/Cip1/Sdi1 mediates retinoblastoma protein degradation. Oncogene. 2007;26(48):6954–6958. doi: 10.1038/sj.onc.1210516. [DOI] [PubMed] [Google Scholar]
- 39.Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282(5393):1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 40.Gillis LD, Leidal AM, Hill R, Lee PWK. p21Cip1/WAF1 mediates cyclin B1 degradation in response to DNA damage. Cell Cycle. 2009;8(2):253–256. doi: 10.4161/cc.8.2.7550. [DOI] [PubMed] [Google Scholar]
- 41.Chuang LSH, Ian HI, Koh TW, Ng HH, Xu G, Li BFL. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997;277(5334):1996–2000. doi: 10.1126/science.277.5334.1996. [DOI] [PubMed] [Google Scholar]
- 42.Baylin SB. Tying it all together: epigenetics, genetics, cell cycle, and cancer. Science. 1997;277(5334):1948–1949. doi: 10.1126/science.277.5334.1948. [DOI] [PubMed] [Google Scholar]
- 43.Chang BD, Watanabe K, Broude EV, et al. Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: implications for carcinogenesis, senescence, and age-related diseases. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(8):4291–4296. doi: 10.1073/pnas.97.8.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delavaine L, La Thangue NB. Control of E2F activity by p21Waf1/Cip1. Oncogene. 1999;18(39):5381–5392. doi: 10.1038/sj.onc.1202923. [DOI] [PubMed] [Google Scholar]
- 45.Kitaura H, Shinshi M, Uchikoshi Y, et al. Reciprocal regulation via protein-protein interaction between c-Myc and p21cip1/waf1/sdi1 in DNA replication and transcription. Journal of Biological Chemistry. 2000;275(14):10477–10483. doi: 10.1074/jbc.275.14.10477. [DOI] [PubMed] [Google Scholar]
- 46.Devgan V, Mammucari C, Millar SE, Brisken C, Dotto GP. p21Waf1/Cip1 is a negative transcriptional regulator of Wnt4 expression downstream of Notch1 activation. Genes and Development. 2005;19(12):1485–1495. doi: 10.1101/gad.341405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75(4):817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 48.Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Experimental Cell Research. 1994;211(1):90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 49.Shay JW. At the end of the millennium, a view of the end. Nature Genetics. 1999;23(4):382–383. doi: 10.1038/70480. [DOI] [PubMed] [Google Scholar]
- 50.Chen QM, Prowse KR, Tu VC, Purdom S, Linskens MHK. Uncoupling the senescent phenotype from telomere shortening in hydrogen peroxide-treated fibroblasts. Experimental Cell Research. 2001;265(2):294–303. doi: 10.1006/excr.2001.5182. [DOI] [PubMed] [Google Scholar]
- 51.Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene. 2004;23(16):2919–2933. doi: 10.1038/sj.onc.1207518. [DOI] [PubMed] [Google Scholar]
- 52.Herbig U, Jobling WA, Chen BPC, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Molecular Cell. 2004;14(4):501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 53.Freedman DA, Folkman J. CDK2 translational down-regulation during endothelial senescence. Experimental Cell Research. 2005;307(1):118–130. doi: 10.1016/j.yexcr.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 54.Crescenzi E, Palumbo G, De Boer J, Brady HJM. Ataxia telangiectasia mutated and p21CIP1 modulate cell survival of drug-induced senescent tumor cells: implications for chemotherapy. Clinical Cancer Research. 2008;14(6):1877–1887. doi: 10.1158/1078-0432.CCR-07-4298. [DOI] [PubMed] [Google Scholar]
- 55.Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21CIP1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nature Cell Biology. 2001;3(3):245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 56.Zhou BP, Hung MC. Novel targets of Akt, p21Cip1/WAF1, and MDM2. Seminars in Oncology. 2002;29(3):62–70. doi: 10.1053/sonc.2002.34057. [DOI] [PubMed] [Google Scholar]
- 57.Zhou BP, Hung MC. Dysregulation of cellular signaling by HER2/neu in breast cancer. Seminars in Oncology. 2003;30(5):38–48. doi: 10.1053/j.seminoncol.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki A, Tsutomi Y, Yamamoto N, Shibutani T, Akahane K. Mitochondrial regulation of cell death: mitochondria are essential for procaspase 3-p21 complex formation to resist Fas-mediated cell death. Molecular and Cellular Biology. 1999;19(5):3842–3847. doi: 10.1128/mcb.19.5.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sohn D, Essmann F, Schulze-Osthoff K, Jänicke RU. p21 blocks irradiation-induced apoptosis downstream of mitochondria by inhibition of cyclin-dependent kinase-mediated caspase-9 activation. Cancer Research. 2006;66(23):11254–11262. doi: 10.1158/0008-5472.CAN-06-1569. [DOI] [PubMed] [Google Scholar]
- 60.Le HV, Minn AJ, Massagué J. Cyclin-dependent kinase inhibitors uncouple cell cycle progression from mitochondrial apoptotic functions in DNA-damaged cancer cells. Journal of Biological Chemistry. 2005;280(36):32018–32025. doi: 10.1074/jbc.M504689200. [DOI] [PubMed] [Google Scholar]
- 61.Zhang F, Zhang T, Teng ZH, Zhang R, Wang JB, Mei QB. Sensitization to γ-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biology and Therapy. 2009;8(9):823–831. doi: 10.4161/cbt.8.9.8143. [DOI] [PubMed] [Google Scholar]
- 62.Zhang T, Jiang T, Zhang F, et al. Involvement of p21Waf1/Cip1 cleavage during roscovitine-induced apoptosis in non-small cell lung cancer cells. Oncology Reports. 2010;23(1):239–245. [PubMed] [Google Scholar]
- 63.Javelaud D, Besançon F. Inactivation of p21WAF1 sensitizes cells to apoptosis via an increase of both p14ARF and p53 levels and an alteration of the Bax/Bcl-2 ratio. Journal of Biological Chemistry. 2002;277(40):37949–37954. doi: 10.1074/jbc.M204497200. [DOI] [PubMed] [Google Scholar]
- 64.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92(6):725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 65.Pomerantz J, Schreiber-Agus N, Liégeois NJ, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92(6):713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 66.Pang LY, Scott M, Hayward RL, et al. p21WAF1 is component of a positive feedback loop that maintains the p53 transcriptional program. Cell Cycle. 2011;10(6):932–950. doi: 10.4161/cc.10.6.15012. [DOI] [PubMed] [Google Scholar]
- 67.Zhang XP, Liu F, Cheng Z, Wang W. Cell fate decision mediated by p53 pulses. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(30):12245–12250. doi: 10.1073/pnas.0813088106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang XP, Liu F, Wang W. Two-phase dynamics of p53 in the DNA damage response. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(22):8990–8995. doi: 10.1073/pnas.1100600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Batchelor E, Mock CS, Bhan I, Loewer A, Lahav G. Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Molecular Cell. 2008;30(3):277–289. doi: 10.1016/j.molcel.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang XH, Sladek TL, Liu X, Butler BR, Froelich CJ, Thor AD. Reconstitution of caspase 3 sensitizes MCF-7 breast cancer cells to doxorubicin- and etoposide-induced apoptosis. Cancer Research. 2001;61(1):348–354. [PubMed] [Google Scholar]
- 71.Eriksson D, Löfroth PO, Johansson L, Riklund K, Stigbrand T. Apoptotic signalling in HeLa Hep2 cells following 5 Gy of cobalt-60 gamma radiation. Anticancer Research. 2009;29(11):4361–4366. [PubMed] [Google Scholar]
- 72.Kim BC, Han NK, Byun HO, et al. Time-dependently expressed markers and the characterization for premature senescence induced by ionizing radiation in MCF7. Oncology Reports. 2010;24(2):395–403. doi: 10.3892/or_00000872. [DOI] [PubMed] [Google Scholar]
- 73.Gil YG, Kang MK. Capsaicin induces apoptosis and terminal differentiation in human glioma A172 cells. Life Sciences. 2008;82(19-20):997–1003. doi: 10.1016/j.lfs.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 74.Banno Y, Takuwa Y, Akao Y, et al. Involvement of phospholipase D in sphingosine 1-phosphate-induced activation of phosphatidylinositol 3-kinase and Akt in Chinese hamster ovary cells overexpressing EDG3. Journal of Biological Chemistry. 2001;276(38):35622–35628. doi: 10.1074/jbc.M105673200. [DOI] [PubMed] [Google Scholar]
- 75.Albert JM, Kim KW, Cao C, Lu B. Targeting the Akt/mammalian target of rapamycin pathway for radiosensitization of breast cancer. Molecular Cancer Therapeutics. 2006;5(5):1183–1189. doi: 10.1158/1535-7163.MCT-05-0400. [DOI] [PubMed] [Google Scholar]
- 76.Lim LY, Vidnovic N, Ellisen LW, Leong CO. Mutant p53 mediates survival of breast cancer cells. British Journal of Cancer. 2009;101(9):1606–1612. doi: 10.1038/sj.bjc.6605335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harbor perspectives in biology. 2010;2(2) doi: 10.1101/cshperspect.a001107.a001107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mogi A, Kuwano H. TP53 mutations in nonsmall cell lung cancer. Journal of Biomedicine and Biotechnology. 2011;2011:9 pages. doi: 10.1155/2011/583929.583929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frank AK, Pietsch EC, Dumont P, Tao J, Murphy ME. Wild-type and mutant p53 proteins interact with mitochondrial caspase-3. Cancer Biology and Therapy. 2011;11(8):740–745. doi: 10.4161/cbt.11.8.14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Deb SP, Munoz RM, Brown DR, Subler MA, Deb S. Wild-type human p53 activates the human epidermal growth factor receptor promoter. Oncogene. 1994;9(5):1341–1349. [PubMed] [Google Scholar]
- 81.Frazier MW, He X, Wang J, Gu Z, Cleveland JL, Zambetti GP. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Molecular and Cellular Biology. 1998;18(7):3735–3743. doi: 10.1128/mcb.18.7.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Preuss U, Kreutzfeld R, Scheidtmann K-H. Tumor-derived p53 mutant c174y is a gain-of-function mutant which activates the fos promoter and enhances colony formation. International Journal of Cancer. 2000;88(2):162–171. doi: 10.1002/1097-0215(20001015)88:2<162::aid-ijc3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 83.Sampath J, Sun D, Kidd VJ, et al. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. Journal of Biological Chemistry. 2001;276(42):39359–39367. doi: 10.1074/jbc.M103429200. [DOI] [PubMed] [Google Scholar]
- 84.Stein WD. Kinetics of the multidrug transporter (P-glycoprotein) and its reversal. Physiological Reviews. 1997;77(2):545–590. doi: 10.1152/physrev.1997.77.2.545. [DOI] [PubMed] [Google Scholar]
- 85.Thottassery JV, Zambettii GP, Arimori K, Schuetz EG, Schuetz JD. p53-dependent regulation of MDR1 gene expression causes selective resistance to chemotherapeutic agents. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(20):11037–11042. doi: 10.1073/pnas.94.20.11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Adorno M, Cordenonsi M, Montagner M, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell. 2009;137(1):87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 87.Strano S, Munarriz E, Rossi M, et al. Physical and functional interaction between p53 mutants and different isoforms of p73. Journal of Biological Chemistry. 2000;275(38):29503–29512. doi: 10.1074/jbc.M003360200. [DOI] [PubMed] [Google Scholar]
- 88.Talos F, Nemajerova A, Flores ER, Petrenko O, Moll UM. p73 suppresses polyploidy and aneuploidy in the absence of functional p53. Molecular Cell. 2007;27(4):647–659. doi: 10.1016/j.molcel.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 89.Hui L, Zheng Y, Yan Y, Bargonetti J, Foster DA. Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene. 2006;25(55):7305–7310. doi: 10.1038/sj.onc.1209735. [DOI] [PubMed] [Google Scholar]
- 90.Ventura A, Kirsch DG, McLaughlin ME, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 91.Skinner HD, Sandulache VC, Ow TJ, et al. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clinical Cancer Research. 2012;18(1):290–300. doi: 10.1158/1078-0432.CCR-11-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sabin RJ, Anderson RM. Cellular Senescence—its role in cancer and the response to ionizing radiation. Genome Integrity. 2011;2, article 7 doi: 10.1186/2041-9414-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tsai KKC, Stuart J, Chuang YYE, Little JB, Yuan ZM. Low-dose radiation-induced senescent stromal fibroblasts render nearby breast cancer cells radioresistant. Radiation Research. 2009;172(3):306–313. doi: 10.1667/RR1764.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rajaraman R, Rajaraman MM, Rajaraman SR, Guernsey DL. Neosis—a paradigm of self-renewal in cancer. Cell Biology International. 2005;29(12):1084–1097. doi: 10.1016/j.cellbi.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 95.Gewirtz DA. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy. 2009;5(8):1232–1234. doi: 10.4161/auto.5.8.9896. [DOI] [PubMed] [Google Scholar]
- 96.Mirzayans R, Murray D. Pharmacological modulation of p53 function in cancer therapy. Current Signal Transduction Therapy. 2008;3(3):183–194. [Google Scholar]
- 97.Foster DA, Xu L. Phospholipase D in cell proliferation and cancer. Molecular Cancer Research. 2003;1(11):789–800. [PubMed] [Google Scholar]
- 98.Zheng Y, Rodrik V, Toschi A, et al. Phospholipase D couples survival and migration signals in stress response of human cancer cells. Journal of Biological Chemistry. 2006;281(23):15862–15868. doi: 10.1074/jbc.M600660200. [DOI] [PubMed] [Google Scholar]
- 99.Foster DA. Phospholipase D survival signals as a therapeutic target in cancer. Current Signal Transduction Therapy. 2006;1:295–303. [Google Scholar]
- 100.Hui L, Rodrik V, Pielak RM, Knirr S, Zheng Y, Foster DA. mTOR-dependent suppression of protein phosphatase 2A is critical for phospholipase D survival signals in human breast cancer cells. Journal of Biological Chemistry. 2005;280(43):35829–35835. doi: 10.1074/jbc.M504192200. [DOI] [PubMed] [Google Scholar]
- 101.Chen Y, Rodrik V, Foster DA. Alternative phospholipase D/mTOR survival signal in human breast cancer cells. Oncogene. 2005;24(4):672–679. doi: 10.1038/sj.onc.1208099. [DOI] [PubMed] [Google Scholar]
- 102.Fang Y, Park IH, Wu AL, et al. PLD1 regulates mTOR signaling and mediates Cdc42 activation of S6K1. Current Biology. 2003;13(23):2037–2044. doi: 10.1016/j.cub.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 103.Hui L, Abbas T, Pielak RM, Joseph T, Bargonetti J, Foster DA. Phospholipase D elevates the level of MDM2 and suppresses DNA damage-induced increases in p53. Molecular and Cellular Biology. 2004;24(13):5677–5686. doi: 10.1128/MCB.24.13.5677-5686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Toschi A, Lee E, Thompson S, et al. Phospholipase D-mTOR requirement for the Warburg effect in human cancer cells. Cancer Letters. 2010;299(1):72–79. doi: 10.1016/j.canlet.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gadir N, Lee E, Garcia A, Toschi A, Foster DA. Suppression of TGF-β signaling by phospholipase D. Cell Cycle. 2007;6(22):2840–2845. doi: 10.4161/cc.6.22.4921. [DOI] [PubMed] [Google Scholar]
- 106.Shi M, Zheng Y, Garcia A, Xu L, Foster DA. Phospholipase D provides a survival signal in human cancer cells with activated H-Ras or K-Ras. Cancer Letters. 2007;258(2):268–275. doi: 10.1016/j.canlet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Castedo M, Ferri KF, Kroemer G. Mammalian target of rapamycin (mTOR): pro- and anti-apoptotic. Cell Death and Differentiation. 2002;9(2):99–100. doi: 10.1038/sj.cdd.4400978. [DOI] [PubMed] [Google Scholar]
- 108.Lu Y, Wang H, Mills GB. Targeting PI3K-AKT pathway for cancer therapy. Reviews in Clinical and Experimental Hematology. 2003;7(2):205–228. [PubMed] [Google Scholar]
- 109.Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anti-Cancer Drugs. 2005;16(8):797–803. doi: 10.1097/01.cad.0000173476.67239.3b. [DOI] [PubMed] [Google Scholar]
- 110.Valdez BC, Murray D, Ramdas L, et al. Altered gene expression in busulfan-resistant human myeloid leukemia. Leukemia Research. 2008;32(11):1684–1697. doi: 10.1016/j.leukres.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nature Reviews Cancer. 2010;10(8):537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Takai H, Xie Y, De Lange T, Pavletich NP. Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes and Development. 2010;24(18):2019–2030. doi: 10.1101/gad.1956410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yin X, Zhang H, Lundgren K, Wilson L, Burrows F, Shores CG. BIIB021, a novel Hsp90 inhibitor, sensitizes head and neck squamous cell carcinoma to radiotherapy. International Journal of Cancer. 2010;126(5):1216–1225. doi: 10.1002/ijc.24815. [DOI] [PubMed] [Google Scholar]
- 114.Erenpreisa J, Ivanov A, Cragg M, Selivanova G, Illidge T. Nuclear envelope-limited chromatin sheets are part of mitotic death. Histochemistry and Cell Biology. 2002;117(3):243–255. doi: 10.1007/s00418-002-0382-6. [DOI] [PubMed] [Google Scholar]
- 115.Ianzini F, Kosmacek EA, Nelson ES, et al. Activation of meiosis-specific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Research. 2009;69(6):2296–2304. doi: 10.1158/0008-5472.CAN-08-3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Erenpreisa J, Cragg MS, Salmina K, Hausmann M, Scherthan H. The role of meiotic cohesin REC8 in chromosome segregation in γ irradiation-induced endopolyploid tumour cells. Experimental Cell Research. 2009;315(15):2593–2603. doi: 10.1016/j.yexcr.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 117.Warner SL, Bearss DJ, Han H, Von Hoff DD. Targeting Aurora-2 kinase in cancer. Molecular cancer therapeutics. 2003;2(6):589–595. [PubMed] [Google Scholar]
- 118.Erenpreisa J, Ivanov A, Wheatley SP, et al. Endopolyploidy in irradiated p53-deficient tumour cell lines: persistence of cell division activity in giant cells expressing Aurora-B kinase. Cell Biology International. 2008;32(9):1044–1056. doi: 10.1016/j.cellbi.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tao Y, Zhang P, Girdler F, et al. Enhancement of radiation response in p53-deficient cancer cells by the Aurora-B kinase inhibitor AZD1152. Oncogene. 2008;27(23):3244–3255. doi: 10.1038/sj.onc.1210990. [DOI] [PubMed] [Google Scholar]
- 120.Kaelin WG, Thompson CB. Q and A: cancer: clues from cell metabolism. Nature. 2010;465(7298):562–564. doi: 10.1038/465562a. [DOI] [PubMed] [Google Scholar]
- 121.Sahra IB, Laurent K, Giuliano S, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Research. 2010;70(6):2465–2475. doi: 10.1158/0008-5472.CAN-09-2782. [DOI] [PubMed] [Google Scholar]
- 122.Aft RL, Zhang FW, Gius D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. British Journal of Cancer. 2002;87(7):805–812. doi: 10.1038/sj.bjc.6600547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lin X, Zhang F, Bradbury CM, et al. 2-Deoxy-D-glucose-induced cytotoxicity and radiosensitization in tumor cells is mediated via disruptions in thiol metabolism. Cancer Research. 2003;63(12):3413–3417. [PubMed] [Google Scholar]
- 124.Maschek G, Savaraj N, Priebe W, et al. 2-Deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Research. 2004;64(1):31–34. doi: 10.1158/0008-5472.can-03-3294. [DOI] [PubMed] [Google Scholar]
- 125.Egler V, Korur S, Failly M, et al. Histone deacetylase inhibition and blockade of the glycolytic pathway synergistically induce glioblastoma cell death. Clinical Cancer Research. 2008;14(10):3132–3140. doi: 10.1158/1078-0432.CCR-07-4182. [DOI] [PubMed] [Google Scholar]
- 126.Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. British Journal of Cancer. 2008;99(7):989–994. doi: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cao W, Yacoub S, Shiverick KT, et al. Dichloroacetate (DCA) sensitizes both wild-type and over expressing bcl-2 prostate cancer cells in vitro to radiation. Prostate. 2008;68(11):1223–1231. doi: 10.1002/pros.20788. [DOI] [PubMed] [Google Scholar]
- 128.Wong JYY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecologic Oncology. 2008;109(3):394–402. doi: 10.1016/j.ygyno.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Monovich L, Mugrage B, Quadros E, et al. Optimization of halopemide for phospholipase D2 inhibition. Bioorganic and Medicinal Chemistry Letters. 2007;17(8):2310–2311. doi: 10.1016/j.bmcl.2007.01.059. [DOI] [PubMed] [Google Scholar]
- 130.Mabuchi S, Altomare DA, Cheung M, et al. RAD001 inhibits human ovarian cancer cell proliferation, enhances cisplatin-induced apoptosis, and prolongs survival in an ovarian cancer model. Clinical Cancer Research. 2007;13(14):4261–4270. doi: 10.1158/1078-0432.CCR-06-2770. [DOI] [PubMed] [Google Scholar]
- 131.Wang M, Morsbach F, Sander D, et al. EGF receptor inhibition radiosensitizes NSCLC cells by inducing senescence in cells sustaining DNA double-strand breaks. Cancer Research. 2011;71(19):6261–6269. doi: 10.1158/0008-5472.CAN-11-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]