

Abstract
HIV-1 protease recognizes and cleaves more than 12 different substrates leading to viral maturation. While these substrates share no conserved motif, they are specifically selected for and cleaved by protease during viral life cycle. Drug resistant mutations evolve within the protease that compromise inhibitor binding but allow the continued recognition of all these substrates. While the substrate envelope defines a general shape for substrate recognition, successfully predicting the determinants of substrate binding specificity would provide additional insights into the mechanism of altered molecular recognition in resistant proteases. We designed a variant of HIV protease with altered specificity using positive computational design methods and validated the design using X-ray crystallography and enzyme biochemistry. The engineered variant, Pr3 (A28S/D30F/G48R), was designed to preferentially bind to one out of three of HIV protease's natural substrates; RT–RH over p2-NC and CA-p2. In kinetic assays, RT–RH binding specificity for Pr3 increased threefold compared to the wild-type (WT), which was further confirmed by isothermal titration calorimetry. Crystal structures of WT protease and the designed variant in complex with RT–RH, CA-p2, and p2-NC were determined. Structural analysis of the designed complexes revealed that one of the engineered substitutions (G48R) potentially stabilized heterogeneous flap conformations, thereby facilitating alternate modes of substrate binding. Our results demonstrate that while substrate specificity could be engineered in HIV protease, the structural pliability of protease restricted the propagation of interactions as predicted. These results offer new insights into the plasticity and structural determinants of substrate binding specificity of the HIV-1 protease.
Keywords: HIV-1 protease, positive design, substrate specificity, X-ray crystallography, isothermal titration calorimetry, substrate envelope
Introduction
Human immunodeficiency virus type-1 (HIV-1) protease recognizes and cleaves many natural substrates composed of diverse peptide sequences.1–3 Although studies have investigated the substrate binding and cleavage properties of proteases,4–8 the factors that determine the specificity of substrate binding remain largely uncharacterized. Studies of HIV-1 protease in complex with substrates and inhibitors indicate that the drug-resistant mutants of this enzyme have altered molecular recognition abilities; they still recognize the enzyme's substrates but binding to inhibitors is disrupted.9–12 The mechanisms involved in substrate recognition and binding of these drug-resistant mutants have yet to be fully elucidated. To gain insight into these processes, an improved understanding of which residues dictate substrate binding specificity and how these sites evolve with the emergence of drug resistance is required.
The “substrate envelope” concept states that the substrate specificity for HIV-1 protease is based on a conserved shape and not on the amino acid sequence of the substrate. This concept has been successfully used in designing robust protease inhibitors and in predicting the sites of drug-resistant mutations.12–14 Additionally, recent studies examined substrates from patient samples and found that the sequences of the substrates coevolved with major drug-resistant mutations in the protease.15,16 These findings suggest that for resistant protease variants, there is an alteration of substrate specificity that is selected for in the coevolving substrate sites.
Although extensive mutational studies have been done on HIV-1 protease, determining the basis of substrate specificity has proved challenging due to the enzyme's plasticity. HIV protease is known to exhibit great structural plasticity in both backbone and side chains. Mutational analyses of HIV protease and other proteases, such as those associated with Rous sarcoma virus or avian myeloblastosis virus,17–20 have shown that substitutions in some sites can affect both the binding and catalytic efficiency of the enzyme. For example, replacing glycine with glutamic acid at position 48 in the flap region affects the specificity of HIV-1 protease.8,21
Several structural,4–6,11,13 kinetic,7,22–24 and binding studies conducted on both active and inactive protease variants in complex with different substrate peptides25–28 have provided insights into the mode of substrate binding and the role of primary and compensatory mutations in altered molecular recognition by resistant proteases. However, a thorough understanding of substrate recognition would require the ability to predict and modulate substrate specificity and computational protein design can be used as a technique to probe what determines specificity.
To investigate the determinants of substrate binding specificity in HIV-1 protease, we redesigned the enzyme to preferentially bind to one substrate, RT–RH, over two other substrates, p2-NC and CA-p2. HIV-1 protease is a unique enzyme because its symmetrical binding region recognizes and cleaves asymmetrical substrates. Although highly specific, this protease recognizes and hydrolyzes sequences that exhibit little sequence homology (Table I). Studies on substrate binding have shown that specificity is driven by steric complementarity of the peptide side chains. Hydrogen bonds between enzyme and substrate side chains are rarely formed, and those that do form are not conserved between substrates. All the conserved hydrogen bonds occur between the enzyme and the substrate backbone, forming an extensive network that locks the substrate backbone into the proper conformation for peptide hydrolysis.13 Because hydrogen bonds are considered crucial for substrate specificity, HIV protease's broad substrate specificity could be due to the lack of specific hydrogen bonds to peptide side chains.13 If this hypothesis is correct, then variants that can form specific hydrogen bonds to wild-type (WT) substrates should exhibit an increase in specificity.29
Table I.
Sequences of Peptide Substrates Hydrolyzed by Wild-Type HIV-1 Protease
| Residue position | ||||||||
|---|---|---|---|---|---|---|---|---|
| Substrate | P4 | P3 | P2 | P1 | P1′ | P2′ | P3′ | P4′ |
| MA-CA | Ser | Gln | Asn | Tyr | Pro | Ile | Val | Gln |
| CA-p2 | Ala | Arg | Val | Leu | Ala | Glu | Ala | Met |
| p2-NC | Ala | Thr | Ile | Met | Met | Gln | Arg | Gly |
| NC-p1 | Arg | Gln | Ala | Asn | Phe | Leu | Gly | Lys |
| p1-p6 | Pro | Gly | Asn | Phe | Leu | Gln | Ser | Arg |
| TF-PR | Ser | Phe | Asn | Phe | Pro | Gln | Ile | Thr |
| PR-RT | Thr | Leu | Asn | Phe | Pro | Ile | Ser | Pro |
| RT-RH | Ala | Glu | Thr | Phe | Tyr | Val | Asp | Gly |
| RT-IN | Arg | Lys | Ile | Leu | Phe | Leu | Asp | Gly |
P1–P1′ is the scissile bond.
The question of specificity has been addressed in the computational design of other proteins. While electrostatic interactions are important in discriminating between side chains with opposite charges, and van der Waals forces can be used to select for side chains with specific geometries, optimizing for hydrogen bonding can be considered a purely positive design approach. Positive design is when a particular property is being selected for, rather than designing against a particular property, which would be considered negative design. Positive design proved successful in the computational design of calmodulin (CaM) to produce mutants with increased specificity toward a target peptide.30–32 Similarly, PDZ domains were effectively reengineered to bind novel sequence targets.33
In addition to designing variants with increased specificity towards peptides, computational protein design has been used to engineer proteins with improved protein–protein specificities. Focusing on positive design resulted in more stable complexes, whereas including negative design provided specificity at the cost of stability.34 Another example is the redesign of a protein complex between colicin E7 DNAse and Im7 immunity protein, where a new hydrogen bond network was predicted at the interface that provided increased specificity for the cognate dimer over the noncognate dimer.35 The success of the redesign was attributed to using positive design on an ensemble and designing against the native complex.
Previous attempts to engineer protease specificity have largely focused on the rational design of trypsin.36–39 Trypsin is a hydrolase that is highly specific for Lys- and Arg-containing peptides, whereas chymotrypsin favors peptides with aromatic residues such as Phe, Tyr, and Trp. A trypsin mutant was successfully engineered with specificity similar to that of chymotrypsin.40 The work presented here uses positive design to increase the specificity of HIV-1 protease.
Our redesign of HIV-1 protease considers the entire binding region, which is composed of eight binding pockets. The computational approach was aimed at reengineering the binding pockets to increase specificity for one of HIV-1 protease's natural substrates, RT–RH. Unlike CaM or PDZ domains, HIV protease is catalytic. Hence, the designs were intended to preserve the proteolytic activity in addition to increasing specificity for RT–RH, thereby, bridging the gap between the computational design of specificity and catalytic activity.41 One protease variant, Pr3 (A28S, D30F, G48R), was designed to bind RT–RH with increased specificity, and was tested experimentally using a combination of structural, functional, and thermodynamic binding assays. While WT HIV-1 protease is a symmetric dimer, the three mutations predicted by the positive design were in the same monomer generating an asymmetric dimer. Therefore, the predicted substrate specificity was based on the asymmetry of the designed protease. The kinetic assays revealed that the designed Pr3 variant (Fig. 1) indeed exhibits specificity toward RT–RH that is increased three- and fourfold compared to the p2-NC and CA-p2 substrates, respectively (Table II). ITC studies indicated that RT–RH binding is consistent with the kinetic data for RT–RH (Table III). Crystal structures were obtained for the WT and specificity designed variant Pr3 in complex with RT–RH, CA-p2, and p2-NC, respectively. The structural analysis, however, revealed that the intended asymmetry was rendered ineffective due to the plasticity of the enzyme, and the interactions predicted by positive design were only partially formed. G48R substitution led to dramatic changes in the backbone and side chain flexibility of the protease flap tips, causing the flap conformation to alter in a manner not predicted by the design. While certain design algorithms, notably Rosetta43–45 include backbone flexibility in the prediction algorithms, the energy functions and the sampling methods used to enable accurate prediction of backbone structures of remodeled proteins is limited. Accurate prediction of the extensive impact of G48R on the structure by any of the current techniques is unlikely. Hence, rather than being a predictor of the structure, the design served as a guide for screening the substrate specificity determinants of HIV protease.
Figure 1.
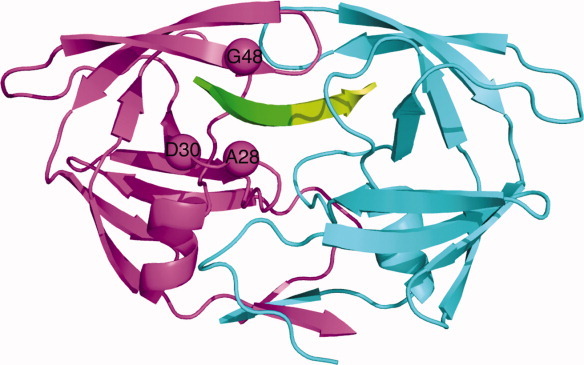
The predicted sites of mutations in the specificity designed Pr3 (A28S, D30F, G48R) HIV-1 protease. In this single-chain HIV protease structure, the sequences corresponding to the two monomers are colored differently: monomer A is in magenta and monomer B is in cyan. The substrate is shown in green with respect to the scissile bond; N terminal residues colored green and C terminal residues colored limon green.
Table II.
Experimental Kinetic Values for WT HIV-1 Protease and Specificity Designed Pr3 Variant for the Cleavage of Three Peptide Substrates
| RT-RH | p2-NC | CA-p2 | ||||
|---|---|---|---|---|---|---|
| Protease | Vmax/KM (s−1) | Normalized | Vmax/KM (s−1) | Normalized | Vmax/KM (s−1) | Normalized |
| Wild-type | 1.65E−03 | 1.00 | 3.70E−04 | 0.22 | 1.34E−03 | 0.81 |
| Pr3 | 1.13E−03 | 1.00 | 1.00E−04 | 0.09 | 1.00E−04 | 0.09 |
Table III.
Thermodynamic Parameters for Binding of RT-RH to Inactive Variants of Single-Chain WT and the Specificity Designed Pr3 HIV-1 Protease Variant
| Protease | Kd (μM) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) | ΔG (kcal mol−1) |
|---|---|---|---|---|
| Single-chain WT | 4.43 ± 0.9 | 0.198 ± 0.06 | −7.25 | −7.05 ± 0.122 |
| Pr3 | 2.52 ± 0.4 | 2.49 ± 0.2 | −9.86 | −7.37 ± 0.089 |
| Dimeric WTa | 6.2 ± 2.1 | 2.4 ± 0.4 | −9.3 | −6.9 ± 0.2 |
Values from previously published data.52
Errors on thermodynamic parameters are derived from the fitting error after repeating the experiments at least three times.
Results and Discussion
Protease design calculations and prediction of mutants
HIV protease was chosen as a target to alter specificity, as the enzyme naturally recognizes diverse substrates with distinguishable sequences and crystal structures are available for many of these substrates.13 Three crystal structures of HIV protease, each bound to a different native substrate, were used in the design (PDB ID: 1F7A, 1KJG, and 1KJ7).13 The entire binding region of HIV protease comprising eight binding pockets was included in the design. Because of the large size of the binding region, only side chains directly contacting the substrate were considered. To conserve function, the catalytic aspartates at positions 25 and 125 were kept in their crystallographic conformations. Early design calculations indicated that the RT–RH-bound structure (1KJG) was the most promising template, and was selected for optimization.
The design was initially divided into eight small calculations, one for each binding pocket. Given that HIV protease, a symmetrical dimer, binds to asymmetrical peptides, the twofold symmetry of the binding region was not conserved. For each pocket, residues within 4.2 Å of a substrate side chain were defined as being in the first shell, and residues within 4.2 Å of any first shell residue were defined as being in the second shell. First shell residues were allowed to mutate, whereas second shell residues and the substrate's side chains were allowed to change conformation but not amino acid identity. Optimization of interactions within the binding region was expected to increase specificity.46 The individual pocket designs (Fig. 2) were carried out in the following sequential order:
Figure 2.

Schematic representation illustrating the binding pocket notation of substrate–protease complex in stereo. The substrate interaction sites are designated as S4–S4′ pockets and the equivalent sites for protease are referred to as P4–P4′ sites. The substrate is represented in green, protease monomer A is in magenta and monomer B is in cyan. The scissile bond is between P1 and P1′ residues.
Mutations predicted from preceding calculations were carried over to the next design. As a result, the final design on the S4′ binding pocket contained all the mutations from preceding designs. One of the major drawbacks with designing individual pockets is their discrete nature. To mitigate this problem, positions that predicted reasonable mutations in the individual pocket calculations were simultaneously designed in the context of the entire binding region.
Based on the results from discrete pocket predictions, four positions (30, 48, 82, and 130) were selected for simultaneous design in the 1KJG crystal structure. All the other positions that had been considered in individual binding pocket calculations were floated, that is, allowed to sample alternate rotamers for that amino acid. This simultaneous design predicted specificity-optimized mutations at all four of the selected positions. Position 30 was mutated to Phe due to improved van der Waals interactions with AlaP4, and position 48 replaced Gly with Arg to form a salt bridge with GluP3. The mutations predicted at positions 82 (V→I) and 130 (D→N) were fairly conservative, and were expected to have little impact on specificity for the target substrate, RT–RH. To test the robustness of the calculation, solvation model was changed from a solvent exclusion-based one to a surface area-based one and the individual pocket designs were repeated.47 This resulted in an additional change from Ala to Ser mutation at position 28; Ser predicted to form a hydrogen bond with the P2 Thr of the substrate (Fig. 3). After considering the mutations from all the preceding designs, a four-point mutant (A28S/D30F/G48R/V82I) was selected for further evaluation.
Figure 3.
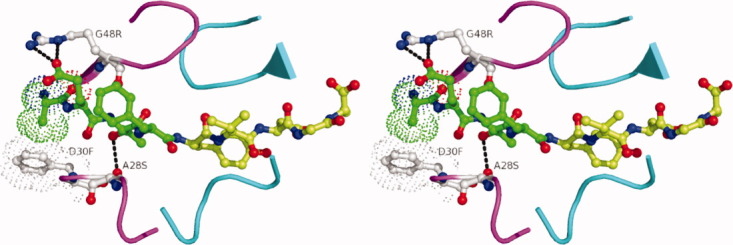
Predicted interactions of the designed mutations in the HIV-1 protease variant Pr3 (A28S/D30F/G48R) with the substrate RT–RH displayed in stereo. In the predicted structure, G48R, forms a salt bridge with Glu at the P3 position in RT-RH, there are improved van der Waals interactions between D30F and Ala at P4 in RT-RH and A28S makes a hydrogen bond with Thr at P2 in RT-RH. RT-RH is shown in green (residues N and C terminal with respect to the scissile bond are colored green and limon green, respectively) and the two monomers are colored magenta and cyan, respectively.
This combination of mutations was evaluated for specificity toward the RT–RH sequence compared with the other substrate sequences. Side-chain placement calculations were carried out on the four-point mutant and on the WT protease using three substrate-bound crystal structures (1KJG, 1F7A, and 1KJ7) in which the protease is bound to RT–RH, CA-p2, or p2-NC peptide, respectively. Energy analysis of the side-chain placement results indicated that the four-point mutant would stabilize the RT–RH substrate by 7.8 kcal mol−1 relative to the WT sequence. In contrast, a much smaller increase in stability (1.96 kcal mol−1) was predicted for the p2-NC peptide, and a decrease in stability was predicted for the CA-p2 peptide (Supporting Information Table I). The majority of increase in stability for RT–RH is expected to be due to a new hydrogen bond and salt bridge that are predicted to form as a result of the A28S and G48R mutations. The G48R mutation appears to stabilize interactions with RT–RH, while disfavoring binding of CA-p2 and p2-NC. Arg at position 48 is able to form a salt bridge with Glu at the P3 position on RT–RH (Fig. 3). The absence of Glu at P3 in the other two substrates prevents a similar interaction (Table I). Position P3 in p2-NC is a Thr, which is too small to accommodate even a hydrogen bond. CA-p2 contains an Arg at P3, which would form a repulsive interaction between the two Arginines, but due to the twofold symmetry in the binding site of HIV protease, the CA-p2 substrate is likely to bind in the opposite orientation where P3Arg can interact with Gly148 instead. Similarly, for p2-NC, although there are no unfavorable interactions predicted between Arg48 and P3Thr, binding in the alternate conformation would result in Arg48 having direct contact with P3′Arg (Table I). As a result, the mutation to an Arg at position 48 contains negative design features as well as positive design attributes. The V82I and D130N mutations were predicted to be beneficial in binding all the three substrates considered, and thus were dropped from experimental validation as not altering the specificity for the RT–RH target sequence. Previous studies have demonstrated that the engineered single chain dimers of HIV protease preserve wildtype structure48–50 and thus, the final single chain construct designed for experimental validation was Pr3 (A28S/D30F/G48R) with the second monomer maintaining a wildtype sequence.
Experimental determination of binding and kinetic parameters for specificity designed protease variant
To assess the accuracy of the computational predictions, the designed protease variant Pr3 and WT protease were tested for their catalytic efficiency for three substrates: RT–RH, CA-p2, and p2-NC. Because the designs allowed asymmetrical mutations, in constructing the WT and mutant proteins, a tethered dimer was used to ensure a heterodimer complex. Therefore, all experimental data was collected using single chain HIV-1 protease in which the two monomers were tethered with a five amino acid flexible linker (GGSSG).48–51 Kinetic experiments for the WT and Pr3 protease were carried out and resulting kinetic parameters for substrate cleavage were compared. Because of minimal peptide solubility, limited data was obtained for substrates with KMs >50 μM. Nevertheless, Vmax/KM values were successfully obtained using hydrolysis rates at low substrate concentrations. A significantly different specificity profile was observed for Pr3 compared to WT protease (Table II). Although the Vmax/KM (s−1) for RT–RH was relatively unaffected, the values for p2-NC and CA-p2 decreased significantly. The normalized values show that, relative to WT, specificity toward the RT–RH substrate increased twofold and ninefold over the p2-NC and CA-p2 substrates, respectively. The experimental kinetic results indicated that positive design resulted in a mutant with effectively increased substrate specificity.
To complement the results on enzyme kinetics, ITC was performed to measure the binding parameters for the inactive (D25N) variant of the designed protease Pr3. Data could only be collected for RT–RH, possibly due to the extremely weak binding affinities of the other two substrates (p2-NC and CA-p2). For RT–RH, single chain WT protease exhibited a dissociation constant (Kd) of 4.4 μM, while binding 1.75 fold more tightly to Pr3 (Table III). When the binding affinity of the specificity designed variant Pr3 was compared to the previously published results,52 Pr3 exhibited 1.6-fold tighter binding affinity to (RT-RH) compared to the dimeric WT-protease. Hence, the designed protease Pr3 binds the target substrate with higher affinity compared to the WT protease.
Crystal structures of the specificity designed HIV protease–substrate complexes
To address whether the interactions predicted by the protein design methodology are an accurate representation of the physiological mode of substrate binding to the specificity-designed protease, crystal structures of catalytically inactive variants of Pr3 and single-chain WT protease were determined in complex with the three substrates: RT–RH, CA-p2, and p2-NC, respectively (with the exception of Pr3 in complex with CA-p2, which did not crystallize). The crystallographic and refinement statistics are listed in (Table IV).
Table IV.
Crystallographic Statistics for the RT-RH, CA-p2, and p2-NC Substrate Complexes of Single-Chain WT and Pr3 Proteases
| Variable | PrWT-RTRHa | PrWT-p2NCb | PrWT-CAp2b | Pr3-RTRHc | Pr3-p2NCb |
|---|---|---|---|---|---|
| Resolution (Å) | 1.9 | 1.68 | 1.8 | 1.8 | 1.7 |
| Temperature (K) | 100 | 100 | 100 | 100 | 100 |
| Space group | p212121 | p212121 | p212121 | p212121 | p212121 |
| Unit cell parameters | |||||
| a (Å) | 51.2 | 51.0 | 51.0 | 51.1 | 51.08 |
| b (Å) | 58.7 | 58.8 | 58.9 | 59.3 | 59.2 |
| c (Å) | 61.8 | 61.8 | 61.5 | 62.1 | 61.9 |
| Z | 1 | 1 | 1 | 1 | 1 |
| Z′ | 4 | 4 | 4 | 4 | 4 |
| Rmerge (%) | 7.9 | 4.9 | 6.8 | 6.8 | 4.3 |
| Completeness (%) | 91.8 | 95.4 | 99.7 | 99.4 | 99.1 |
| Total reflections | 92118 | 152868 | 121051 | 110383 | 122038 |
| Unique reflections | 13957 | 21538 | 17300 | 18038 | 21064 |
| Redundancy (%) | 6.6 | 7.1 | 7.0 | 6.1 | 5.8 |
| I/σ | 12.2 | 14.5 | 13.5 | 14.8 | 16.2 |
| RMSD (Å) | |||||
| Bond length (Å) | .011 | .009 | .009 | .011 | .008 |
| Bond angles | 1.47 | 1.35 | 1.35 | 1.46 | 1.37 |
| Rfactor (%) | 20.61 | 19.8 | 18.9 | 19.9 | 22.0 |
| Rfree (%) | 26.06 | 22.7 | 22.7 | 25.2 | 26.9 |
| Waters | 116 | 69 | 62 | 76 | 70 |
Data collected in-house on Rigaku RAXIS IV.
Data collected by Annie Heroux (mail-in-crystal program @ BNL).
Data collected at 14-BMC, BioCARS @ Argonne Photon Source. Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4EP2, 4EPJ, 4EP3,4EQJ and 4EQ0).
The overall three-dimensional structure of the single-chain WT protease in complex with RT–RH was similar to the dimeric WT protease–RT–RH structure determined previously.13 In the substrate-bound crystal structures of Pr3, however, the 2Fo − Fc and Fo − Fc maps showed ambiguous densities at the sites of engineered substitutions, particularly for A28S substitution (Fig. 4). The side-chain density for the A28S substitution was present on both sides; hence, A128 also showed partial occupancy of serine [Fig. 4(A)]. As a result, Ser side chain is modeled at both the positions; 28 and 128 in the crystal structures of Pr3 in complex with RT-RH and p2-NC, respectively. These ambiguities in the electron density maps suggested that the protease–substrate complex was not uniquely oriented in the crystal lattice. The observed electron density is a composite of four differently oriented forms of the same complex, which is averaged over the crystal lattice. Despite the structures being averages, R48 was seen predominantly only on one side of the dimeric protease and was used to distinguish the two monomers in these tethered dimer structures. All the structural analyses and alignments are thus defined based on this orientation. The crystallographic waters were also analyzed (Table IV) and no significant differences were observed in the Pr3 complex with respect to the WT protease.
Figure 4.

Stereo view of the representative 2Fo − Fc electron density maps from Pr3-RTRH complex showing averaged electron density maps contoured at 1σ. (A) The A28S and A128S residue positions where both Ala and Ser side chains were modeled with partial occupancy for each side chain. (B) RT–RH density with RT–RH modeled in both possible orientations with respect to the protease dimer.
Comparison of predicted and experimental structures
Structural analysis revealed a partially open or ajar flap conformation for one of the flaps in the p2–NC and RT–RH complexes of Pr3 protease, as we previously observed.42 The designed mutation G48R possibly allowed several low energy flap conformations to be accessed. Analysis of crystal packing showed that there is enough space within the crystal lattice to allow either conformation of the flap to be present.
The orientations of the substrates are also complicated in the complexes. The sequence of RT-RH is highly symmetric in shape about the scissile bond (Table I) and binds in both the WT and Pr3 variant complexes in two orientations [Fig. 4(B)]. The sequence of p2-NC is asymmetric (Table I) and is uniquely oriented in the complex, although the single-chain dimer is averaged [Fig. 5(A,B)]. Interestingly, in the complex with p2-NC, the flap on the P′ side of the substrate was partially open compared to the flap conformation over the P side [Fig. 5(A)]. This suggests that the flap interacting with P3-P1 residues of the substrate may engage first, possibly more tightly, and that these designed proteases have stabilized a structural intermediate of substrate recognition.42
Figure 5.
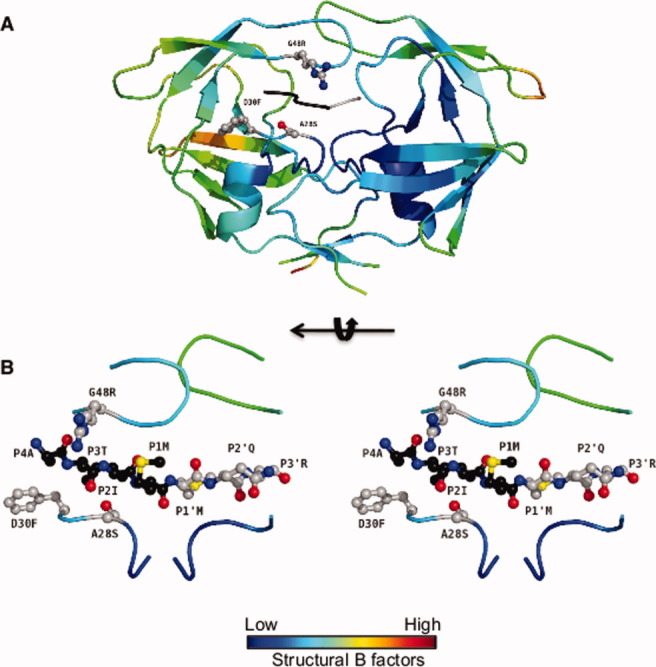
Pr3 in complex with p2-NC. (A) The protease molecule is colored according to the B factor values and represents an average structure of both possible protein orientations. One of the flaps is less ordered as reflected in light green color representing higher B factor values. (B) Stereo representation: The substrate p2-NC is uniquely oriented in the structure. The N and C terminal residues p2-NC, with respect to the scissile bond are colored black and gray, respectively.
Because the G48R mutation in Pr3 introduced an aspect of negative design in substrate binding for the sequences of CA-p2 and p2-NC, these substrates were predicted to bind in only one of the two possible orientations with respect to the designed proteases. However, in the p2-NC complex with Pr3, both possible orientations of substrate binding were observed, as indicated by the averaged electron density for protease chain (evident in the observed electron density of Ser side chain not only at position 28 but also at position 128 in the crystal structure). In the Pr3 crystal structures, R48 was in a different conformation than what was predicted by design (Figs. 3, 5, and 6). The observed conformation of R48 in these complexes eliminated any possibility of electrostatic clashes with Arg in p2-NC and possibly in CA-p2, thereby allowing substrate binding in both possible orientations. The interaction predicted for A28S and D30F substitutions also only partially agreed with the observed interactions in the crystal structures (Fig. 6). The degree of flexibility exhibited by the designed protease Pr3 was hard to anticipate, and resulted in the structural accommodation of engineered mutations in the crystallized complexes. Therefore, while the kinetic and binding data successfully demonstrates that targeted alterations in specificity can be engineered in proteins based on computational designs, inherent structural pliability restricted the structural validation of predictions in the model system of HIV protease.
Figure 6.

Stereo view of the predicted versus observed interactions for the residues G48R, D30F, and A28S in the Pr3 protease–RT–RH complex. RT–RH is modeled in both orientations. The conformation of the G48R side chain observed in the crystal structures differs from the predicted orientation and P3 Glu side chain could not be modeled in the structure due to incomplete density. D30F was predicted to make improved van der Waals interactions with Ala at P4 in RT-RH. While the P4 Ala side chain could not be modeled in the Pr3-RTRH structures due to incomplete density, the interaction distance could support the predicted improvement in van der Waals interactions. A28S was predicted to make a hydrogen bond with Thr at P2 in RT-RH. Ambiguous side chain density was observed for the p2-Thr side in the Pr3-RTRH crystal structure, however, for one possible P2-Thr conformation, the predicted hydrogen bond could likely form. The substrate, protease monomers and van der Waals surface representation is in lighter shades for the predicted RTRH complex and corresponding darker shades for experimental Pr3-RTRH structure. RT-RH is colored green and the two monomers are colored magenta and cyan, respectively.
Materials and Methods
Computational positive design
The crystal structure of HIV-1 protease bound to the RT–RH substrate (PDB code 1KJG) was used as the positive design scaffold. The crystal structure was subjected to 50 steps of minimization to relax van der Waals interactions, atomic bonds, and angles. Residues within 4.2 Å of a substrate side chain were selected for design (first shell residues); residues within 8.4 Å not selected to be in the first shell (second shell residues) and substrate side chains were floated (allowed to change conformation but not amino acid identity). Five conserved water molecules, residues hydrogen bonding to the waters (8, 29, 87, 108, 129, and 187) and catalytic residues (25 and 125) were fixed in their crystallographic conformations. In addition, proline-containing positions (81 and 181) were only allowed to change conformation.
A Dunbrack and Cohen-based backbone-dependent rotamer library was used for side-chain optimization. c1 and c2 values were expanded one standard deviation for all amino acids. In addition, the crystallographic rotamer at every design position was included. Either a solvent exclusion-based or an atomic surface area-based solvation potential was used. A rotamer probability scale factor of 0.3 proportionally penalized side-chain conformations based on their precalculated probabilities. All other parameters and potential functions used have been described previously.53–56 An optimization algorithm based on the dead-end elimination (DEE) theorem was used in the design of individual pockets.57 Designs on the entire binding region required the use of the FASTER algorithm to achieve convergence.58 A combination of energy analysis and visual inspection of the predicted low-energy conformations was used to identify promising mutations.
Mutagenesis, protein purification, and crystallization
As described previously,15,59 HIV protease variants were expressed from a pET11a Escherichia coli plasmid vector. The gene construct coded for two copies of the HIV monomer linked by the nucleotide sequence that codes for Gly-Gly-Ser-Ser-Gly.48,51 The nucleotide sequence that coded for each monomer was unique; the use of two different sequences allowed for site-directed mutagenesis to be targeted to a specific monomer. Cysteines at positions 67, 95, 167, and 195 were mutated to Leu, Met, Leu, and Met, respectively. The QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used to introduce the D25N substitution to inactivate the designed variants for ITC and crystallography studies.
HIV protease variants were expressed in 2-L cultures of E. coli BL21(pLysS) cells at 37°C. Protein expression was initiated at an OD600 of 0.6 by adding IPTG. Cells were harvested after 3 h and lysed using an emulsiflex. The active and inactive variants of the designed proteases were purified and refolded by two different methods. For active variants, the inclusion bodies were isolated and resuspended in 66% acetic acid and diluted 10-fold in water. The soluble fraction was isolated after centrifugation and dialyzed overnight against 100% water. Any precipitate was removed and the resulting sample was purified using cation exchange chromatography. The pure sample was desalted and lyophilized. Active enzyme was produced by taking the lypholized sample in 8M guanidinium and refolding the protein at 0.6 mg mL−1 in 55 mM Tris pH 8.2, 10.56 mM NaCl, 0.44 mM KCl, 0.055% PEG 3350, 550 mM guanidine HCl, 1.1 mM EDTA, 440 mM sucrose, and 1 mM DTT at 0°C. For inactive variants, the inclusion bodies were isolated and resuspended in 50% acetic acid. The soluble fraction was isolated after centrifugation and the resulting sample was purified using Superdex75 gravity gel filtration chromatography in 50% acetic acid. The purified inactive protease, in 50% acetic acid, was then refolded by rapid 10-fold dilution into the refolding buffer, containing 0.05M sodium acetate (pH 5.5), 5% ethylene glycol, 10% glycerol, and 5 mM DTT, over ice. The refolded and diluted sample was concentrated and dialyzed to remove residual acetic acid. Protease intended for crystallization was further purified on a Pharmacia Superdex75 FPLC column equilibrated with refolding buffer. Crystals were grown by the hanging drop vapor diffusion method at room temperature over a reservoir solution consisting of 126 mM phosphate buffer (pH 6.2), 63 mM sodium citrate and 18–24% ammonium sulphate. The protein concentrations used were ∼1.5–2 mg mL−1. The substrates were crystallized in 5–10 molar excess. Substrate stocks were made in DMSO. Small crystals started appearing after about 6–9 days and grew over time at room temperature.
Protease kinetics
Kinetics of protease activity were determined using three DABCYL/EDANS substrates: NH2-D(Edans)-KARVLAEAM-K(Dabcyl)-R-COOH, NH2-D(Edans)-ATIMMQRGN-K(Dabcyl)-R-COOH, and NH2-D(Edans)-AETFYVDGA-K(Dabcyl)-R-COOH, which are derivatives of CA-p2, p2-NC, and RT-RH substrates, respectively.60 Hydrolysis was monitored at 490 nm while exciting at 340 nm in a PTI fluorimeter. The reaction buffer was composed of 0.1M sodium acetate, 0.1M NaCL, 1 mM EDTA, 1 mM DTT, 1 mg mL−1 BSA, and 10% DMSO at a pH of 4.7.
Isothermal titration calorimetry
A VP-ITC (MicroCal, Northampton, MA) isothermal titration calorimeter was used to determine the thermodynamic parameters of peptide binding to the designed proteases. The buffer used for all protease and peptide solutions consisted of 10 mM sodium acetate pH 5.0, 2% DMSO, and 2 mM Tris (2-carboxyethyl) phospine (TCEP). A 750 μM solution of RT-RH was directly titrated into solutions containing 45–67 μM of the D25N (inactive) variants of WT protease and Pr3, respectively. The experiments were performed at 15°C and were repeated at 10 and 25°C for the WT protease, as at 15°C the calculated enthalpy of binding was zero. Each experiment was repeated at least in triplicate and data were processed using the Origin 7 software package from MicroCal.
Crystallographic data collection and processing
Protein crystals were harvested and flash frozen by dipping in liquid nitrogen. All data was collected under cryo conditions from various sources, including an in-house Rigaku X-ray generator with an R-axis IV image plate, a synchrotron radiation source at Argonne National Laboratory (APS, Chicago, IL) BioCARS 14-BMC, and a synchrotron radiation source at Brookhaven National Laboratory (BNL, Upton, NY) X-25 through a mail-in crystal program. Diffraction images were indexed and scaled using HKL2000.61 Complete data collection statistics are listed in (Table IV).
Structure solution and crystallographic refinement
Crystal structures were solved and refined using the programs within the CCP4I suite.62 Structure solution was carried out by molecular replacement using PHASER.63 Molecular replacement phases were further improved by building solvent molecules using ARP/warp.64,65 Refinement was carried out with REFMAC 566,67 in the CCP4 suite with cycles of restrained and TLS (translation, libration, screw rotation) refinement.68 A free R value with 5% of the data was used to limit the possibility of over refinement. Electron density was viewed and interactive model building was carried out by using program COOT.69 Refinement statistics are also shown in (Table IV).
Analysis and comparison to experimental structures
Graphical visualization and analysis was carried out using PyMOL (Delano Scientific LLC).70 All figures were also generated in PyMOL.
Conclusions
A computational design approach was applied to screen the factors determining the substrate binding specificity of HIV-1 protease. Positive design was used to redesign the protease to preferentially bind to one of its natural substrates, RT–RH, relative to two other substrates, p2-NC and CA-p2. Based on this design strategy, one of the predicted protease variants was expressed, purified, and tested experimentally. While the binding and kinetic data agreed well with the design, the predictions were only partially realized in the crystal structures. The crystallographic analysis was complicated by the flexibility of the flaps and the averaged orientation of the molecules in the crystal lattice, which generated complex averaged density maps. The internal twofold symmetry in the primary sequence of RT–RH further complicated the structural analysis of RT–RH complexes and hence the analysis of predicted versus observed interactions. The charged substitution, G48R, exhibited an unpredicted effect on the specificity of protease–substrate interactions, likely due to decreased backbone flexibility but increased conformational flexibility of extended charged side chain. With the homodimeric HIV protease accommodating the design of asymmetry, the positive design method was rendered partially ineffective in predicting the observed substrate binding orientation. Thus, the crystal structures partly supported the design and revealed an unanticipated influence of protease flexibility on the designed protease structure.
Predicting the true impact of introduced hydrogen bonds and van der Waals clashes is difficult in a computational protein design procedure that requires the use of a fixed backbone. Proteins are intrinsically dynamic and can adapt to mutations to prevent van der Waals violations. With the fixed backbone restriction, there is no means of modeling protein motions that might accommodate unfavorable van der Waals energies. In addition, the discrete nature of the rotamer library can exclude side-chain conformations that might prevent atomic clashes. Minimization and the use of multiple crystal structures while useful, quickly becomes computationally expensive, when performed at every step. A good alternative to minimization is the parallel design of multiple static backbone structures that represent a protein's dynamic range. Future studies in computational protein design will most likely incorporate methods that are more accurate than the current techniques in predicting backbone conformations in remodeled proteins. Our knowledge of the adaptability of enzymes and ability to model specificity is very limited, and the complete parameterization of protein flexibility in the known design algorithms is still challenging. The intent of this study was to improve and build upon the current successes of computational design as a tool with enhanced predictive potential. Such computational methodology may be more broadly applicable to other systems, and enable evaluating in silico the basis of fine tuned multi-substrate specificity in proteins.
In summary, this work on understanding how the scaffold of HIV protease provides specificity is of more fundamental significance in defining determinants of protein specificities in general. The results of this study not only present a successful attempt at computationally altering the specificity of HIV protease but also highlight the limitations of the applied algorithms and offer new insights into the structural and conformational flexibility determinants of substrate specificity in HIV-1 protease.
Acknowledgments
The authors thank Dr. Vukica Srajer at BioCARS, sector 14 Advanced Photon Source at Argonne National Laboratory for help with data collection, Dr. William E. Royer and Dr. Madhavi N. Nalam for assistance with initial refinement, and Dr. Nese Kurt Yilmaz and Ms. Marie Ary for editorial assistance. They thank Annie Heroux, beam line scientist at the Macromolecular Crystallography Research Resource (PXRR) of the Brookhaven National Laboratory, for collecting some of the data at beamline X25 of the National Synchrotron Light Source through the mail-in crystal program.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Krausslich HG, Ingraham RH, Skoog MT, Wimmer E, Pallai PV, Carter CA. Activity of purified biosynthetic proteinase of human immunodeficiency virus on natural substrates and synthetic peptides. Proc Natl Acad Sci USA. 1989;86:807–811. doi: 10.1073/pnas.86.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner BG, Summers MF. Structural biology of HIV. J Mol Biol. 1999;285:1–32. doi: 10.1006/jmbi.1998.2354. [DOI] [PubMed] [Google Scholar]
- 3.de Oliveira T, Engelbrecht S, Janse van Rensburg E, Gordon M, Bishop K, zur Megede J, Barnett SW, Cassol S. Variability at human immunodeficiency virus type 1 subtype C protease cleavage sites: an indication of viral fitness? J Virol. 2003;77:9422–9430. doi: 10.1128/JVI.77.17.9422-9430.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber IT, Wu J, Adomat J, Harrison RW, Kimmel AR, Wondrak EM, Louis JM. Crystallographic analysis of human immunodeficiency virus 1 protease with an analog of the conserved CA-p2 substrate—interactions with frequently occurring glutamic acid residue at P2′ position of substrates. Eur J Biochem. 1997;249:523–530. doi: 10.1111/j.1432-1033.1997.00523.x. [DOI] [PubMed] [Google Scholar]
- 5.Mahalingam B, Louis JM, Hung J, Harrision RW, Weber IT. Structural implications of drug-resistant mutants of HIV-1 protease: high-resolution crystal structures of the mutant protease/substrate analogue complexes. Proteins. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 6.Tie Y, Boross PI, Wang YF, Gaddis L, Liu F, Chen X, Tozser J, Harrison RW, Weber IT. Molecular basis for substrate recognition and drug resistance from 1.1 to 1.6 angstroms resolution crystal structures of HIV-1 protease mutants with substrate analogs. FEBS J. 2005;272:5265–5277. doi: 10.1111/j.1742-4658.2005.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pettit SC, Lindquist JN, Kaplan AH, Swanstrom R. Processing sites in the human immunodeficiency virus type 1 (HIV-1) Gag-Pro-Pol precursor are cleaved by the viral protease at different rates. Retrovirology. 2005;2:66. doi: 10.1186/1742-4690-2-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moody MD, Pettit SC, Shao W, Everitt L, Loeb DD, Hutchison CA, III, Swanstrom R. A side chain at position 48 of the human immunodeficiency virus type-1 protease flap provides an additional specificity determinant. Virology. 1995;207:475–485. doi: 10.1006/viro.1995.1107. [DOI] [PubMed] [Google Scholar]
- 9.Martinez-Picado J, Savara AV, Sutton L, D'Aquila RT. Replicative fitness of protease inhibitor-resistant mutants of human immunodeficiency virus type 1. J Virol. 1999;73:3744–3752. doi: 10.1128/jvi.73.5.3744-3752.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Croteau G, Doyon L, Thibeault D, McKercher G, Pilote L, Lamarre D. Impaired fitness of human immunodeficiency virus type 1 variants with high-level resistance to protease inhibitors. J Virol. 1997;71:1089–1096. doi: 10.1128/jvi.71.2.1089-1096.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. How does a symmetric dimer recognize an asymmetric substrate? A substrate complex of HIV-1 protease. J Mol Biol. 2000;301:1207–1220. doi: 10.1006/jmbi.2000.4018. [DOI] [PubMed] [Google Scholar]
- 12.Prabu-Jeyabalan M, Nalivaika EA, King NM, Schiffer CA. Viability of a drug-resistant human immunodeficiency virus type 1 protease variant: structural insights for better antiviral therapy. J Virol. 2003;77:1306–1315. doi: 10.1128/JVI.77.2.1306-1315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prabu-Jeyabalan M, Nalivaika EA, Schiffer CA. Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure. 2002;10:369–381. doi: 10.1016/s0969-2126(02)00720-7. [DOI] [PubMed] [Google Scholar]
- 14.King NM, Prabu-Jeyabalan M, Nalivaika EA, Schiffer CA. Combating susceptibility to drug resistance: lessons from HIV-1 protease. Chem Biol. 2004;11:1333–1338. doi: 10.1016/j.chembiol.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Prabu-Jeyabalan M, Nalivaika EA, King NM, Schiffer CA. Structural basis for coevolution of a human immunodeficiency virus type 1 nucleocapsid-p1 cleavage site with a V82A drug-resistant mutation in viral protease. J Virol. 2004;78:12446–12454. doi: 10.1128/JVI.78.22.12446-12454.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolli M, Stawiski E, Chappey C, Schiffer CA. Human immunodeficiency virus type 1 protease-correlated cleavage site mutations enhance inhibitor resistance. J Virol. 2009;83:11027–11042. doi: 10.1128/JVI.00628-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cameron CE, Ridky TW, Shulenin S, Leis J, Weber IT, Copeland T, Wlodawer A, Burstein H, Bizub-Bender D, Skalka AM. Mutational analysis of the substrate binding pockets of the Rous sarcoma virus and human immunodeficiency virus-1 proteases. J Biol Chem. 1994;269:11170–11177. [PubMed] [Google Scholar]
- 18.Grinde B, Cameron CE, Leis J, Weber IT, Wlodawer A, Burstein H, Bizub D, Skalka AM. Mutations that alter the activity of the Rous sarcoma virus protease. J Biol Chem. 1992;267:9481–9490. [PubMed] [Google Scholar]
- 19.Grinde B, Cameron CE, Leis J, Weber IT, Wlodawer A, Burstein H, Skalka AM. Analysis of substrate interactions of the Rous sarcoma virus wild type and mutant proteases and human immunodeficiency virus-1 protease using a set of systematically altered peptide substrates. J Biol Chem. 1992;267:9491–9498. [PubMed] [Google Scholar]
- 20.Konvalinka J, Horejsi M, Andreansky M, Novek P, Pichova I, Blaha I, Fabry M, Sedlacek J, Foundling S, Strop P. An engineered retroviral proteinase from myeloblastosis associated virus acquires pH dependence and substrate specificity of the HIV-1 proteinase. EMBO J. 1992;11:1141–1144. doi: 10.1002/j.1460-2075.1992.tb05154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loeb DD, Swanstrom R, Everitt L, Manchester M, Stamper SE, Hutchison CA., III Complete mutagenesis of the HIV-1 protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- 22.Tozser J, Blaha I, Copeland TD, Wondrak EM, Oroszlan S. Comparison of the HIV-1 and HIV-2 proteinases using oligopeptide substrates representing cleavage sites in Gag and Gag-Pol polyproteins. FEBS Lett. 1991;281:77–80. doi: 10.1016/0014-5793(91)80362-7. [DOI] [PubMed] [Google Scholar]
- 23.Ohtaka H, Schon A, Freire E. Multidrug resistance to HIV-1 protease inhibition requires cooperative coupling between distal mutations. Biochemistry. 2003;42:13659–13666. doi: 10.1021/bi0350405. [DOI] [PubMed] [Google Scholar]
- 24.Gulnik SV, Suvorov LI, Liu B, Yu B, Anderson B, Mitsuya H, Erickson JW. Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry. 1995;34:9282–9287. doi: 10.1021/bi00029a002. [DOI] [PubMed] [Google Scholar]
- 25.King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, de Bethune MP, Schiffer CA. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol. 2004;78:12012–12021. doi: 10.1128/JVI.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bardi JS, Luque I, Freire E. Structure-based thermodynamic analysis of HIV-1 protease inhibitors. Biochemistry. 1997;36:6588–6596. doi: 10.1021/bi9701742. [DOI] [PubMed] [Google Scholar]
- 27.Luque I, Todd MJ, Gomez J, Semo N, Freire E. Molecular basis of resistance to HIV-1 protease inhibition: a plausible hypothesis. Biochemistry. 1998;37:5791–5797. doi: 10.1021/bi9802521. [DOI] [PubMed] [Google Scholar]
- 28.Velazquez-Campoy A, Muzammil S, Ohtaka H, Schon A, Vega S, Freire E. Structural and thermodynamic basis of resistance to HIV-1 protease inhibition: implications for inhibitor design. Curr Drug Targets Infect Disord. 2003;3:311–328. doi: 10.2174/1568005033481051. [DOI] [PubMed] [Google Scholar]
- 29.Wells JA, Powers DB, Bott RR, Graycar TP, Estell DA. Designing substrate specificity by protein engineering of electrostatic interactions. Proc Natl Acad Sci USA. 1987;84:1219–1223. doi: 10.1073/pnas.84.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shifman JM, Mayo SL. Exploring the origins of binding specificity through the computational redesign of calmodulin. Proc Natl Acad Sci USA. 2003;100:13274–13279. doi: 10.1073/pnas.2234277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shifman JM, Mayo SL. Modulating calmodulin binding specificity through computational protein design. J Mol Biol. 2002;323:417–423. doi: 10.1016/s0022-2836(02)00881-1. [DOI] [PubMed] [Google Scholar]
- 32.Green DF, Dennis AT, Fam PS, Tidor B, Jasanoff A. Rational design of new binding specificity by simultaneous mutagenesis of calmodulin and a target peptide. Biochemistry. 2006;45:12547–12559. doi: 10.1021/bi060857u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reina J, Lacroix E, Hobson SD, Ballester G, Rybin V, Schwab MS, Serrano L, Gonzalez C. Computer-aided design of a PDZ domain to recognize new target sequences. Nat Struct Biol. 2002;9:621–627. doi: 10.1038/nsb815. [DOI] [PubMed] [Google Scholar]
- 34.Bolon DN, Grant RA, Baker TA, Sauer RT. Specificity versus stability in computational protein design. Proc Natl Acad Sci USA. 2005;102:12724–12729. doi: 10.1073/pnas.0506124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joachimiak LA, Kortemme T, Stoddard BL, Baker D. Computational design of a new hydrogen bond network and at least a 300-fold specificity switch at a protein–protein interface. J Mol Biol. 2006;361:195–208. doi: 10.1016/j.jmb.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Kurth T, Grahn S, Thormann M, Ullmann D, Hofmann HJ, Jakubke HD, Hedstrom L. Engineering the S1′ subsite of trypsin: design of a protease which cleaves between dibasic residues. Biochemistry. 1998;37:11434–11440. doi: 10.1021/bi980842z. [DOI] [PubMed] [Google Scholar]
- 37.Kurth T, Ullmann D, Jakubke HD, Hedstrom L. Converting trypsin to chymotrypsin: structural determinants of S1′ specificity. Biochemistry. 1997;36:10098–10104. doi: 10.1021/bi970937l. [DOI] [PubMed] [Google Scholar]
- 38.Hung SH, Hedstrom L. Converting trypsin to elastase: substitution of the S1 site and adjacent loops reconstitutes esterase specificity but not amidase activity. Protein Eng. 1998;11:669–673. doi: 10.1093/protein/11.8.669. [DOI] [PubMed] [Google Scholar]
- 39.Page MJ, Wong SL, Hewitt J, Strynadka NC, MacGillivray RT. Engineering the primary substrate specificity of Streptomyces griseus trypsin. Biochemistry. 2003;42:9060–9066. doi: 10.1021/bi0344230. [DOI] [PubMed] [Google Scholar]
- 40.Hedstrom L, Szilagyi L, Rutter WJ. Converting trypsin to chymotrypsin: the role of surface loops. Science. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 41.Wilson C, Mace JE, Agard DA. Computational method for the design of enzymes with altered substrate specificity. J Mol Biol. 1991;220:495–506. doi: 10.1016/0022-2836(91)90026-3. [DOI] [PubMed] [Google Scholar]
- 42.Prabu-Jeyabalan M, Nalivaika EA, Romano K, Schiffer CA. Mechanism of substrate recognition by drug-resistant human immunodeficiency virus type 1 protease variants revealed by a novel structural intermediate. J Virol. 2006;80:3607–3616. doi: 10.1128/JVI.80.7.3607-3616.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das R, Baker D. Macromolecular modeling with rosetta. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 44.Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 45.Huang P-S, Ban Y-EA, Richter F, Andre I, Vernon R, Schief WR, Baker D. RosettaRemodel: a generalized framework for flexible backbone protein design. PLoS One. 2011;6:e24109. doi: 10.1371/journal.pone.0024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park S, Morley KL, Horsman GP, Holmquist M, Hult K, Kazlauskas RJ. Focusing mutations into the P. fluorescens esterase binding site increases enantioselectivity more effectively than distant mutations. Chem Biol. 2005;12:45–54. doi: 10.1016/j.chembiol.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 47.Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 48.Bhat TN, Baldwin ET, Liu B, Cheng YS, Erickson JW. Crystal structure of a tethered dimer of HIV-1 proteinase complexed with an inhibitor. Nat Struct Biol. 1994;1:552–556. doi: 10.1038/nsb0894-552. [DOI] [PubMed] [Google Scholar]
- 49.Bhat TN, Baldwin ET, Liu B, Cheng YS, Erickson JW. X-ray structure of a tethered dimer for HIV-1 protease. Adv Exp Med Biol. 1995;362:439–444. doi: 10.1007/978-1-4615-1871-6_56. [DOI] [PubMed] [Google Scholar]
- 50.Das A, Rao DR, Hosur MV. X-ray structure of HIV-1 protease tethered dimer complexed to ritonavir. Protein Pept Lett. 2007;14:565–568. doi: 10.2174/092986607780989930. [DOI] [PubMed] [Google Scholar]
- 51.Cheng YS, Yin FH, Foundling S, Blomstrom D, Kettner CA. Stability and activity of human immunodeficiency virus protease: comparison of the natural dimer with a homologous, single-chain tethered dimer. Proc Natl Acad Sci USA. 1990;87:9660–9664. doi: 10.1073/pnas.87.24.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altman MD, Nalivaika EA, Prabu-Jeyabalan M, Schiffer CA, Tidor B. Computational design and experimental study of tighter binding peptides to an inactivated mutant of HIV-1 protease. Proteins. 2008;70:678–694. doi: 10.1002/prot.21514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dahiyat BI, Gordon DB, Mayo SL. Automated design of the surface positions of protein helices. Protein Sci. 1997;6:1333–1337. doi: 10.1002/pro.5560060622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dahiyat BI, Mayo SL. Probing the role of packing specificity in protein design. Proc Natl Acad Sci USA. 1997;94:10172–10177. doi: 10.1073/pnas.94.19.10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 56.Dahiyat BI, Sarisky CA, Mayo SL. De novo protein design: towards fully automated sequence selection. J Mol Biol. 1997;273:789–796. doi: 10.1006/jmbi.1997.1341. [DOI] [PubMed] [Google Scholar]
- 57.Gordon DB, Hom GK, Mayo SL, Pierce NA. Exact rotamer optimization for protein design. J Comput Chem. 2003;24:232–243. doi: 10.1002/jcc.10121. [DOI] [PubMed] [Google Scholar]
- 58.Desmet J, Spriet J, Lasters I. Fast and accurate side-chain topology and energy refinement (FASTER) as a new method for protein structure optimization. Proteins. 2002;48:31–43. doi: 10.1002/prot.10131. [DOI] [PubMed] [Google Scholar]
- 59.Hui JO, Tomasselli AG, Reardon IM, Lull JM, Brunner DP, Tomich CS, Heinrikson RL. Large scale purification and refolding of HIV-1 protease from Escherichia coli inclusion bodies. J Protein Chem. 1993;12:323–327. doi: 10.1007/BF01028194. [DOI] [PubMed] [Google Scholar]
- 60.Matayoshi ED, Wang GT, Krafft GA, Erickson J. Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science. 1990;247:954–958. doi: 10.1126/science.2106161. [DOI] [PubMed] [Google Scholar]
- 61.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 62.The CCP4 suite. The CCP4 suite: programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 63.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morris RJ, Perrakis A, Lamzin VS. ARP/wARP's model-building algorithms. I. The main chain. Acta Cryst. 2002;D58:968–975. doi: 10.1107/s0907444902005462. [DOI] [PubMed] [Google Scholar]
- 65.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Potterton L, McNicholas S, Krissinel E, Gruber J, Cowtan K, Emsley P, Murshudov GN, Cohen S, Perrakis A, Noble M. Developments in the CCP4 molecular-graphics project. Acta Cryst. 2004;D60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 67.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 68.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Cryst. 2006;D62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 69.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 70.DeLano WL. The PyMol user's manual. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.