

Abstract
Reactive oxygen species (ROS) are mutagenic and may thereby promote cancer1. Normally, ROS levels are tightly controlled by an inducible antioxidant program that responds to cellular stressors and is predominantly regulated by the transcription factor Nrf2 and its repressor protein Keap12-5. In contrast to the acute physiological regulation of Nrf2, in neoplasia there is evidence for increased basal activation of Nrf2. Indeed, somatic mutations that disrupt the Nrf2-Keap1 interaction to stabilize Nrf2 and increase the constitutive transcription of Nrf2 target genes were recently identified, suggesting that enhanced ROS detoxification and additional Nrf2 functions may in fact be pro-tumorigenic6. Here, we investigated ROS metabolism in primary murine cells following the expression of endogenous oncogenic alleles of K-Ras, B-Raf and Myc, and find that ROS are actively suppressed by these oncogenes. K-RasG12D, B-RafV619E and MycERT2 each increased the transcription of Nrf2 to stably elevate the basal Nrf2 antioxidant program and thereby lower intracellular ROS and confer a more reduced intracellular environment. Oncogene-directed increased expression of Nrf2 is a novel mechanism for the activation of the Nrf2 antioxidant program, and is evident in primary cells and tissues of mice expressing K-RasG12D and B-RafV619E, and in human pancreatic cancer. Furthermore, genetic targeting of the Nrf2 pathway impairs K-RasG12D-induced proliferation and tumorigenesis in vivo. Thus, the Nrf2 antioxidant and cellular detoxification program represents a previously unappreciated mediator of oncogenesis.
To examine the role of ROS in cellular transformation and tumorigenesis, we utilized an endogenous and conditional oncogenic LSL-K-RasG12D allele7. K-RasG12D/+ and K-RasLSL/+ MEFs were compared to MEFs and NIH3T3 fibroblasts (NIH3T3s) transduced with K-Ras4BG12D and H-RasV12 (Fig. 1a), since ectopic Ras introduction was previously reported to increase ROS production8. In contrast to ectopic overexpression of oncogenic Ras, K-RasG12D/+ MEFs demonstrated lower levels of hydrogen peroxide, superoxide, and mitochondrial ROS compared to K-RasLSL/+ MEFs (Fig. 1a, Supplementary Fig. 2a,b). Accordingly, the level of 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxo-dGuo), one of the major products of DNA oxidation, was decreased in MEFs expressing endogenous K-RasG12D/+, but increased by ectopic oncogenic Ras (Fig. 1b). The levels of ROS and 8-oxo-dGuo were also lower in p53-/-; K-RasG12D/+ MEFs compared to p53-/-; K-RasLSL/+ MEFs (Supplementary Fig. 2c,d), supporting that these differences are not due to differential activation of a senescence program and demonstrating that increased ROS production is not required for full transformation7,9.
Figure 1. Physiological expression of oncogenes lowers ROS.
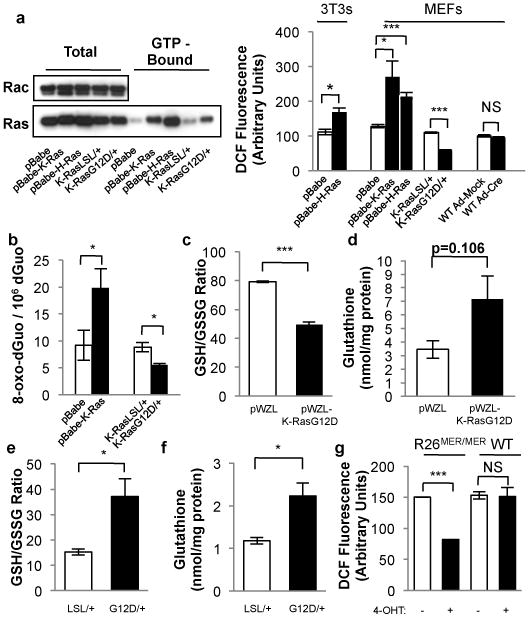
NIH3T3s and MEFs were transduced with retroviral vectors and evaluated 6 days later: control vector (pBabe), pBabe-H-RasG12V (p-Babe-H-Ras), or pBabe-K-RasG12D (p-Babe-K-Ras). Alternatively, LSL-K-RasG12D MEFs were infected with Ad-mock (K-RasLSL/+) or Ad-cre (K-RasG12D/+) and evaluated 4 days later. Wild-type MEFs were infected with Ad-mock (WT Ad-Mock) or Ad-cre (WT Ad-Cre) and used as controls. a, (Left) Western blot of total and GTP-bound Ras in MEFs expressing endogenous and ectopic Ras, with Rac used as a loading control. (Right) ROS levels following expression of oncogenic Ras, as determined by 2′,7′-dichlorofluorescein diacetate (DCF) staining. b, 8-oxo-dGuo levels following ectopic and endogenous expression of K-RasG12D. c-f, Determination of the GSH/GSSG ratios and total cellular glutathione in cells overexpressing ectopic K-RasG12D (c,d), or expressing endogenous K-RasG12D (e,f). g, ROS levels following activation of MycERT2. R26MER/MER MEFs were treated with DMSO or 100nM 4-OHT and assayed after 24 hours. Data are representative of 3 or more independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 and error bars represent ± SEM here and for all figures.
Because alterations of ROS can affect the intracellular redox state10, the ratio of reduced/oxidized glutathione (GSH/GSSG) was examined in cells expressing ectopic and endogenous K-RasG12D. While both elevated the level of total glutathione (Fig. 1d,f and ref. 11), only the endogenous expression of K-RasG12D elevated the GSH/GSSG ratio to promote a more reduced intracellular environment (Fig. 1c,e). The production of ROS by ectopic oncogenic Ras is chiefly regulated by NADPH-oxidase (Nox)12. Only the ectopic overexpression of oncogenic Ras increased Nox activity and mRNA, providing a mechanistic explanation for these differing results (Supplementary Fig. 3). Similar to oncogenic Ras, ectopic expression of Myc has also been shown to increase the production of ROS in NIH3T3 fibroblasts13 and accordingly we sought to determine whether near-physiological expression of this oncogene had a similar effect. Treatment of R26MER/MER MEFs, which express a homozygous MycERT2 allele under the control of the Rosa26 promoter14, with 4-OHT resulted in a substantial reduction of ROS (Fig. 1g). However, this was not a characteristic feature of all oncogenic stimuli as expression of activated Notch1 and β-catenin did not lower ROS (Supplementary Fig. 4). Therefore, endogenous expression of oncogenic K-Ras or MycERT2 lowers the level of cellular ROS, in contrast to the ectopic overexpression of these oncogenes.
As the intracellular redox state is chiefly regulated by Nrf2, we examined Nrf2 protein levels and activity in K-RasLSL/+ and K-RasG12D/+ MEFs by western blot, reporter assay and chromatin immunoprecipitation analysis of the Nrf2 target genes Hmox1 and Nqo1. Expression of K-RasG12D resulted in an approximately two-fold increase in Nrf2 protein and binding activity compared to control MEFs (Fig. 2a,b, Supplementary Fig. 5a), which was not attributable to altered regulation by Keap12 (Supplementary Fig. 6). K-RasG12D/+ MEFs demonstrated increased Nrf2 mRNA and increased expression of Hmox1, Nqo1, Gclc, Gclm and Ggt1 mRNA and protein (Fig. 2c, Supplementary Fig. 5b). Increased expression of Nrf2 and its target genes was also observed following K-RasG12D expression in p53-/- MEFs, and following ectopic expression of K-RasG12D and H-RasV12 in primary MEFs, but not following expression of activated Notch1 or β-catenin (Supplementary Figs. 5c-g). However, expression of K-RasG12D in Nrf2-deficient MEFs failed to elevate total glutathione and resulted in a more oxidized intracellular environment (Fig. 2d,e). Neither the cell culture conditions employed to express K-RasG12D nor the gene dosage of wild-type K-Ras affected the expression of Nrf2 target genes (Supplementary Fig. 7a,b). Additionally, ROS metabolism in wild-type MEFs was sensitive to acute changes in the levels of Keap1 and Nrf2, further supporting a causal relationship between Nrf2 and ROS (Supplementary Fig. 7c-e). Furthermore, acute knockdown of Nrf2 attenuated the reduction in ROS by K-RasG12D (Fig. 2f), and the effects of Nrf2 depletion on ROS were dosage-dependent, supporting the importance of the level of Nrf2 mRNA for ROS control (Supplementary Fig. 7f,g). Similar to K-RasG12D, activation of c-MycERT2 (with 4-OHT) promoted an increase in the mRNA and protein levels of Nrf2 and its target genes (Fig. 2g,h, Supplementary Fig. 7h). Furthermore, ChIP-seq data from the ENCODE consortium demonstrated direct binding of Myc to the Nrf2 locus (Supplementary Fig. 8a)15. Therefore, the K-Ras and Myc oncogenes can constitutively increase the transcription of Nrf2 to elevate the basal activity of the antioxidant and cellular detoxification program.
Figure 2. Physiological expression of oncogenes activates the Nrf2 antioxidant program.
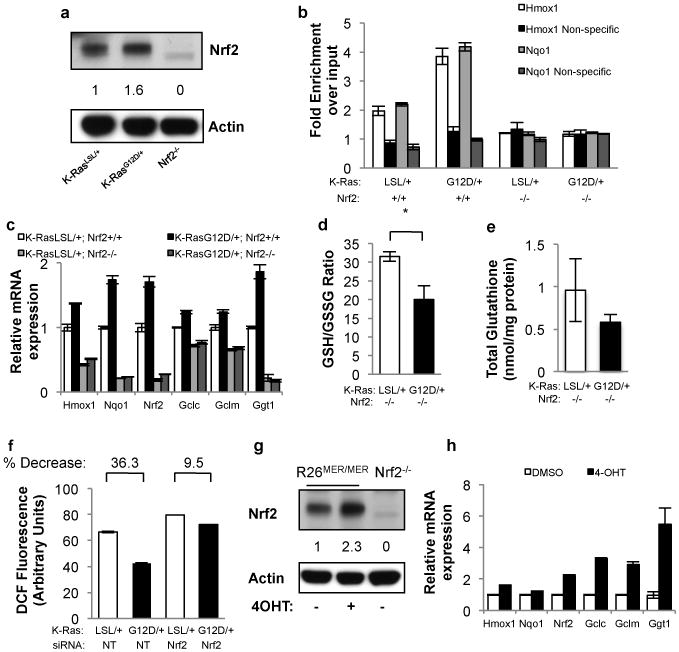
a, Western blot demonstrates a 60% increase in Nrf2 protein following expression of endogenous K-RasG12D. Antibody specificity was confirmed using Nrf2-/- MEFs. b, Nrf2 ChIP followed by q-PCR for the Hmox1 and Nqo1 promoters. Control non-specific primers amplified regions of DNA located 50Kb from the Hmox1 and Nqo1 promoters. c, Expression of Nrf2 and Nrf2 target genes Nqo1, Hmox1, Gclm, Gclc and Ggt1 upon K-RasG12D expression in Nrf2+/+ and Nrf2-/- MEFs. Nrf2 mRNA is relatively unstable but still detectable at low levels in Nrf2-/- MEFs. d-e, Determination of the GSH/GSSG ratio (d) and total glutathione (e) upon K-RasG12D expression in Nrf2-/- MEFs. f, ROS levels following Nrf2 depletion with siRNA. LSL-K-RasG12D MEFs were transfected with non-targeting (NT) or Nrf2 siRNA, infected with Ad-mock or Ad-cre and assayed after 48 hours for DCF oxidation. g, Western blot of Nrf2 protein levels following induction of MycERT2 by 4-OHT. Densitometry shows a 2.3-fold increase. h, Analysis of Nrf2 antioxidant program gene expression following activation of MycERT2. R26MER/MER MEFs were treated with DMSO or 100nM 4-OHT for 24 hours and assayed for antioxidant gene expression. Data is representative of 3 independent experiments.
To investigate the mechanism of Nrf2 activation by K-RasG12D, the roles of the Raf/MEK/ERK and p38alpha MAPK pathways were investigated. First, cells were treated with a potent and specific inhibitor of MEK, AZD6244 (ARRY-142886) (Supplementary Fig. 9a-c), which restored the ROS level of K-RasG12D/+ cells nearly to the level of K-RasLSL/+ cells (Fig. 3a). Additionally, AZD6244 treatment resulted in decreased induction of Nrf2 and its target genes (Fig. 3b). Furthermore, endogenous expression of B-RafV619E (corresponding to human B-RafV600E)16 resulted in increased phospho-ERK levels, a decrease in ROS, and an increase in Nrf2 mRNA and antioxidant gene expression (Supplementary Fig. 9d-f). As previously reported17, we found that p38alpha MAPK kinase did not activate Nrf2 (Supplementary Fig. 9g-i). To determine the mechanism of increased Nrf2 expression, transcription factors downstream of MAPK signaling were examined. Accordingly, knockdown of Jun, Fra1, and Myc, but not JunD or Elk1, decreased the Nrf2 mRNA in K-RasG12D/+ cells, with almost complete rescue achieved with Jun (Fig. 3c). siRNA efficiency was confirmed by real-time PCR and western blot (Supplementary Fig. 10a,b). Importantly, K-RasG12D/+ MEFs and B-RafV619E MEFs demonstrated elevated Jun protein levels compared to control MEFs, and the elevated Jun level in K-RasG12D/+ MEFs was rescued by treatment with AZD6244 (Fig. 3d). Furthermore, depletion of Jun with siRNA prevented the decrease in ROS following expression of K-RasG12D (Fig. 3e, Supplementary Fig. 10c). Our results reinforce a prior finding that antioxidant response elements that closely resemble AP-1 sites help regulate the Nrf2 promoter18. Finally, ChIP-seq revealed that the transcriptional start site of the Nrf2 locus is a direct binding target of Jun (Supplementary Fig. 8b)15. Thus, K-RasG12D and B-RafV619E stimulate transcription of Nrf2 via Jun and Myc.
Figure 3. Activation of Nrf2 by K-RasG12D occurs via the Raf-MEK-ERK-Jun pathway.
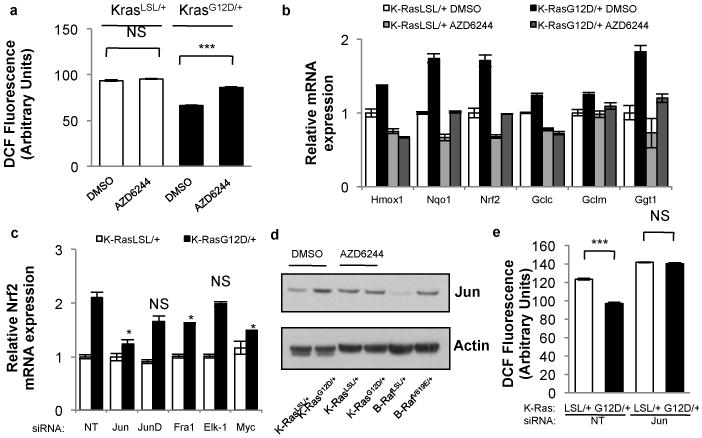
a, ROS levels following treatment of K-RasG12D/+ MEFs with AZD6244. LSL-K-RasG12D MEFs were treated with DMSO or 0.1uM AZD6244, infected with Ad-mock (K-RasLSL/+) or Ad-cre (K-RasG12D/+) and assayed after 72 hours. b, Analysis of antioxidant gene expression following treatment of K-RasLSL/+ and K-RasG12D/+ MEFs with AZD6244 for 24 hours. c, Control of Nrf2 transcription by AP-1 family members. K-RasLSL/+ and K-RasG12D/+ MEFs were transfected with siRNA and assayed for Nrf2 expression after 48 hours. d, Western blot of Jun and actin protein levels in LSL-K-RasG12D and LSL-B-RafV619E MEFs. K-RasLSL/+ and K-RasG12D/+ MEFs were treated with DMSO or 0.1uM AZD6244 for 24 hours. e, ROS levels following Jun depletion with siRNA. LSL-K-RasG12D MEFs were transfected with non-targeting (NT) or Jun siRNA, infected with Ad-mock or Ad-cre and assayed after 48 hours for DCF oxidation. Data are representative of 3 independent experiments.
We next examined whether these oncogenes promote activation of the Nrf2 antioxidant and cellular detoxification program in vivo. Elevated protein expression of the Nrf2 target gene Nqo1 and decreased immunoreactivity for 8-oxo-dGuo were evident in K-Ras mutant murine19 and human preinvasive pancreatic intraepithelial neoplasia (PanIN) and pancreatic ductal adenocarcinoma (PDA), and in murine lung adenomas expressing B-RafV619E (Fig. 4a, Supplementary Figs. 11a, 12, 13, 14a). PanIN also demonstrated reduced immunoreactivity for the lipid peroxidation adduct malondialdehyde (MDA) (Supplementary Fig. 11b). Furthermore, K-RasG12D/+ pancreatic epithelial cells demonstrated activation of Nrf2 and decreased ROS compared to K-RasLSL/+, while treatment of human PDA tumor lines with K-Ras siRNA resulted in decreased Nrf2 and Nqo1 mRNA and an increase in ROS (Supplementary Fig. 15). The activation of Nrf2 in human pancreatic cancer could not be explained by somatic mutations in NRF2 or KEAP1; we sequenced over 100 samples of human PDA and identified only one case with a concomitant KEAP1 and K-RAS mutation (Supplemental material and Supplementary Fig. 14b). Importantly, Nrf2-deficient murine PanIN were negative for Nqo1 and demonstrated similar levels of 8-oxo-dGuo and MDA in PanIN compared to neighboring morphologically normal ductal cells, supporting a role for Nrf2 in ROS detoxification in vivo (Fig. 4b, Supplementary Fig. 11b,c). Nrf2-deficient salivary gland did not demonstrate elevated 8-oxo-dGuo immunoreactivity, suggesting that K-RasG12D-expressing cells are more reliant on Nrf2 for ROS detoxification (Supplementary Fig. 16a). Therefore, an antioxidant program is operant during tumorigenesis, consistent with our findings that link oncogenic K-Ras and B-Raf expression with the activation of Nrf2.
Figure 4. Evidence for Nrf2 antioxidant program in pancreatic cancer.
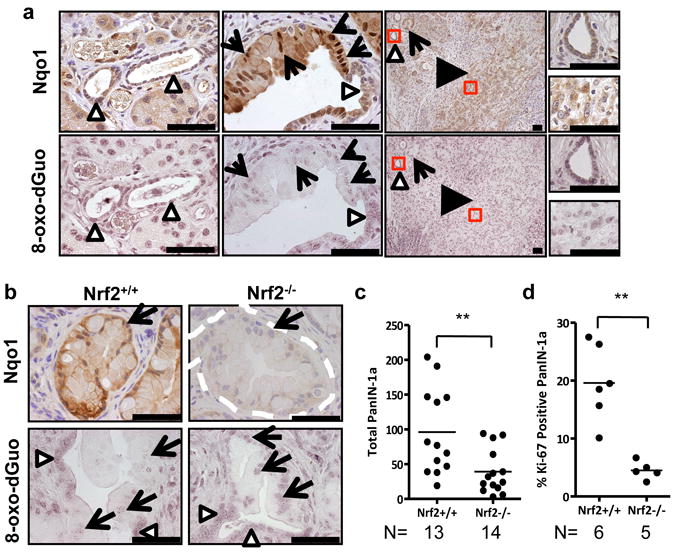
a, Immunohistochemical detection of Nqo1 (brown staining) and 8-oxo-dGuo (purple staining) in mouse PanIN and PDA (similar patterns observed for 11/11 of cases examined) in comparison to morphologically normal ducts. PanIN (arrows), PDA (black arrowheads), normal ducts (white arrowheads) here and for all figures. Scale bar = 56 μm. b, Immunohistochemical detection of Nqo1 and 8-oxo-dGuo in Nrf2-/- PanIN compared to Nrf2+/+ PanIN (similar patterns observed for 5/5 of each genotype examined, PanIN outlined by white dashes). Scale bar = 56 μm. c, Nrf2-/- and Nrf2+/+ PanIN-1a incidence. Whole pancreata were sectioned at 100-micron intervals and total numbers of PanIN-1a were counted. d, Proliferation of PanIN-1a cells in Nrf2-/- and Nrf2+/+ mice, as determined by Ki-67 immunostaining.
To investigate whether activation of Nrf2 promotes K-RasG12D–initiated carcinomas, the effects of Nrf2 ablation were examined in established mouse models of pancreatic and lung cancer. Nrf2-deficient pancreata were noted to contain fewer PanIN (Fig. 4c). While the proliferation of the salivary gland was unchanged (Supplementary Fig. 16b), PanIN from Nrf2-deficient mice were significantly less proliferative and demonstrated an increased content of cells exhibiting senescence-associated β-galactosidase activity (Fig. 4d, Supplementary Fig. 17a). No significant differences were observed in PanIN cell apoptosis or DNA damage (Supplementary Fig. 17b,c). Senescence was also observed in K-RasG12D/+ MEFs following acute depletion of Nrf2 with siRNA and was dependent on expression of p53 (Supplementary Fig. 17d). The Nrf2 dependent proliferation defect noted in PanIN was also observed in MEFs, and both could be rescued by the antioxidant N-acetyl cysteine (Supplementary Fig. 18a,b). Furthermore, treatment of Nrf2+/+ PanIN mice with a glutathione synthesis inhibitor resulted in a significant decrease in the proliferation of PanIN but not adjacent acinar cells (Supplementary Fig. 18c-e). Additionally, the role of Nrf2 in lung cancer development was investigated. Nrf2 deficiency resulted in a significant reduction in disease burden and proliferation, and an increase in median survival (Supplementary Fig. 19). While we cannot exclude the potential involvement of non-cell autonomous effects arising from constitutive Nrf2 deficiency, the major impact appeared to be restricted to tumor initiation and proliferation of nascent preneoplastic cells. These results demonstrate that Nrf2 promotes K-RasG12D-initiated pancreatic and lung tumorigenesis and proliferation.
ROS can stimulate tumorigenesis through the oxidation of DNA and subsequent mutation of genes that promote carcinogenesis. In contrast, we found that activating an ROS-detoxification program contributed to tumorigenesis, although our study does not exclude a role for other Nrf2 targets such as drug-metabolizing enzymes and efflux pumps, heat shock proteins, 26S proteasome subunits, growth factors and receptors6. Future studies will be necessary to determine the role of these Nrf2 functions in tumors with activated Nrf2. Increased expression of Nrf2 target genes and increased stability of Nrf2 caused by somatic mutations in Nrf2 and Keap1 are well documented in human cancer, providing one mechanism for enhanced Nrf2 activity during tumorigenesis. Our work describes oncogenic signaling as an alternative mechanism to activate Nrf2 transcription during tumorigenesis (Supplementary Fig. 1), and suggests that modulation of the redox state is uniformly important in cancer and may therefore represent a therapeutic opportunity. Thus, constitutively elevated Nrf2 activity in cancer cells occurs through two distinct mechanisms: diminished Nrf2 turnover and augmented Nrf2 mRNA levels.
Methods
Chromatin Immunoprecipitation
Chromatin Immunoprecipitation was performed as described25. 5×106 cells were fixed at 37°C in DMEM + 1% Formaldehyde for 10 minutes and lysed in 1% SDS, 10mM EDTA, 50 mM Tris-HCl pH 8.1 plus protease inhibitors and sonicated for 20 minutes in a cold water bath until DNA was an average size of 1Kb. Input was saved and lysate was diluted in IP buffer (1% Triton, 2mM EDTA, 150mM NaCl, 20 mM Tris-HCl pH 8.1) and mixed with beads (Dynal Protein A, Invitrogen) that were pre-bound overnight with Nrf2 antibody (H-300, Santa Cruz). Chromatin was immunoprecipitated overnight, and beads were washed 6 times with RIPA buffer (50mM HEPES pH 7.6, 1mM EDTA, 0.7% Na deoxycholate, 1% NP-40, 0.5M LiCl) and twice with TE. Beads were incubated with 1% SDS, 0.1M NaHCO3 for 30 minutes at room temperature, and then crosslinks were reversed on both the input and the IP by heating overnight in a 65°C water bath. DNA was purified with a QIAquick spin kit (Qiagen) and Q-PCR was performed in triplicate with power sybr mastermix (Applied Biosystems) on an Applied Biosystems 7900HT with the following primers26,27: Nqo1-F 5′-GCAGTTTCTAAGAGCAGAAC-3′, Nqo1-R 5′-GTAGATTAGTCCTCACTCAGCCG-3′, Nqo1 Non-specific-F 5′-AGGAGATGGAAGGCAGGAAG-3′, Nqo1 Non-specific-R 5′-GGGCGCACTATTGTCATCTT-3′, Hmox-1-F 5′-GGGCTAGCATGCGAAGTGAG-3′, Hmox-1-R 5′-AGACTCCGCCCTAAGGGTTC-3′, Hmox-1 Non-specific-F 5′-GGCAGGTATGGACCTTCAAA-3′, Hmox-1 Non-specific-R 5-AAAGGAGTCAGGGAGGGAGA-3′.
ChIP-seq (ENCODE)
These data were generated and analyzed by the labs of Michael Snyder, Mark Gerstein and Sherman Weissman at Yale University; Peggy Farnham at UC Davis; and Kevin Struhl at Harvard using the K562 cell line. The following antibodies were used: c-Jun (sc-1694, Santa Cruz), c-Myc (sc-764, Santa Cruz). The data can be accessed at http://genome.ucsc.edu/ENCODE/
Human pancreatic cancer cell culture
MiaPaCa2 and Hs766T cells were obtained from CRUK Clare Hall Laboratories. Both lines were grown in DMEM + 10% FBS. siRNA transfections were performed with DharmaFECT 1.
Immunohistochemistry
Human tissue was donated for research purposes by patients undergoing pancreatic surgery at a tertiary hepatopancreaticobiliary referral center (Addenbrookes Teaching Hospitals NHS Trust, Cambridge, UK). Ethical approval was granted by the local research and ethics committee (LREC Number: 08/H0306/32). Paraffin embedded mouse and human specimens were deparaffinized and rehydrated, followed by antigen retrieval in boiling citrate buffer. The following primary antibodies were used: Ki-67 (SP-6, Neomarkers), Nqo1 (Abcam, Sigma Aldrich), 8-oxo-dGuo ([N45.1], Abcam), γ -H2AX (Millipore), Cleaved caspase 3 (Cell Signaling), anti-Malondialdehyde (Abcam). HRP-based detection reagents were used for all IHC except 8-oxo-dGuo, for which AP-based reagents were used. 8-oxo-dGuo specificity was verified by treating sections for 1 hour with 1U/ml DNase at 37°C. For quantification of 8-oxo-dGuo, mean signal intensity was calculated using Adobe Photoshop CS. Immunohistochemistry images were converted to grayscale images, inverted, and mean pixel 8-oxo-dGuo intensity analyzed. For murine Nqo1 IHC, the Sigma Aldrich anti-Nqo1 antibody was used, and the staining pattern was confirmed to be reproducible with the Abcam antibody (not shown). The Abcam Nqo1 antibody was used for human tissue.
Luciferase assay
LSL-K-RasG12D and Nrf2-/-; K-RasG12D MEFs were plated in 12 well dishes at a density of 5 × 104 cells/well and infected with a Cignal Lenti ARE Reporter (Qiagen) at an MOI of 10. After 6 hours the cells were split 1:2 and infected the next day with Ad-mock and Ad-cre. After 4 days the cells were plated at 70% confluence and analyzed for luciferase activity using the Dual-Luciferase Reporter Assay System (Promega) with a luminometer. Lysates were analyzed in duplicate and cells lacking the ARE reporter were included as a control.
Mitochondrial ROS
Cells were incubated in serum- and phenol red-free DMEM with 5uM MitoSOX or 100nM Mitotracker Green FM (both Invitrogen) for 30 minutes and analyzed by flow cytometry. Mitochondrial ROS was plotted as the mitochondrial ROS signal (MitoSOX) normalized to mitochondrial mass (Mitotracker Green FM).
Nox Activity assay
Cells were washed twice with ice cold PBS, scraped in PBS, and centrifuged to obtain a cell pellet. The pellet was dounce homogenized in PBS + 1mM MgCl2 + 1mM EGTA + protease inhibitors and 100ul of homogenate was added to 100ul of 2× reaction buffer (PBS + 1mM MgCl2 + 1mM EGTA + 300mM sucrose + 10uM lucigenin [Sigma Aldrich] + 200uM NADPH [Sigma Aldrich]) and relative light units were measured with a luminometer every minute for 10 minutes. Specificity was confirmed by adding the specific Nox inhibitor diphenylene iodonium (10uM, Sigma Aldrich). Graphs show one representative measurement in the linear range (2-3 minutes) and are expressed as relative light units per μg protein.
Nrf2 stability
MEFs were treated with 25ug/ml cycloheximide (Acros Organics) for indicated time points and total cell lysates extracted and separated by SDS-PAGE. Gels were transferred to nitrocellulose (Biorad), and membranes were probed with Anti-Nrf2 antibody, affinity purified as described.28
Quantification of proliferation and neoplasms
Values for proliferation were obtained by counting at least 5 fields of greater than 50 cells. For PanIN studies, only cells contained in PanIN-1a were included in the analysis (surrounding stromal, acinar or immune cells were all excluded). Proliferation is determined as a percentage and therefore represents the number of Ki-67 positive PanIN-1a cells /total PanIN-1a cells. For quantification of lung disease burden, total lung areas and neoplasm areas were calculated using Image J29 and disease burden was represented as a percent of total area. For characterization of individual neoplasms, data was represented as neoplasms/lung area. For quantification of PanIN per mouse, paraffin embedded pancreata were sectioned at 100 micron intervals and individual PanIN were counted.
Ras-GTP activity assays
Ras-GTP levels were determined according to the manufacturer's instructions (Millipore). Membranes were also blotted with anti-Rac antibody (Millipore) as a loading control.
Reagents
4-hydroxy-tamoxifen (4-OHT) was obtained from Sigma Aldrich. AZD6244 (ARRY-142886) was obtained from Symansis.
Senescence-associated b-galactosidase staining
Cells were fixed in 2% formaldehyde/0.2% glutaraldehyde in PBS for 5 minutes and stained in staining solution (Citric acid/Phosphate buffer pH 6.0, 150mM NaCl, 2mM MgCl, 5mM Potassium Ferricyanide, 5mM Potassium Ferrocyanide, and 1mg/ml X-gal) overnight at 37°C. Fields of at least 50 cells were counted, in triplicate. For tissue, fresh pancreas from mice harboring PanIN was cut into small pieces (approximately 1-3 mm cubes) and fixed for 2 hours in 4% PFA in PBS on ice, and incubated in staining solution overnight at 37°C. The pieces were then fixed in formalin overnight, followed by a quick processing and embedding in paraffin wax. Sections were cut with a thickness of 10 microns, dewaxed with minimal exposure to xylene, and counterstained with nuclear fast red. Five fields of at least 50 cells were counted per mouse.
Sequencing of KEAP1 and NRF2 in patient samples
Xenograft enriched pancreatic cancers were created as described previously30. Cell lines were obtained from the ATCC (Aspc-1, CAPAN1, CFPAC1, Hs766T, MiaPACA2, Panc-1 and Su86.86) or from other sources. PK8 and PK9 were kindly provided by Dr. A. Horii31. PL11, PL19, PL23, (kindly provided by Dr. E. Jaffee), and XPA1 (kindly provided by Dr. A. Maitra) were also used. Genomic DNA from immortalized peripheral blood lymphocytes of 29 patients with familial pancreatic cancer were obtained from the National Familial Pancreatic Tumor Registry (NFPTR). The acquisition of human tissues from resection specimens or the NFPTR was approved by the Institutional Review Board at the Johns Hopkins Hospital. The coding sequences of exons 2-6, encompassing the entire coding sequence of KEAP1, and exon 2 of NRF2 (official current gene name, NFE2L2), encompassing the KEAP1 binding domain, were amplified from genomic DNAs using intronic primers flanking each exon. Primers were designed using Primer3 online software (http://frodo.wi.mit.edu/primer3). PCR-amplified products were sequenced using nested primers and an ABI Prism model 3700 Applied Biosystems, Foster City, CA). Sequence analysis employed Sequencher™ version 4.8 software (Gene Codes, Ann Arbor, MI). Identified variants were verified by independent PCR amplification and reverse sequencing of the amplified products. The somatic or germline nature of each variant was determined by comparing their respective sequences to matched normal DNA available from the same patients. The following primers were used:
| Location | Forward | Reverse |
|---|---|---|
| Keap1 Exon 2 | ATCAGGTCGGGGAAGTTTG | AGCCCAGAACCTCCTTTTTC |
| Keap1 Exon 3 | GTCAGCGGCAGTGATAAGTTAC | TGACAGTCCCCTAAGCATTTC |
| Keap1 Exon 4 | TCCACGAAGGTCAGCTATAATG | TCCAGGGCTTCTGTGGTTAC |
| Keap1 Exon 5 | TCTCTCCCCGCTTCATTTC | GCAAAAGCAGTCCACAAAAG |
| Keap1 Exon 6 | GACCATCCCTTCTGTTCTTC | GCTTTGGACTTCTTTTGAGATG |
| Nrf2 Exon 2 | CCACCATCAACAGTGGCATA | AAGGCAAAGCTGGAACTCAA |
Statistical Analyses
All data are expressed as the mean ± SEM. Results are representative examples of three or more individual experiments. Statistical analyses were performed with the Mann-Whitney U test or Student's T-test (*p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant). Graph Pad Prism was used for both analyses.
Transfections
DNA transfections were performed with Fugene 6 (Roche) or Lipofectamine 2000 (Invitrogen). siRNA transfections were performed with ON-TARGETplus SMARTpool siRNA using DharmaFECT 1 reagent (both Dharmacon) according to the manufacturer′s instructions. The following siRNA sequences were used:
| Gene | Target sequence (sense strand), pool of 4 |
|---|---|
| Non-targeting | 5′-UGGUUUACAUGUCGACUAA-3′ ; 5′-UGGUUUACAUGUUGUGUGA-3′ |
| 5′-UGGUUUACAUGUUUUCUGA-3′ ; 5′-UGGUUUACAUGUUUUCCUA-3′ | |
| Elk1 | 5′-GGAAUGAAUACAUGCGCUC-3′ ; 5′-CCAAGGUGGCUUAGCACGA-3′ |
| 5′-GGGAUGGUGGUGAGUUCAA-3′ ; 5′-ACCAAAGGGUGCAGGAAUG-3′ | |
| Fra1 | 5′-GAACCGGAAGCACUGCAUA-3′ ; 5′-AGGCGGAGACCGACAAAUU-3′ |
| 5′-GAACCUUGCUCCUCCGCUC-3′ ; 5′-GCUAAGUGCAGAAACCGAA-3′ | |
| Jun | 5′-CCAAGAACGUGACCGACGA-3′ ; 5′-GCAGAGAGGAAGCGCAUGA-3′ |
| 5′-GAAACGACUUCUACGACG-3′ ; 5′-GAACAGGUGGCACAGCUUA-3′ | |
| JunD | 5′-GAACAAACGUUGGUUGCGU-3′ ; 5′-AGCGCAAGCUGGAGCGUAU-3′ |
| 5′-CCACAUUCCUGUUCCGUAA-3′; 5′-AAGUCUUCGUUACGCCAAA-3′ | |
| Keap1 | 5′-GCGCCAAUGUUGACACGGA-3′ ; 5′-GAUAUGAGCCAGAGCGGGA-3′ |
| 5′-GGAUGAUCACACCGAUGAA-3′ ; 5′-GUUCGAGCCUGCAGCGACU-3′ | |
| K-Ras (mouse) | 5′-GAACAGUAGACACGAAACA-3′ ; 5′-AGCAAGGAGUUACGGGAUU-3′ |
| 5′-GGUUGGAGCUGGUGGCGUA-3′ ; 5′-GGUGUACAGUUAUGUGAAU-3′ | |
| K-Ras (human) | 5′-GGAGGGCUUUCUUUGUGUA-3′ ; 5′-UCAAAGACAAAGUGUGUAA-3′ |
| 5′-GAAGUUAUGGAAUUCCUUU-3′ ; 5′-GAGAUAACACGAUGCGUAU-3′ | |
| Myc | 5′-GAAACGACGAGAACAGUUG-3′ ; 5′-CCACUCACCAGCACAACUA-3′ |
| 5′-GGACACACAACGUCUUGGA-3′ ; 5′-UCGAAACUCUGGUGCAUAA-3′ | |
| Nrf2 | (1) 5′-ACUCAAAUCCCACCUUAAA-3′ ; (2) 5′-UGGAGUAAGUCGAGAAGUG-3′ |
| (3) 5′-CAUGUUACGUGAUGAGGAU-3′ (4) 5′-GGACAGCAAUUACCAUUUU-3′ |
Supplementary Material
Acknowledgments
We thank Jeffrey Johnson for providing the Nrf2-/- mice; Gerard Evan, Owen Sansom, Charles Murtaugh, Juan-Jose Ventura and Erwin Wagner for MEFs; Ed Schmidt for Nrf2 antiserum; Elizabeth Jaffee, Anirban Maitra and Arata Horii for human PDA cell lines; Beverley Haynes, Susan Davies, and Natalie Cook for human PDA tissue samples; Caryn Ross-Innes, Kelly Holmes and Jason Carroll for advice with the ChIP assay; and the ENCODE Consortium for ChIP-seq studies. We thank Frances Connor, Carla Martins and other members of the Tuveson lab for assistance and advice, and the animal care staff and histology core at CRI. This research was supported by the University of Cambridge and Cancer Research UK, The Li Ka Shing Foundation and Hutchison Whampoa Limited, the NIHR Cambridge Biomedical Research Centre, and the NIH (grants CA101973, CA111294, CA084291, and CA105490 to DAT; CA62924 and CA128920 to SEK and CID; and CA106610 to CID). Additional support was obtained from the Abramson Cancer Center of the University of Pennsylvania Pilot Grant IRG 78-002-26 (DAT), Emerald Foundation (ESC), the Marjorie Kovler Fund (SEK) and the Ruth L. Kirschstein National Research Service Award F32CA123887-01 (KF). We regret that many primary references have been omitted due to space limitations.
Footnotes
Author Contributions: GMD, FAK, TH, AG and KF performed cell culture and mouse experiments. CW, DM, KHY and IAB performed 8-oxo-dGuo and glutathione assays. CJY, ESC, FS, JMW, RHH, CID and SEK performed Nrf2 and Keap1 sequencing. GMD and DAT designed the study and wrote the manuscript, and all authors commented on it.
References
- 1.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 2.Itoh K, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wakabayashi N, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–3156. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 6.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Tuveson DA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 8.Lee AC, et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274:7936–7940. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- 9.Mitsushita J, Lambeth JD, Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004;64:3580–3585. doi: 10.1158/0008-5472.CAN-03-3909. [DOI] [PubMed] [Google Scholar]
- 10.Cakir Y, Ballinger SW. Reactive species-mediated regulation of cell signaling and the cell cycle: the role of MAPK. Antioxid Redox Signal. 2005;7:726–740. doi: 10.1089/ars.2005.7.726. [DOI] [PubMed] [Google Scholar]
- 11.Recktenwald CV, Kellner R, Lichtenfels R, Seliger B. Altered detoxification status and increased resistance to oxidative stress by K-ras transformation. Cancer Res. 2008;68:10086–10093. doi: 10.1158/0008-5472.CAN-08-0360. [DOI] [PubMed] [Google Scholar]
- 12.Irani K, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka H, et al. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-kappaB activity that facilitates MnSOD-mediated ROS elimination. Mol Cell. 2002;9:1017–1029. doi: 10.1016/s1097-2765(02)00522-1. [DOI] [PubMed] [Google Scholar]
- 14.Murphy DJ, et al. Distinct thresholds govern Myc's biological output in vivo. Cancer Cell. 2008;14:447–457. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karreth FA, DeNicola GM, Winter SP, Tuveson DA. C-Raf inhibits MAPK activation and transformation by B-Raf(V600E) Mol Cell. 2009;36:477–486. doi: 10.1016/j.molcel.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 17.Keum YS, et al. Mechanism of action of sulforaphane: inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer research. 2006;66:8804–8813. doi: 10.1158/0008-5472.CAN-05-3513. [DOI] [PubMed] [Google Scholar]
- 18.Kwak MK, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol. 2002;22:2883–2892. doi: 10.1128/MCB.22.9.2883-2892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 20.Jackson EL, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawaguchi Y, et al. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- 23.Park JH, et al. Evidence for the aldo-keto reductase pathway of polycyclic aromatic trans-dihydrodiol activation in human lung A549 cells. Proc Natl Acad Sci U S A. 2008;105:6846–6851. doi: 10.1073/pnas.0802776105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu P, Oe T, Blair IA. Determination of cellular redox status by stable isotope dilution liquid chromatography/mass spectrometry analysis of glutathione and glutathione disulfide. Rapid Commun Mass Spectrom. 2008;22:432–440. doi: 10.1002/rcm.3380. [DOI] [PubMed] [Google Scholar]
- 25.Carroll JS, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 26.He X, Chen MG, Lin GX, Ma Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 × Keap1 × Cul3 comple × and recruiting Nrf2 × Maf to the antioxidant response element enhancer. J Biol Chem. 2006;281:23620–23631. doi: 10.1074/jbc.M604120200. [DOI] [PubMed] [Google Scholar]
- 27.Sun J, et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002;21:5216–5224. doi: 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suvorova ES, et al. Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS One. 2009;4:e6158. doi: 10.1371/journal.pone.0006158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rasband WS. ImageJ. U. S. National Institutes of Health; Bethesda, Maryland, USA: 1997-2008. [Google Scholar]
- 30.Hahn SA, et al. Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res. 1995;55:4670–4675. [PubMed] [Google Scholar]
- 31.Sun C, et al. Characterization of the mutations of the K-ras, p53, p16, and SMAD4 genes in 15 human pancreatic cancer cell lines. Oncol Rep. 2001;8:89–92. doi: 10.3892/or.8.1.89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.