

Abstract
The function of the anti-apoptotic Bcl-2 family member Bcl2a1/Bfl-1/A1 is poorly understood due to the lack of appropriate loss-of-function mouse models and redundant effects with other Bcl-2 pro-survival proteins upon overexpression. Expression analysis of A1 suggests predominant roles in leukocyte development, their survival upon viral or bacterial infection, as well as during allergic reactions. In addition, A1 has been implicated in autoimmunity and the pathology and therapy resistance of hematological as well as solid tumors that may aberrantly express this protein. In this review, we aim to summarize current knowledge on A1 biology, focusing on its role in the immune system and compare it to that of other pro-survival Bcl-2 proteins.
Keywords: Bcl-2 protein family, A1/Bfl-1, Hematopoiesis, Apoptosis
Introduction
Two convergent pathways are involved in the induction of apoptosis: the “extrinsic” and the “intrinsic” pathway. The extrinsic pathway, also called the “death receptor” (DR) pathway, is initiated by the ligation of Tumor Necrosis Factor Receptor (TNF-R) family members that contain a cytoplasmic death domain (DD) with a specific “death ligand” of the TNF-family. This triggers the formation of the death-inducing signaling complex (DISC) that leads to the activation of apoptotic proteases of the caspase family, i.e. the initiator pro-caspase-8 and/or, in humans, also pro-caspase-10 [1–3].
The intrinsic pathway, also known as “mitochondrial” pathway, is responsible for the induction of apoptosis in response to, e.g. DNA damaging agents, altered redox status or endoplasmatic reticulum (ER) stress. Importantly, also cytokine- or growth factor-withdrawal as well as high-affinity ligation of antigen receptors during negative selection processes in the developing immune system triggers mitochondrial apoptosis [4]. All these stimuli cause increased mitochondrial permeability, often referred to as mitochondrial outer membrane permeabilization (MOMP), which induces the release of cytochrome C from the inter-membrane space, which binds the WD-40 repeats of the cytoplasmic adaptor protein Apaf-1. Upon binding to cytochrome C and using dATP/ATP as a cofactor, Apaf-1 undergoes a conformational change that recruits pro-caspase-9 molecules via caspase recruitment domain (CARD)-dependent interaction. This in turn mediates the full activation of initiator pro-caspase-9 molecules by autoprocessing in the apoptosome complex [3]. In addition the permeabilization of the mitochondrial outer membrane leads to the release of the apoptogenic proteins such as SMAC/DIABLO and HtrA2/Omi that inhibit caspase-inhibitors of the “inhibitor of apoptosis” (IAP) family, mainly XIAP, that blocks caspase-9 and caspase-3 activity, amplifying apoptotic signals [5]. MOMP ultimately also associates with the release of additional cell death promoting molecules such as endo-G or AIF that both possess nuclease activities but may be more critical for non-apoptotic cell death paradigms [6,7].
In certain cell types, including hepatocytes and pancreatic beta cells, as well as in certain tumors, the intrinsic pathway can connect and enhance the pro-apoptotic potential of the extrinsic pathway via caspase-8-mediated proteolysis of the Bcl-2 family protein Bid, that can trigger MOMP in its processed truncated form, tBid [2]. tBid has also been implicated in a special form of cell death that does not easily fall in one of the two aforementioned categories of apoptosis. In the immune system, the perforin/granzyme pathway can be considered as an additional special paradigm of apoptosis. It is induced in infected target cells by cytotoxic T cells through the activation of apoptotic proteases of the caspase-family at a level where usually both, the extrinsic and intrinsic apoptosis pathways, converge, i.e. the direct activation of caspase-3/7 zymogens. NK cells can make use of the same cell death pathway attacking transformed cells lacking MHC-class-I expression on their cell surface. Granzyme-B-mediated processing of Bid may also amplify the death signal, similar to that seen upon death receptor ligation in hepatocytes. However, here species-specific differences seem to apply as only the human enzyme processes Bid effectively, while mouse granzyme B cleaves Bid only with very low efficacy [8].
The Bcl-2 family
Key-players of the intrinsic apoptotic pathway are the Bcl-2 family proteins. Bcl-2 was first identified as a protein upregulated in human follicular B cell lymphoma, following the t(14;18) chromosomal translocation of the BCL-2 gene juxtaposing it to the Ig heavy-chain locus [9,10]. The Bcl-2 family members show sequence and structural similarity in their “Bcl-2 homology regions” (referred to as BH-domains) and can be divided into two subgroups according to their function: anti- and pro-apoptotic proteins. The pro-apoptotic members can be further subdivided into the Bax/Bak-like proteins, i.e. Bax, Bak and Bok, which share three BH-regions with their pro-survival relatives (therefore sometimes also referred to as BH-123 proteins) and the BH3-only proteins (Bad, Bid, Bik, Bim, Bmf, Hrk, Noxa and Puma) that only contain the BH3-domain but are otherwise only poorly conserved amongst each other and all other Bcl-2 family members [2,3]. Bax and Bak proteins regulate the permeabilization of the mitochondrial outer membrane and their promiscuity in cell death signaling has been confirmed by gene targeting studies in mice showing redundant function in cell death initiation [11]. In contrast, the role of Bok, if any in this process, is still unknown. Once activated, Bax and Bak proteins can form homo-oligomers in the outer mitochondrial membrane that leads to the formation of a pore-like structure which then triggers the subsequent disruption of the membrane integrity and the release of cytochrome C, followed by caspase activation. The precise structure of the Bax/Bak pore, however, remains to be determined [2,3]. In healthy cells the anti-apoptotic members of the Bcl-2 family (Bcl-2, Bcl-xL, Bcl-w, Mcl-1, A1/Bfl-1) sequester Bak and neutralize or restrain activated Bax in the cytosol, thereby preventing MOMP. However, it remains to be determined if all these proteins prevent MOMP by exactly the same molecular mechanism(s). Upon different stress stimuli, sentinel BH3-only proteins become activated or transcribed/translated and initiate the apoptotic cascade leading to Bax/Bak activation [12]. The molecular mechanism underlying the BH3-only protein-mediated activation of Bax/Bak is still debated. In the so-called “affinity model”, BH3-only proteins are able to bind to a hydrophobic groove on the surface of the anti-apoptotic Bcl-2 family members, preventing them from counteracting the apoptotic activity of Bax/Bak. This interaction is dependent on the characteristics of the BH3-domain. Hence each BH3-only protein has a certain preference for one or more proteins of the anti-apoptotic family. In an alternative model, referred to as “direct activation” model, all the BH3 only proteins compete for the binding to the anti-apoptotic members of the family, but only Bid, Bim and probably also Puma can induce direct conformational changes in Bax or Bak that promote their targeting (in the case of Bax) to and oligomerization in the outer mitochondrial membrane. Recently, evidence has been provided that both mechanisms are able to cooperate in cell death induction [13,14].
The apoptotic threshold is tightly regulated by the balance between pro- and anti-apoptotic Bcl-2 family proteins present in a given cell type [12]. In healthy cells, pro-survival pathways including those triggered upon growth factor or cytokine-mediated activation of protein kinase networks, such as ERK or AKT, can stimulate the transcriptional induction of anti-apoptotic Bcl-2 family members. Expression levels of Bax and Bak are usually stable in most cells and barely subjected to transcriptional regulation but may also undergo posttranslational modifications for priming. On the other hand, BH3-only proteins are held in check by very diverse mechanisms. For example, Noxa and Puma are frequently controlled at the transcriptional level. Bim, Bmf or Bad abundance and/or activity are most frequently regulated post-transcriptionally, e.g. by IRES-dependent translation [15], subcellular sequestration or phosphorylation events, e.g. by above-mentioned kinases or their effectors, controlling their interaction capacity with Bcl-2 pro-survival homologues [12,16]. As indicated above, proteolysis of Bid, by caspases or granzyme-B, regulates its pro-apoptotic activity [8].
Each BH3-only protein is considered to transmit a distinct set of death signals, albeit redundancy and cell-type specific responses are frequently observed. For example loss of Bim impairs the response of lymphocytes to cytokine deprivation or high-affinity ligation of antigen receptors as well as calcium-flux or ER-stress while Puma is most frequently required for cell death induced by DNA-damage, but also acts in concert with Bim upon growth factor deprivation [17]. Of note, both Bim and Puma effectively limit survival upon aberrant oncogene activation [18,19]. The role of other BH3-only proteins appears more redundant with or auxiliary to that of Bim or Puma with possible additional roles in non-apoptotic cell death modalities [20,21].
Anti-apoptotic Bcl-2 family members
Bcl-2 was the first described member of the family as being overexpressed in B cell lymphoma and acting as an oncogene by inhibiting apoptosis [22,23]. The other anti-apoptotic Bcl-2 family members, Bcl-xL, Bcl-w, Mcl-1 and A1/Bfl-1 share up to four Bcl-2 homology (BH) domains (Fig. 2) and secondary and tertiary structures [2]. These proteins can bind to Bax/Bak-like proteins although certain preferences exist. As pointed out above, most BH3-only proteins interact only with a subset of Bcl-2 pro-survival homologues, but the overexpression of any one of these proteins can protect cells from apoptosis induced by a broad range of stimuli. This probably reflects their functional overlap, in particular when highly abundant in cells (reviewed in [2]). The phylogenetic analysis of these proteins, however, enlightens two distinct sub-groups of pro-survival homologues: Bcl-2, Bcl-xL and Bcl-w on one side and Mcl-1, A1/Bfl-1 on the other. Interestingly, Bcl-B/BOO/DIVA, a more distantly related Bcl-2 family member initially assigned with pro-apoptotic properties, clusters with Mcl-1 and A1/Bfl-1 [24] (Fig. 1) suggesting that these genes may have developed form a common ancestor.
Fig. 2.

ClustalW2 alignment of mouse Bcl-2 prosurvival homologues http://www.ebi.ac.uk Mouse proteins: A1-a NP_033872, A1-b NP_031560, A1-d NP_031562, Bcl-2 AAH95964, Bcl-xLAAA51039, Bcl-w NP_031563, Mcl1 NP_032588.1 and Boo/Diva AAH52690. Highlighted in bold the glutamate 78 of A1 and the corresponding residue of the other anti-apoptotic proteins.
Fig. 1.

PhyloTree: unrooted phylogenetic tree http://www.cbrg.ethz.ch/services/AllAll Mouse proteins: A1-a NP_033872, A1-b NP_031560, A1-d NP_031562, Bcl-2 AAH95964, Bcl-xLAAA51039, Bcl-w NP_031563, Mcl1 NP_032588.1 and Boo/Diva AAH52690.
A further distinction within the pro-survival group is their preferred sub-cellular localization. Bcl-2 is a membrane bound protein that localizes to the ER, but prefers the mitochondrial outer membrane, albeit association with nuclear membranes has also been reported [25]. In contrast, its homologues Bcl-xL and Bcl-w as well as Mcl-1 are partially cytosolic and often need to translocate to organelle membranes after cells were exposed to cytotoxic stimuli [26–28]. While Bcl-xL appears to prefer the ER-membrane, Bcl-w seems only loosely attached to the outer mitochondrial membrane and needs to insert its carboxy-terminus into it, in response to cell stress [26]. Little however is known about A1/Bfl-1 in that regard but species specific differences seem to apply as mouse A1 is preferentially localized in the cytosol, while human A1/Bfl-1 can be found at the mitochondria. Hence the pro-survival function of A1 seems to be independent of its localization when overexpressed at high levels [29]. Nonetheless, deletion or mutation of the C-term end of either mouse or human A1 negatively interferes with its anti-apoptotic potential [30,31]. This indicates that in mice A1's site of anti-apoptotic action may be imposed by its interaction partners, i.e., the BH3-only proteins that again can locate to mitochondria [15,32]. In contrast, human A1 preferentially targets that organelle directly [30,31].
Also the regulation of gene expression differs significantly among these anti-apoptotic proteins. Bcl-2 expression appears to be quite robust in most cell types but in lymphocytes it frequently changes between different maturation stages. For example, Bcl-2 is high in mature lymphocytes, but low in immature subsets that undergo active selection processes, such as CD4+8+ thymocytes or T1 transitional B cells, where Bcl-xL and Mcl-1 seem to be the preferred pro-survival proteins [33,34] (pers. observations). Accordingly, different post-transcriptional modes of regulating Bcl-2 protein abundance and function, including IRES-mediated translation [35] as well as inactivating phosphorylation events by “pro-death kinases” such as ASK1 or JNK have been reported [36]. Bcl-xL can be transcriptionally induced by cytokines [37] and growth factors activating the JAK-STAT pathway [38] or members of the TNF family such as BAFF, e.g. in B cells [39], to name just a few examples. Mcl-1 expression is transcriptionally regulated by cytokines such as Il-7 or G-CSF in leukocytes, by IGF-1 signaling in neurons [40] or by EGF-receptor family members ErbB1 and ErbB2 in 3T3 cells [41]. Beyond rapid transcriptional activation it becomes also rapidly degraded via the proteasome pathway in response to several apoptotic stimuli, including UV-radiation or cytokine deprivation [27,42]. Prolonged M-phase arrest in response to microtubule poisoning leads to the activation of the spindle check-point and the gradual decline of Mcl-1 seems to act as an apoptotic timer in the absence of gene-transcription in mitosis [43].
Albeit highly redundant when overexpressed in tissue culture, gene-targeting studies of individual Bcl-2 pro-survival genes demonstrated independent roles and functions in tissue homeostasis and development. Loss of Bcl-2 causes abnormal death of renal epithelial cells and melanocyte progenitors as well as mature B and T lymphocytes [44,45]. Bcl-xL is critical for the survival of erythroid progenitors as well as neuronal cells during embryogenesis and its absence causes embryonic death around E14 [46]. Bcl-w is essential for spermatogenesis [47] and Mcl-1 is already required for implantation of the early embryo [48]. Conditional deletion of Mcl-1 revealed key roles for the survival of hematopoietic stem cells, immature and mature lymphocytes [49,50], mast cells [51] as well as neurons [52] or hepatocytes [53].
A1/Bfl-1 discovery
Murine A1 was originally identified in 1991 as a granulocyte and macrophage colony stimulating factor (GM-CSF) early response gene. In a screening approach stimulating bone marrow derived macrophages with GM-CSF, Orlofsky et al. identified A1 among six other novel genes showing early transcriptional induction in response to this cytokine [54]. In 1993 Lin et al. documented A1 expression in thymus, spleen, bone marrow, myeloid and T-lymphoid cell lines, but not in non-hematopoietic lineages, and confirmed the induction of A1 in GM-CSF and LPS-stimulated primary macrophages [55]. Hatakeyama and colleagues mapped multiple A1 genes on murine chromosome 9 (A1-a, A1-b and A1-d) and identified also an A1 pseudogene (A1-c) [56]. Of note, rats and humans express A1 only from one gene locus. All mouse A1 genes consist of two exons. Exon 1 contains the 82% of the total open reading frame and exon 2 encodes for the remaining 18% resulting in a 172 amino acid protein. Among the three murine A1 genes, the exon sequences display 96% of sequence identity at the nucleotide level and 97% of identity at the protein level, and also the intronic region (4.7 kb for A1-a; 7.8 kb for A1-b and A1-d) is very well conserved with 96% sequence identity [56]. A1-c is a pseudogene containing a frame-shift mutation caused by a single nucleotide insertion resulting in a truncated version of A1 protein that would only encode for the BH1-domain. Interestingly, the different isoforms of A1 present a different pattern of expression in the hematopoietic compartment. For example, in DN4 pre-T cells, A1-d is the major isoform detected [57]. However, another study reports that A1-b is the predominant form in total thymocytes, as in resting T and B cells. A1-a seems least abundant and one study reports its induction in TCR-activated CD8+ T cells, together with A1-d [37]. In peritoneal macrophages an expression pattern similar to that noted in T cells was reported, while in neutrophils, all A1 isoforms appear expressed at comparable levels [56]. These cell type specific differences may indicate delicate regulation and different functions of the individual mouse A1 isoforms or, maybe more likely, different accessibility of the individual A1 promoter regions in multiple cell types conferred by their different epigenetic states. However, genetic background differences in the different mouse strains used for analysis may also contribute and a direct comparison of the efficacies of these isoforms in cell death inhibition is actually lacking,
The human A1 gene shares about 72% of sequence identity at the protein level with mouse A1-a discovered in the early 90s by three groups. First, it was identified in fetal livers at week 22 of gestation and thus called Bcl-2 related gene in fetal liver (Bfl-1), and discussed to be involved in stomach cancer development and progression [58]. In a second report, Karsan et al. demonstrated that the expression of human A1 is not only restricted to the hematopoietic compartment, as reported before for its murine homologue, but can be detected in vascular epithelial cells where it is induced in response to TNF-α and IL-1β, suggesting a role for A1 in protecting endothelial cells during inflammation [59]. In the third independent study A1 was discovered as a gene involved in a genomic translocation in patients with CML leukemia. In one patient, the fibroblast growth factor 4 (FGF-4) gene was truncated and rearranged with the A1 gene, called in this study Glasgow rearranged sequence (GRS). GRS and A1 sequences differ only at two positions (N72T and Q107H). High expression levels of GRS were also observed in hematopoietic malignancies, such as chronic myeloid leukemia and the Burkitt lymphoma cell line Raji [60]. The human A1 gene is located on chromosome 15q24 and consists of three exons giving rise to two different splice variants [61,62]. The most abundant mRNA is generated from exon 1 and exon 2 and is translated into a 175 amino acid long protein that shares 73% of sequence identity with murine A1-a. The second transcript variant contains all three exons. This splicing combination causes a shift in the open reading frame, which leads to premature translational termination after 163 amino acids. The polypeptide encoded is referred to as Bfl-1_short (s). Bfl-1s contains a hydrophilic C-terminus, which consists of 23 amino acids and includes four consecutive positively charged amino acids (KKRK), which are recognized as nuclear localization sequence. However, the nuclear function of Bfl-1s is still poorly understood. Its mRNA was found in high levels in lymph nodes, B leukemia cell lines, myeloid cell lines and at lower levels in the spleen, but it is lacking in primary T cells and cell lines. Additionally, overexpression of Bfl-1s in an acute lymphoblastic leukemia cell line protected against treatment with chemotherapeutic agents [63]. However, its relative abundance at the protein level is unexplored, as no discriminating antibodies are available at present.
A1/Bfl-1 mode of action
A1 shares all four BH-domains with Bcl-2. Of these, the BH1 and BH2 domains are highly conserved while the BH3 and in particular the BH4-domain have little homology with those of the cognate anti-apoptotic proteins. Similar to the other anti-apoptotic Bcl-2 proteins, A1 consists of 8 α-helixes. In particular, the helixes α4, α5 and α6, corresponding to the BH3, BH1 and BH2 domains respectively, form a groove on the protein surface that is able to interact with the BH3 domain of the pro-apoptotic Bcl-2 family proteins [64]. Despite the overall structural homology, A1 differs from all the other anti-apoptotic Bcl-2 family members in its C-terminal end [65]. While Bcl-2, Bcl-xL, Bcl-w and Mcl-1 localize predominantly to inner cellular membranes via their hydrophobic region at the C-terminal end, the C-terminal portion of A1 contains a hydrophilic stretch responsible for ubiquitination and degradation via the proteosome pathway. Interestingly among all the pro-survival family members only A1 and Mcl-1 show such a rapid turnover rate. The half-life of A1 and Mcl-1 is estimated to be as short as 30 min while the half-life of Bcl-2 is estimated to be around 24 h [29]. Due to the hydrophilic C-terminal portion of the protein, mouse A1 cannot be anchored at the mitochondrial outer membrane but can be found associated with membranes, as well as in the cytosol [29,66]. Whether A1 membrane-association can be equated with insertion, as reported for Bcl-w, remains to be investigated. Of note, A1 may interact with all BH-123 proteins, including Bok, as well as several BH3-only proteins. Yet, there are conflicting reports describing the interaction between A1 with these proteins. An initial yeast two-hybrid screen identified A1 as a binding partner of both, Bax and Bak [67]. In a second study using in vitro translated protein, human A1/Bfl-1 was found to co-immunoprecipitate only with Bax when its transmembrane domain was deleted [68]. Other studies performed in a more physiological context, reported a strong association between both human and murine A1 and endogenous Bak but not with Bax [69,70]. The interaction of A1 with Bax or Bak reflects a preferential binding affinity that can be found also in the other pro-survival members of the family. In general the anti-apoptotic proteins can be subdivided depending on their ability to interact strongly with either Bax or Bak. The BH3-only peptide of Bax is able to interact with high affinity with Bcl-2 and Bcl-w, while Bak peptide binds potently Bcl-xL, Mcl-1 and A1 [71,72]. A1 interacts with different affinity also to the BH3-only proteins. Competitive binding assays using BH3-peptides show that A1, similarly to Mcl-1, has no affinity for Bad whereas it binds very tightly to Bim, Bid and Puma; while Bcl-2, Bcl-xL and Bcl-w interact potently with Bad, Bmf, Bim and Puma [73]. The interaction between A1 and Bid or Bim has been confirmed also independently in immunoprecipitation assays [29,66,69]. The specific interaction pattern of A1 is probably due to the presence of an acidic residue, glutamate 78, within the binding groove that serves as an interaction surface for the BH3-domains of the pro-apoptotic Bcl-2 family members (Fig. 3). In contrast, all other anti-apoptotic proteins have a hydrophobic or slightly positively charged binding groove [64]. Moreover, A1 can be stabilized by the interaction with Bim [29]. The ability to be stabilized by BH3-only proteins and its cytosolic localization suggest that mouse A1 might act as a first barrier against premature or faulty activation of the apoptotic machinery. However, this feature does not seem to be conserved, as human A1 can target mitochondria, although it is unclear if it could also do so in the absence of pro-apoptotic Bcl-2 family proteins.
Fig. 3.
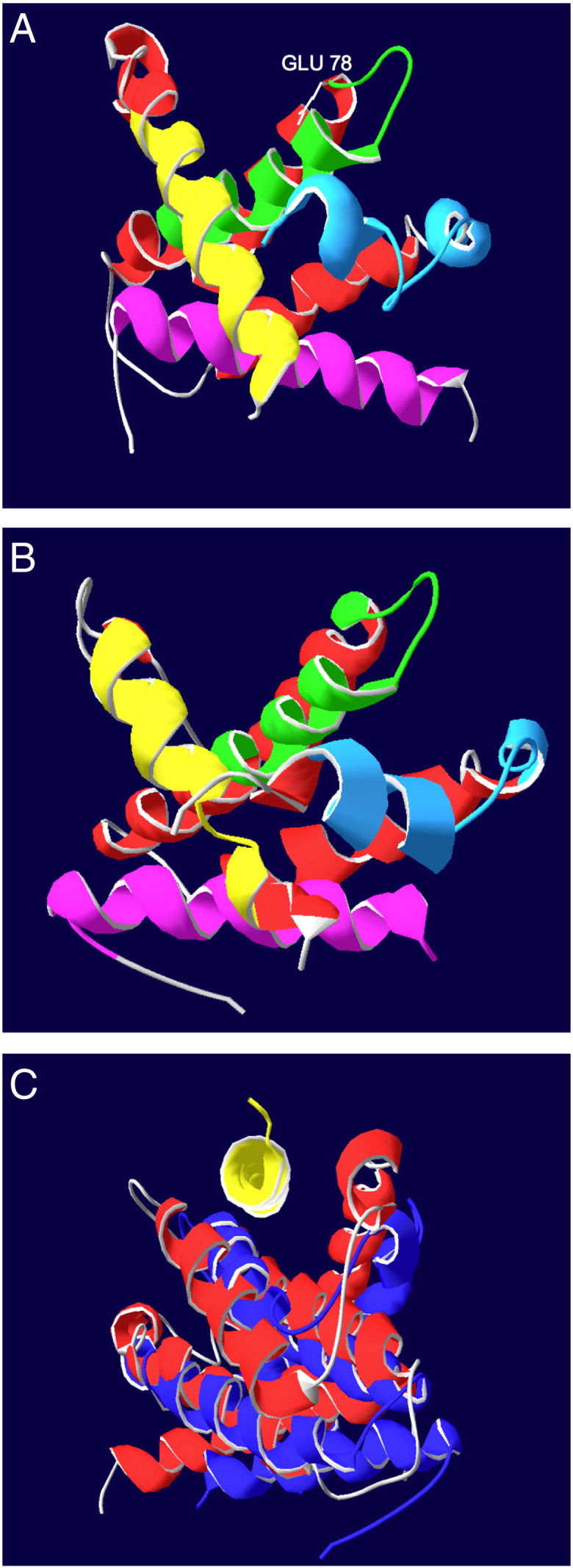
Ribbon structure of (A) Bfl-1 structure pdb file 2vm6[65] using Swiss-PdbViewer 4.0.4 act on ribbon showing the secondary structure. BH1 domain in green, BH2 in blue, BH3 in yellow BH4 in purple and highlighted the glutamate 78 in the binding groove in gray. (B) Bcl-2 structure pdb file 2xa0[72] using Swiss-PdbViewer 4.0.4 act on ribbon showing the secondary structure. BH1-domain in green, BH2-domain in blue, BH3-domain in yellow and BH4-domain in purple. (C) Alignments using magic fit in Swiss-PdbViewer 4.0.4 on backbone atoms only. Bfl-1 pdb file 2vm6[65] red Bcl-2 pdb file 2xa0[72] blue BH3 only peptide Bim in yellow pdb file 2vm6[65].
A lot of speculation relates to a putative interaction of A1 with Noxa that binds preferentially to Mcl-1, and, with lower but still significant affinity, to A1 [73]. As commercially available antibodies for both, Noxa and A1, are of limited quality, interaction at the endogenous levels has never been successfully demonstrated. However, a significant body of circumstantial evidence suggests that interaction of A1 with Noxa can contribute to cell death decisions, as continuous activation of cell death signals engaging Bcl-2, such as the BH3-mimetic ABT-737, leads to compensatory upregulation of A1, promoting tumor cell survival and drug resistance [74,75].
Surprisingly, human A1/Bfl-1 can apparently be converted into a pro-apoptotic factor by a proteolytic cleavage by μ-calpain or a similar protease activity in response to TNF in the presence of cycloheximide in FL5.12 pro-B cells [30]. Although controversial, A1 is not the only anti-apoptotic protein of the Bcl-2 family reported to be able to trigger apoptosis. Processing of Bcl-2 [76] or Bcl-x [77] has been reported to give rise to a pro-apoptotic polypeptide and a truncated version of Bcl-x, generated by alternative splicing, Bcl-xS, has also been reported to exert apoptosis enhancing properties [78]. However, in many of these cases physiological settings leading to the generation of these fragments have not been defined and data available from the different mouse mutants lacking these proteins and the associated phenotypes do not really support a pro-apoptotic function of any of these genes. Accordingly, the physiological relevance of the cleaved A1 polypeptide is not understood at present and has not been investigated further.
Beside the role in apoptosis, A1, like Bcl-2, Mcl-1 or Bcl-xL can interact with Beclin1, thereby inhibiting induction of autophagy, possibly via the disruption of Beclin1/hVps34 complexes [79]. Interestingly, the interaction between Bcl-2 and Beclin1 is mediated by the BH1 and BH2 domains, which are highly conserved between A1 and Bcl-2 [64]. Some evidence supports the interaction between A1 and Beclin1, as A1 knockdown seems to enhance phagosome–lysosome fusion, thereby limiting growth of Mycobacertium tuberculosis infected macrophages [80]. However, interaction of endogenous A1 with Beclin1 remains to be formerly demonstrated.
A1/Bfl-1 in cancer
A1 transgenic mice fail to develop lymphoma [81], suggesting that, unlike Bcl-2 [82], A1 is not an oncogene. However, ubiquitination resistant A1 mutants predispose mice to lymphomagenesis when co-expressed with a dominant-negative mutant of p53 and A1 overexpression accelerated Myc-induced lymphomagenesis in mice [83,84].
A number of hematopoietic malignancies, including B chronic lymphocytic leukemia (B CLL) [85,86] and acute myeloid leukemia (AML), are characterized by elevated levels of A1/Bfl-1 and its expression strongly correlates with chemoresistance and poor prognosis. Interestingly, AML patients with high levels of Wilms Tumor protein (WT1) show increased A1 expression and show the worst prognosis and resistance to chemotherapeutic agents [87]. Differentiation of AML cells and acute promyelocytic leukemia (APL) cells by all-trans retinoic acid (ATRA) is also accompanied by A1 induction via the activation of the transcription and myeloid lineage commitment factor PU.1 [88]. Also the human promyelocytic leukemia HL-60 cell line differentiates in neutrophils when treated with ATRA and thereby rapidly induces A1 and Mcl-1, while the expression of Bcl-2 and Bcl-xL is downregulated [89]. Hence it will be interesting to see if ablation of A1 expression or function can increase the efficacy of these treatments. Of note, aberrant A1 expression has been documented also in a number of non-hematological malignancies, including breast and stomach cancers and reportedly associates with metastasizing disease in HCC and melanoma.
Most importantly for current drug-development efforts, A1 seems to be a primary resistance factor for hematological malignancies treated with new-generation compounds mimicking the action of BH3-only proteins, i.e. BH3-mimetics like ABT-737 and its follow up compound, ABT-263. Continuous exposure to ABT-737 induced compensatory upregulation of A1 and Mcl-1 in DLBCL lymphoma lines [75] while in primary B CLL cells derived from lymph nodes upregulation of Bcl-xL and A1 rapidly induced about 1000-fold resistance to ABT-737 treatment [74]. Hence, understanding the molecular mechanisms involved in A1 transcription and post-translational regulation is critical to improve the efficacy of these compounds. For a more detailed overview on the role of A1 in cancer and drug resistance we want to refer to an excellent recent review by Meike Vogler [90].
A1/Bfl-1 in lymphocyte development and function
Murine A1 is expressed in multiple organs in the embryo including the developing nervous system, liver, heart and cartilaginous primordial skeleton from embryonic day 15.5. However, in adult mice A1 expression seems to be restricted to the hematopoietic compartment (Fig. 4). In the T cell lineage A1 can be first detected in double negative thymocytes, where its expression is induced via the PLCγ/PKC/NF-kB pathway upon pre-TCR signaling [91]. More precisely, A1 is expressed during the transition from the DN3 (CD44−CD25+CD4−CD8−) to the DN4 (CD44−CD25−CD4−CD8−) stage of thymocyte development where “beta-selection” occurs. The functional importance of A1 at this stage was demonstrated by two findings: (i) A1 overexpression promotes survival of Rag-1−/− pre-TCR thymocytes [57] and (ii) reduction of A1 expression in A1-knockdown mice leads to a significantly reduced percentage of DN4 and DP cells with a relative increase in DN2 and DN3 thymocytes. In this animal model, we have expressed an shRNA targeting all A1 isoforms, embedded in the microRNA-30 (miR30) backbone, under control of the hematopoiesis-specific Vav-gene promoter. In a second model, we have tested an alternative shRNA sequence targeting all mouse A1 isoforms under control of the tetracycline responsive TRE-CMVmin promoter and expressed it conditionally using a Tet-transactivator (tTA), driven by the Vav-gene promoter, yielding similar results (Ottina et al., in revision). This strongly indicates that A1 is necessary and sufficient for further differentiation of DN3 thymocytes. While IL-7-mediated Mcl-1 expression is necessary at the DN2 stage of thymocyte development [50] and Bcl-2 overexpression can restore thymocyte development in Il7R−/− mice [92], A1 seems to play a unique role in promoting survival of thymocytes receiving signals via their pre-TCR.
Fig. 4.

A1 in leukocyte development. Schematic overview of hematopoiesis with key-check points indicated in which developing leukocytes rely on A1 expression for progression and/or survival. Abbreviations used: hematopoietic stem cell (HSC), common myeloid progenitor (CMP), common lymphoid progenitor (CLP), B cell (B), germinal center (GC), double negative thymocyte (DN), double positive (DP), single positive (SP), T cell (T).
Once cells become CD4+8+ double positive (DP) thymocytes, A1 is down regulated and then re-induced in single positive (SP) thymocytes in particular in CD4+ cells where A1 expression is up to 30 fold higher compared to the DP subgroup [37]. In line with this observation A1 knockdown mice have a reduced proportion of CD4+ and increasing amounts of CD8+ thymocytes (Ottina et al. in revision), functionally showing that the high A1 expression in this thymic subset is indeed required for their survival. Enforced expression of A1 driven from a CD2-minigene in F5-TCR transgenic Rag−/− mice induces accumulation of DP thymocytes, but does not seem to affect negative selection by exogenous administration of the NP68 peptide of influenza [93]. A1 seems to be also involved in the survival of Th1/Th2 T lymphocytes. Retinoid X receptor (RXR) agonist treated CD4+ thymocytes induced A1-d and A1-b expression specifically during Th2 differentiation that depends on IL-4, IL-12 and IFN-γ stimulation [94]. In lymph node derived T cells A1 expression is not detectable under steady state conditions, but it is strongly upregulated in response to TCR engagement, similar to Noxa, which may need to be antagonized early in the immune response allowing survival of high-affinity TCR-bearing antigen-specific T cell clones [95]. TCR stimulation alone induces NF-kB and MEK/ERK signaling which can trigger A1 expression, along with PI3K-dependent Bcl-xL induction, and proliferation of lymphocytes [37,96]. In agreement with these findings, A1 is also induced within 6 h by ConA activation in splenocytes and then rapidly decreases to basal levels by day 3, while Bcl-2 mRNA levels were induced only after 24 h post activation and remained high till day 4 post activation. Interestingly, A1 expression in CD8+ activated T cells is not dependent on cytokines such as IL-2, IL-4, IL-7 and IL-15, which in turn are required to increase Bcl-2 expression [37]. In contrast to Bcl-2, A1 overexpression in lck-A1-a transgenic mice protects splenic T cells from apoptosis without inhibiting their proliferation [97], but cannot prevent activated T cells from cell death induced by re-stimulation of the TCR that is actually CD95-dependent [1]. Together, these findings show that A1 is an early regulator of TCR-mediated survival of T lymphocytes and, since its expression does not inhibit cell proliferation, allows an efficient accumulation of T cells upon antigen stimulation.
During B cell development in the bone marrow A1 is expressed at very low levels, but apparently might have a role in the survival of early B cells as transgenic mice expressing A1-a under the control of the IgH gene enhancer in the B cell lineage (Eμ-A1-a) show an accumulation of pro-B cells and proportional decrease of class-switched mature B cells in the periphery. However, A1 overexpression in SCID mice did neither promote V(D)J rearrangement nor rescue pre-B cells, as seen in Eμ-Bcl-2 transgenic SCID mice [81]. Similar to Bcl-2, A1 is up-regulated in peripheral B cells [98,99], which is in contrast to Mcl-1 and Bcl-xL that are already found expressed in pro/pre-B cells, but at lower levels in mature B cells [100–102]. In particular, A1 is upregulated during differentiation from transitional (T) B cells (IgMhigh, CD24+, CD21−) to follicular (FO) B cells (IgMlow, IgD+, CD24+, CD21+) in the spleen. At this stage, FO B cells become also dependent on the TNF family cytokine BAFF for survival, but results related to BAFF-dependent induction of A1 are contradictory [99,103]. Nevertheless, induction of A1 is dependent on the PLC-γ2 pathway and consequent activation of Nf-κB via the BCR [104], considered to be essential for B cells to fully mature. Consistently, A1 overexpression is sufficient to rescue survival of mature FO B cells in the absence of PLC-γ2 [105]. Since A1 expression is strongly induced upon BCR signals on FO B cells, but not on transitional type 1 (T1) B cells, the latter can still undergo clonal deletion in response to autoreactive BCR signals while follicular B cells become less susceptible to this trigger [105]. Accordingly, the immature WEHI231 mouse B cell lymphoma line undergoes apoptosis in response to BCR stimulation, which can be prevented by co-stimulatory CD40 signals known to strongly upregulate A1 [106]. Consistent with these findings, B cells from SLE patients show increased levels of A1/Bfl-1 [107]. These results indicate the essential role of A1 upon antigen stimulation of mature B cells, but the role of A1 on B cell effector function is still unclear. Our own results using B cells derived from A1 knockdown mice support a role for A1 as a key survival factor for mature FO B cells as their percentage is decreased in these animals. This finding correlated with increased sensitivity of transitional type 2 (T2) and FO B cells to BCR ligation-mediated killing ex vivo and impaired proliferation of splenic B cells upon mitogenic stimulation (Ottina et al., in revision).
A1/Bfl-1 in the development and function of myeloid cells
A1 was originally identified in bone marrow derived macrophages treated with GM-CSF [55]. Since then its role in the myeloid compartment has been widely studied. Under steady state conditions neutrophils are short-lived. However, upon infection and in response to inflammatory cytokines these cells have an extended lifespan allowing the effective clearance of bacteria. A1 is expressed in both murine and human neutrophils and is induced via NF-kB signals in response to bacterial endotoxins like LPS or pro-inflammatory cytokines, such as G-CSF, GM-CSF, TNFα, IFN-γ or IL-8, all extending cell survival [55,89]. Neutrophils derived from mice lacking one of the three functional A1 isoforms, A1-a, have a higher spontaneous apoptosis rate or when exposed to LPS ex vivo do no longer respond with increased survival, indicating that A1 is the main inhibitor of neutrophil apoptosis [108]. However, A1-a−/− mice have normal granulocyte numbers under (i) steady state as well as (ii) inflammatory conditions induced by i.p. injection of peptone. This indicates that bone marrow resident progenitors do still have sufficient clonogenic capacity to produce sufficient neutrophils A1-a knockout mice [108].
Our own results using in vivo RNAi to knockdown all A1 isoforms in transgenic mice show that dependent on the rate of knockdown achieved, granulocytes fail to accumulate in these animals or die most rapidly ex vivo. We also noted that the clonogenic potential of bone marrow derived myeloid progenitors from these mice show reduced capacity to form granulocytic colonies, particularly under conditions of delayed cytokine addition (Ottina et al., in revision). This indicates that A1 may not only be critical for mature granulocyte survival but possibly also for the survival and/or differentiation of their progenitors.
As mentioned above, A1 is constitutively expressed in neutrophils while it is induced by inflammatory cytokines such as INF-γ in macrophages. In particular A1-a is the isoform with maximal fold-induction in inflammatory macrophages [109]. Treatment with LPS or Mycobacterium bovis derived Bacillus Calmette-Guerin (BCG), or Toxoplasma gondii elicts the expression of A1 in the first 8–16 h post infection via the NF-kB signaling pathway, which then decreases by 24 h [109–111]. The expression of A1 correlated with increased macrophage survival upon infection with BCG and nitric oxide resistance, while macrophages derived from A1-a−/− mice fail to be protected [109]. On the one hand A1 seems to be essential for macrophage survival upon infection but on the other hand A1 is possibly a negative regulator of the autophagosome maturation in virulent mycobacteria infected macrophages. In particular, in Mycobacterium tubercolosis infected macrophages, A1 overexpression inhibits the maturation and acidification of mycobacterial phagosomes by the inhibition of the autophagy pathway [111]. Since A1 inhibits the apoptosis of the infected macrophages [80] but also the killing of intracellular mycobacteria in autophagosome maturation, the virulent M. tubercolosis strain is able to survive within the host.
Together with Mcl-1 [51], A1 seems to play a key role in the mediation of mast cell survival and subsequent severity of allergic reactions. Mast cells are long-lived myeloid cells that reside in the tissue and skin where they participate in inflammatory responses to infection and parasites, but they can also cause allergic reactions. These cells are characterized by the presence of granules rich in histamine, heparin, leukotriens, prostaglandin and other inflammatory mediators that can be released via the activation of their IgE receptor, FcεR1, which also promotes their cell survival. De-granulated mast cells are therefore able to re-granulate and be activated repeatedly leading to the perpetuation of the allergic response. Both murine and human A1 are induced in mast cells already 2 h post IgE receptor activation [112] by a mechanism which is dependent on NF-AT activation and calcium release, but NF-kB independent [113,114]. Interestingly, inflammatory stimuli such as GM-CSF, SCF, IL-4 or TNF-α fail to induce A1 in mast cells, contrary to what has been observed in granulocytes and macrophages. Crosslinking of the FcεR1 induces A1 to promote survival, which is diminished in mast cells derived from A1-a−/− mice challenged ex vivo [112].
Non-hematopoietic functions of A1/Bfl-1
Outside the hematopoietic system, A1 expression was observed in vascular endothelial (VEC) and smooth muscle cells (SMC) after stimulation with pro-inflammatory cytokines (IL-1β, TNF-α) or PMA [59]. In endothelial cells, A1 can confer protection to TNF-α and ceramide-induced apoptosis [115]. In addition, high levels of human A1 were detected in the lung, moderate levels in the intestine and testis and lower extent in the heart, placenta, skeletal muscle, pancreas, colon and prostate [59]. However, this widespread expression pattern might actually result from contaminating hematopoietic cells, VEC or SMC.
Atherosclerosis is characterized by increased VSMC proliferation and is accelerated in diabetic patients. Increased A1 expression in VSMC might contribute to the development of vascular disease as it was shown that high glucose levels augment A1 and Bcl-xL expression and cell death protection in coronary artery smooth muscle cells [116].
A protective role for A1 in hyperoxic acute lung injury (HALI), induced by high O2 concentrations after treatment of patients suffering from pulmonary or cardiac diseases was also reported. Additionally, IL-11 and VEGF can mediate protective functions in HALI by inducing A1, which was abolished in A1-a knockout mice [117].
A1 might also be cytoprotective in Alzheimer disease. The n-3 fatty acid, docosahexaenoic acid and its derivative, neuroprotectin D1 (NPD1) that is reduced in the hippocampal cornu ammonis region 1 in Alzheimer patients, induces expression of pro-survival Bcl-2, Bcl-xL and A1 [118].
Finally, conflicting results have been reported on A1 expression in the mammary gland. Augmented A1 levels were observed in mammary tissue upon forced weaning of pregnant mice that might contribute to the delayed involution observed in such animals [119]. However, other reports showed A1 downregulation during involution depending on the genetic background of the mouse strains used [120]. A more detailed analysis of these phenomena is required given a possible role of A1 in breast cancer [90].
Concluding remarks
20 years after its initial description, we have gathered some evidence of A1 function in leukocyte development and hematological disorders. However, we know very little about A1 function outside the hematopoietic system or the E3-ligases involved in its rapid turnover. Generation of reliable antibodies, recognizing endogenous A1, the detailed analysis of recently generated in vivo knockdown mouse models (Ottina et al., in revision) and sophisticated future gene-targeting will help to better define A1 function in normal physiology and disease. Given its proposed role in drug-resistance, tumor progression and metastasis as well as its association with autoimmunity in humans, selective inactivation of its pro-survival action by next generation BH3-mimetics [74,75] or optimized versions of peptide aptamers [121] should help to improve treatment strategies for multiple human pathologies.
Acknowledgments
Research in our laboratory is supported by the Austrian Science Fund (FWF; AV and EO), the Österreichische Krebshilfe-Tirol (EO and DT) and the Daniel Swarovski Fund (DT). MH was funded by a postdoctoral fellowship from the German Research Council (DFG, He 5740/1-1). The authors have no competing financial interests.
References
- 1.Lavrik I.N., Krammer P.H. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012;19:36–41. doi: 10.1038/cdd.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Youle R.J., Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 3.Chipuk J.E., Green D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strasser A. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 2005;5:189–200. doi: 10.1038/nri1568. [DOI] [PubMed] [Google Scholar]
- 5.Gyrd-Hansen M., Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer. 2011;10:561–574. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- 6.Kitagawa K., Niikura Y. Caspase-independent mitotic death (CIMD) Cell Cycle. 2008;7:1001–1005. doi: 10.4161/cc.7.8.5720. [DOI] [PubMed] [Google Scholar]
- 7.Delavallee L., Cabon L., Galan-Malo P., Lorenzo H.K., Susin S.A. AIF-mediated caspase-independent necroptosis: a new chance for targeted therapeutics. IUBMB Life. 2011;63:221–232. doi: 10.1002/iub.432. [DOI] [PubMed] [Google Scholar]
- 8.Afonina I.S., Cullen S.P., Martin S.J. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol. Rev. 2011;235:105–116. doi: 10.1111/j.0105-2896.2010.00908.x. [DOI] [PubMed] [Google Scholar]
- 9.Bakhshi A., Jensen J.P., Goldman P., Wright J.J., McBride O.W., Epstein A.L., Korsmeyer S.J. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- 10.Tsujimoto Y., Finger L.R., Yunis J., Nowell P.C., Croce C.M. Vol. 226. 1984. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation; pp. 1097–1099. (Science (New York, N.Y)). [DOI] [PubMed] [Google Scholar]
- 11.Lindsten T., Ross A.J., King A., Zong W., Rathmell J.C., Shiels H.A., Ulrich E., Waymire K.G., Mahar P., Frauwirth K., Chen Y., Wei M., Eng V.M., Adelman D.M., Simon M.C., Ma A., Golden J.A., Evan G., Korsmeyer S.J., MacGregor G.R., Thompson C.B. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol. Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Labi V., Erlacher M., Kiessling S., Villunger A. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ. 2006;13:1325–1338. doi: 10.1038/sj.cdd.4401940. [DOI] [PubMed] [Google Scholar]
- 13.Merino D., Giam M., Hughes P.D., Siggs O.M., Heger K., O'Reilly L.A., Adams J.M., Strasser A., Lee E.F., Fairlie W.D., Bouillet P. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J. Cell Biol. 2009;186:355–362. doi: 10.1083/jcb.200905153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llambi F., Moldoveanu T., Tait S.W., Bouchier-Hayes L., Temirov J., McCormick L.L., Dillon C.P., Green D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell. 2011;44:517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grespi F., Soratroi C., Krumschnabel G., Sohm B., Ploner C., Geley S., Hengst L., Hacker G., Villunger A. BH3-only protein Bmf mediates apoptosis upon inhibition of CAP-dependent protein synthesis. Cell Death Differ. 2010;17:1672–1683. doi: 10.1038/cdd.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ley R., Ewings K.E., Hadfield K., Cook S.J. Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell Death Differ. 2005;12:1008–1014. doi: 10.1038/sj.cdd.4401688. [DOI] [PubMed] [Google Scholar]
- 17.Erlacher M., Labi V., Manzl C., Bock G., Tzankov A., Hacker G., Michalak E., Strasser A., Villunger A. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J. Exp. Med. 2006;203:2939–2951. doi: 10.1084/jem.20061552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Labi V., Villunger A. PUMA-mediated tumor suppression: a tale of two stories. Cell Cycle. 2010;9:4269–4275. doi: 10.4161/cc.9.21.13666. [DOI] [PubMed] [Google Scholar]
- 19.Pinon J.D., Labi V., Egle A., Villunger A. Bim and Bmf in tissue homeostasis and malignant disease. Oncogene. 2008;27(Suppl. 1):S41–S52. doi: 10.1038/onc.2009.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maiuri M.C., Criollo A., Tasdemir E., Vicencio J.M., Tajeddine N., Hickman J.A., Geneste O., Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3:374–376. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 21.Hitomi J., Christofferson D.E., Ng A., Yao J., Degterev A., Xavier R.J., Yuan J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strasser A., Harris A.W., Bath M.L., Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 23.Vaux D.L., Cory S., Adams J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 24.Lanave C., Santamaria M., Saccone C. Comparative genomics: the evolutionary history of the Bcl-2 family. Gene. 2004;333:71–79. doi: 10.1016/j.gene.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen M., Millar D.G., Wee-Yong V., Korsmeyer S.J., Shore G.C. Targeting of Bcl-2 to the mitochondrial outer membrane by a COOH-terminal signal anchor sequence. J. Biol. Chem. 1993;268:25265–25268. [PubMed] [Google Scholar]
- 26.Wilson-Annan J., O'Reilly L.A., Crawford S.A., Hausmann G., Beaumont J.G., Parma L.P., Chen L., Lackmann M., Lithgow T., Hinds M.G., Day C.L., Adams J.M., Huang D.C. Proapoptotic BH3-only proteins trigger membrane integration of prosurvival Bcl-w and neutralize its activity. J. Cell Biol. 2003;162:877–887. doi: 10.1083/jcb.200302144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nijhawan D., Fang M., Traer E., Zhong Q., Gao W., Du F., Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu Y.-T., Wolter K.G., Youle R.J. Cytosol-to-membrane redistribution of Bax and Bcl-XL during apoptosis. Proc. Natl. Acad. Sci. U. S. A. 1997;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herold M.J., Zeitz J., Pelzer C., Kraus C., Peters A., Wohlleben G., Berberich I. The stability and anti-apoptotic function of A1 are controlled by its C terminus. J. Biol. Chem. 2006;281:13663–13671. doi: 10.1074/jbc.M600266200. [DOI] [PubMed] [Google Scholar]
- 30.Kucharczak J.F., Simmons M.J., Duckett C.S., Gelinas C. Constitutive proteasome-mediated turnover of Bfl-1/A1 and its processing in response to TNF receptor activation in FL5.12 pro-B cells convert it into a prodeath factor. Cell Death Differ. 2005;12:1225–1239. doi: 10.1038/sj.cdd.4401684. [DOI] [PubMed] [Google Scholar]
- 31.Brien G., Debaud A.L., Robert X., Oliver L., Trescol-Biemont M.C., Cauquil N., Geneste O., Aghajari N., Vallette F.M., Haser R., Bonnefoy-Berard N. C-terminal residues regulate localization and function of the antiapoptotic protein Bfl-1. J. Biol. Chem. 2009;284:30257–30263. doi: 10.1074/jbc.M109.040824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber A., Paschen S.A., Heger K., Wilfling F., Frankenberg T., Bauerschmitt H., Seiffert B.M., Kirschnek S., Wagner H., Hacker G. BimS-induced apoptosis requires mitochondrial localization but not interaction with anti-apoptotic Bcl-2 proteins. J. Cell Biol. 2007;177:625–636. doi: 10.1083/jcb.200610148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore N.C., Anderson G., Williams G.T., Owen J.J.T., Jenkinson E.J. Developmental regulation of bcl-2 expression in the thymus. Immunology. 1994;81:115–119. [PMC free article] [PubMed] [Google Scholar]
- 34.Ma A., Pena J.C., Chang B., Margosian E., Davidson L., Alt F.W., Thompson C.B. Bclx regulates the survival of double-positive thymocytes. Proc. Natl. Acad. Sci. U. S. A. 1995;92:4763–4767. doi: 10.1073/pnas.92.11.4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marash L., Liberman N., Henis-Korenblit S., Sivan G., Reem E., Elroy-Stein O., Kimchi A. DAP5 promotes cap-independent translation of Bcl-2 and CDK1 to facilitate cell survival during mitosis. Mol. Cell. 2008;30:447–459. doi: 10.1016/j.molcel.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto K., Ichijo H., Korsmeyer S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G2/M. Mol. Cell. Biol. 1999;19:8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verschelde C., Walzer T., Galia P., Biemont M.C., Quemeneur L., Revillard J.P., Marvel J., Bonnefoy-Berard N. A1/Bfl-1 expression is restricted to TCR engagement in T lymphocytes. Cell Death Differ. 2003;10:1059–1067. doi: 10.1038/sj.cdd.4401265. [DOI] [PubMed] [Google Scholar]
- 38.Grad J.M., Zeng X.R., Boise L.H. Regulation of Bcl-xL: a little bit of this and a little bit of STAT. Curr. Opin. Oncol. 2000;12:543–549. doi: 10.1097/00001622-200011000-00006. [DOI] [PubMed] [Google Scholar]
- 39.Mackay F., Schneider P. Cracking the BAFF code. Nat. Rev. Immunol. 2009;9:491–502. doi: 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J., D'Ercole A.J. Expression of Mcl-1 in cerebellar granule neurons is regulated by IGF-I in a developmentally specific fashion. Brain Res. Dev. Brain Res. 2004;152:255–263. doi: 10.1016/j.devbrainres.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 41.Henson E.S., Gibson E.M., Villanueva J., Bristow N.A., Haney N., Gibson S.B. Increased expression of Mcl-1 is responsible for the blockage of TRAIL-induced apoptosis mediated by EGF/ErbB1 signaling pathway. J. Cell. Biochem. 2003;89:1177–1192. doi: 10.1002/jcb.10597. [DOI] [PubMed] [Google Scholar]
- 42.Zhong Q., Gao W., Du F., Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121:1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 43.Wertz I.E., Kusam S., Lam C., Okamoto T., Sandoval W., Anderson D.J., Helgason E., Ernst J.A., Eby M., Liu J., Belmont L.D., Kaminker J.S., O'Rourke K.M., Pujara K., Kohli P.B., Johnson A.R., Chiu M.L., Lill J.R., Jackson P.K., Fairbrother W.J., Seshagiri S., Ludlam M.J., Leong K.G., Dueber E.C., Maecker H., Huang D.C., Dixit V.M. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 44.Veis D.J., Sorenson C.M., Shutter J.R., Korsmeyer S.J. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- 45.Bouillet P., Cory S., Zhang L.-C., Strasser A., Adams J.M. Degenerative disorders caused by Bcl-2 deficiency are prevented by loss of its BH3-only antagonist Bim. Dev. Cell. 2001;1:645–653. doi: 10.1016/s1534-5807(01)00083-1. [DOI] [PubMed] [Google Scholar]
- 46.Motoyama N., Wang F.P., Roth K.A., Sawa H., Nakayama K., Nakayama K., Negishi I., Senju S., Zhang Q., Fujii S., Loh D.Y. Vol. 267. 1995. Massive cell death of immature hematopoietic cells and neurons in Bcl-x deficient mice; pp. 1506–1510. (Science (New York, N.Y)). [DOI] [PubMed] [Google Scholar]
- 47.Print C.G., Loveland K.L., Gibson L., Meehan T., Stylianou A., Wreford N., de Kretser D., Metcalf D., Köntgen F., Adams J.M., Cory S. Apoptosis regulator Bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc. Natl. Acad. Sci. U. S. A. 1998;95:12424–12431. doi: 10.1073/pnas.95.21.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rinkenberger J.L., Horning S., Klocke B., Roth K., Korsmeyer S.J. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000;14:23–27. [PMC free article] [PubMed] [Google Scholar]
- 49.Opferman J.T., Iwasaki H., Ong C.C., Suh H., Mizuno S., Akashi K., Korsmeyer S.J. Vol. 307. 2005. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells; pp. 1101–1104. (Science (New York, N.Y)). [DOI] [PubMed] [Google Scholar]
- 50.Opferman J.T., Letai A., Beard C., Sorcinelli M.D., Ong C.C., Korsmeyer S.J. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 51.Lilla J.N., Chen C.C., Mukai K., Benbarak M.J., Franco C.B., Kalesnikoff J., Yu M., Tsai M., Piliponsky A.M., Galli S.J. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood. 2011;118(26):6930–6938. doi: 10.1182/blood-2011-03-343962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Germain M., Nguyen A.P., Le Grand J.N., Arbour N., Vanderluit J.L., Park D.S., Opferman J.T., Slack R.S. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 2011;30:395–407. doi: 10.1038/emboj.2010.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weber A., Boger R., Vick B., Urbanik T., Haybaeck J., Zoller S., Teufel A., Krammer P.H., Opferman J.T., Galle P.R., Schuchmann M., Heikenwalder M., Schulze-Bergkamen H. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology. 2010;51:1226–1236. doi: 10.1002/hep.23479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orlofsky A., Berger M.S., Prystowsky M.B. Novel expression pattern of a new member of the MIP-1 family of cytokine-like genes. Cell Regul. 1991;2:403–412. doi: 10.1091/mbc.2.5.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin E.Y., Orlofsky A., Berger M.S., Prystowsky M.B. Characterization of A1, a novel hemopoietic-specific early-response gene with sequence similarity to bcl-2. J. Immunol. 1993;151:1979–1988. [PubMed] [Google Scholar]
- 56.Hatakeyama S., Hamasaki A., Negishi I., Loh D.Y., Sendo F., Nakayama K. Multiple gene duplication and expression of mouse bcl-2-related genes, A1. Int. Immunol. 1998;10:631–637. doi: 10.1093/intimm/10.5.631. [DOI] [PubMed] [Google Scholar]
- 57.Mandal M., Borowski C., Palomero T., Ferrando A.A., Oberdoerffer P., Meng F., Ruiz-Vela A., Ciofani M., Zuniga-Pflucker J.C., Screpanti I., Look A.T., Korsmeyer S.J., Rajewsky K., von Boehmer H., Aifantis I. The BCL2A1 gene as a pre-T cell receptor-induced regulator of thymocyte survival. J. Exp. Med. 2005;201:603–614. doi: 10.1084/jem.20041924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi S.S., Park I.C., Yun J.W., Sung Y.C., Hong S.I., Shin H.S. A novel bcl-2 related gene, bfl-1, is overexpressed in stomach cancer and preferentially expressed in bone marrow. Oncogene. 1995;11:1693–1698. [PubMed] [Google Scholar]
- 59.Karsan A., Yee E., Kaushansky K., Harlan J.M. Cloning of human Bcl-2 homologue: inflammatory cytokines induce human A1 in cultured endothelial cells. Blood. 1996;87:3089–3096. [PubMed] [Google Scholar]
- 60.Kenny J.J., Knobloch T.J., Augustus M., Carter K.C., Rosen C.A., Lang J.C. Grs, a novel member of the Bcl-2 gene family, is highly expressed in multiple cancer cell lines and in normal leukocytes. Oncogene. 1997;14:997–1001. doi: 10.1038/sj.onc.1200898. [DOI] [PubMed] [Google Scholar]
- 61.Lin E.Y., Kozak C.A., Orlofsky A., Prystowsky M.B. The bcl-2 family member, Bcl2a1, maps to mouse chromosome 9 and human chromosome 15. Mamm. Genome. 1997;8:293–294. doi: 10.1007/s003359900418. [DOI] [PubMed] [Google Scholar]
- 62.Choi S.S., Park S.H., Kim U.J., Shin H.S. Bfl-1, a Bcl-2-related gene, is the human homolog of the murine A1, and maps to chromosome 15q24.3. Mamm. Genome. 1997;8:781–782. doi: 10.1007/s003359900567. [DOI] [PubMed] [Google Scholar]
- 63.Ko J.K., Lee M.J., Cho S.H., Cho J.A., Lee B.Y., Koh J.S., Lee S.S., Shim Y.H., Kim C.W. Bfl-1S, a novel alternative splice variant of Bfl-1, localizes in the nucleus via its C-terminus and prevents cell death. Oncogene. 2003;22:2457–2465. doi: 10.1038/sj.onc.1206274. [DOI] [PubMed] [Google Scholar]
- 64.Smits C., Czabotar P.E., Hinds M.G., Day C.L. Structural plasticity underpins promiscuous binding of the prosurvival protein A1. Structure. 2008;16:818–829. doi: 10.1016/j.str.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 65.Herman M.D., Nyman T., Welin M., Lehtio L., Flodin S., Tresaugues L., Kotenyova T., Flores A., Nordlund P. Completing the family portrait of the anti-apoptotic Bcl-2 proteins: crystal structure of human Bfl-1 in complex with Bim. FEBS Lett. 2008;582:3590–3594. doi: 10.1016/j.febslet.2008.09.028. [DOI] [PubMed] [Google Scholar]
- 66.Werner A.B., de Vries E., Tait S.W., Bontjer I., Borst J. Bcl-2 family member Bfl-1/A1 sequesters truncated bid to inhibit is collaboration with pro-apoptotic Bak or Bax. J. Biol. Chem. 2002;277:22781–22788. doi: 10.1074/jbc.M201469200. [DOI] [PubMed] [Google Scholar]
- 67.Tao W.K., Kurschner C., Morgan J.I. Modulation of cell death in yeast by the Bcl-2 family of proteins. J. Biol. Chem. 1997;272:15547–15552. doi: 10.1074/jbc.272.24.15547. [DOI] [PubMed] [Google Scholar]
- 68.Zhang H., Cowan-Jacob S.W., Simonen M., Greenhalf W., Heim J., Meyhack B. Structural basis of BFL-1 for its interaction with BAX and its anti-apoptotic action in mammalian and yeast cells. J. Biol. Chem. 2000;275:11092–11099. doi: 10.1074/jbc.275.15.11092. [DOI] [PubMed] [Google Scholar]
- 69.Simmons M.J., Fan G., Zong W.X., Degenhardt K., White E., Gelinas C. Bfl-1/A1 functions, similar to Mcl-1, as a selective tBid and Bak antagonist. Oncogene. 2008;27:1421–1428. doi: 10.1038/sj.onc.1210771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holmgreen S.P., Huang D.C.S., Adams J.M., Cory S. Survival activity of Bcl-2 homologs Bcl-w and A1 only partially correlates with their ability to bind pro-apoptotic family members. Cell Death Differ. 1999;6:525–532. doi: 10.1038/sj.cdd.4400519. [DOI] [PubMed] [Google Scholar]
- 71.Willis S.N., Chen L., Dewson G., Wei A., Naik E., Fletcher J.I., Adams J.M., Huang D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ku B., Liang C., Jung J.U., Oh B.H. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 2010;21:627–641. doi: 10.1038/cr.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen L., Willis S.N., Wei A., Smith B.J., Fletcher J.I., Hinds M.G., Colman P.M., Day C.L., Adams J.M., Huang D.C. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 74.Vogler M., Butterworth M., Majid A., Walewska R.J., Sun X.M., Dyer M.J., Cohen G.M. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood. 2009;113:4403–4413. doi: 10.1182/blood-2008-08-173310. [DOI] [PubMed] [Google Scholar]
- 75.Yecies D., Carlson N.E., Deng J., Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2011;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cheng E.H.-Y., Kirsch D.G., Clem R.J., Ravi R., Kastan M.B., Bedi A., Ueno K., Hardwick J.M. Vol. 278. 1997. Conversion of Bcl-2 to a Bax-like death effector by caspases; pp. 1966–1968. (Science (New York, N.Y)). [DOI] [PubMed] [Google Scholar]
- 77.Fujita N., Nagahashi A., Nagashima K., Rokudai S., Tsuruo T. Acceleration of apoptotic cell death after the cleavage of Bcl-XL protein by caspase-3-like proteases. Oncogene. 1998;17:1295–1304. doi: 10.1038/sj.onc.1202065. [DOI] [PubMed] [Google Scholar]
- 78.Grillot D.A., Gonzalez-Garcia M., Ekhterae D., Duan L., Inohara N., Ohta S., Seldin M.F., Nunez G. Genomic organization, promoter region analysis, and chromosome localization of the mouse bcl-x gene. J. Immunol. 1997;158:4750–4757. [PubMed] [Google Scholar]
- 79.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dhiman R., Kathania M., Raje M., Majumdar S. Inhibition of bfl-1/A1 by siRNA inhibits mycobacterial growth in THP-1 cells by enhancing phagosomal acidification. Biochim. Biophys. Acta. 2008;1780:733–742. doi: 10.1016/j.bbagen.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 81.Chuang P.I., Morefield S., Liu C.Y., Chen S., Harlan J.M., Willerford D.M. Perturbation of B-cell development in mice overexpressing the Bcl-2 homolog A1. Blood. 2002;99:3350–3359. doi: 10.1182/blood.v99.9.3350. [DOI] [PubMed] [Google Scholar]
- 82.Egle A., Harris A.W., Bath M.L., O'Reilly L., Cory S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood. 2004;103:2276–2283. doi: 10.1182/blood-2003-07-2469. [DOI] [PubMed] [Google Scholar]
- 83.Fan G., Simmons M.J., Ge S., Dutta-Simmons J., Kucharczak J., Ron Y., Weissmann D., Chen C.C., Mukherjee C., White E., Gelinas C. Defective ubiquitin-mediated degradation of antiapoptotic Bfl-1 predisposes to lymphoma. Blood. 2011;115:3559–3569. doi: 10.1182/blood-2009-08-236760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beverly L.J., Varmus H.E. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene. 2009;28:1274–1279. doi: 10.1038/onc.2008.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Olsson A., Norberg M., Okvist A., Derkow K., Choudhury A., Tobin G., Celsing F., Osterborg A., Rosenquist R., Jondal M., Osorio L.M. Upregulation of bfl-1 is a potential mechanism of chemoresistance in B-cell chronic lymphocytic leukaemia. Br. J. Cancer. 2007;97:769–777. doi: 10.1038/sj.bjc.6603951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Morales A.A., Olsson A., Celsing F., Osterborg A., Jondal M., Osorio L.M. High expression of bfl-1 contributes to the apoptosis resistant phenotype in B-cell chronic lymphocytic leukemia. Int. J. Cancer. 2005;113:730–737. doi: 10.1002/ijc.20614. [DOI] [PubMed] [Google Scholar]
- 87.Simpson L.A., Burwell E.A., Thompson K.A., Shahnaz S., Chen A.R., Loeb D.M. The antiapoptotic gene A1/BFL1 is a WT1 target gene that mediates granulocytic differentiation and resistance to chemotherapy. Blood. 2006;107:4695–4702. doi: 10.1182/blood-2005-10-4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jenal M., Batliner J., Reddy V.A., Haferlach T., Tobler A., Fey M.F., Torbett B.E., Tschan M.P. The anti-apoptotic gene BCL2A1 is a novel transcriptional target of PU.1. Leukemia. 2010;24:1073–1076. doi: 10.1038/leu.2010.26. [DOI] [PubMed] [Google Scholar]
- 89.Chuang P.I., Yee E., Karsan A., Winn R.K., Harlan J.M. A1 is a constitutive and inducible Bcl-2 homologue in mature human neutrophils. Biochem. Biophys. Res. Commun. 1998;249:361–365. doi: 10.1006/bbrc.1998.9155. [DOI] [PubMed] [Google Scholar]
- 90.Vogler M. BCL2A1: the underdog in the BCL2 family. Cell Death Differ. 2011;19(1):67–74. doi: 10.1038/cdd.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tomayko M.M., Punt J.A., Bolcavage J.M., Levy S.L., Allman D.M., Cancro M.P. Expression of the Bcl-2 family member A1 is developmentally regulated in T cells. Int. Immunol. 1999;11:1753–1761. doi: 10.1093/intimm/11.11.1753. [DOI] [PubMed] [Google Scholar]
- 92.Akashi K., Kondo M., von Freeden-Jeffry U., Murray R., Weissman I.L. Bcl-2 rescues T lymphopoiesis in interleukin-7 receptor-deficient mice. Cell. 1997;89:1033–1041. doi: 10.1016/s0092-8674(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 93.Verschelde C., Michonneau D., Trescol-Biemont M.C., Berberich I., Schimpl A., Bonnefoy-Berard N. Overexpression of the antiapoptotic protein A1 promotes the survival of double positive thymocytes awaiting positive selection. Cell Death Differ. 2006;13:1213–1221. doi: 10.1038/sj.cdd.4401814. [DOI] [PubMed] [Google Scholar]
- 94.Rasooly R., Schuster G.U., Gregg J.P., Xiao J.H., Chandraratna R.A., Stephensen C.B. Retinoid x receptor agonists increase bcl2a1 expression and decrease apoptosis of naive T lymphocytes. J. Immunol. 2005;175:7916–7929. doi: 10.4049/jimmunol.175.12.7916. [DOI] [PubMed] [Google Scholar]
- 95.Wensveen F.M., van Gisbergen K.P., Derks I.A., Gerlach C., Schumacher T.N., van Lier R.A., Eldering E. Apoptosis threshold set by Noxa and Mcl-1 after T cell activation regulates competitive selection of high-affinity clones. Immunity. 2010;32:754–765. doi: 10.1016/j.immuni.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 96.Lee H.W., Park S.J., Choi B.K., Kim H.H., Nam K.O., Kwon B.S. 4-1BB promotes the survival of CD8 + T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J. Immunol. 2002;169:4882–4888. doi: 10.4049/jimmunol.169.9.4882. [DOI] [PubMed] [Google Scholar]
- 97.Gonzalez J., Orlofsky A., Prystowsky M.B. A1 is a growth-permissive antiapoptotic factor mediating postactivation survival in T cells. Blood. 2003;101:2679–2685. doi: 10.1182/blood-2002-04-1229. [DOI] [PubMed] [Google Scholar]
- 98.Tomayko M.M., Cancro M.P. Long-lived B cells are distinguished by elevated expression of A1. J. Immunol. 1998;160:107–111. [PubMed] [Google Scholar]
- 99.Trescol-Biemont M.C., Verschelde C., Cottalorda A., Bonnefoy-Berard N. Regulation of A1/Bfl-1 expression in peripheral splenic B cells. Biochimie. 2004;86:287–294. doi: 10.1016/j.biochi.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 100.Merino R., Ding L., Veis D.J., Korsmeyer S.J., Nuñez G. Developmental regulation of the Bcl-2 protein and susceptibility to cell death in B lymphocytes. EMBO J. 1994;13:683–691. doi: 10.1002/j.1460-2075.1994.tb06307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vikstrom I., Carotta S., Luthje K., Peperzak V., Jost P.J., Glaser S., Busslinger M., Bouillet P., Strasser A., Nutt S.L., Tarlinton D.M. Mcl-1 is essential for germinal center formation and B cell memory. Science (New York, N.Y) 2010;330:1095–1099. doi: 10.1126/science.1191793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Grillot D.A.M., Merino R., Pena J.C., Fanslow W.C., Finkelman F.D., Thompson C.B., Núñez G. bcl-x exhibits regulated expression during B cell development and activation and modulates lymphocyte survival in transgenic mice. J. Exp. Med. 1996;183:381–391. doi: 10.1084/jem.183.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hsu B.L., Harless S.M., Lindsley R.C., Hilbert D.M., Cancro M.P. Cutting edge: BLyS enables survival of transitional and mature B cells through distinct mediators. J. Immunol. 2002;168:5993–5996. doi: 10.4049/jimmunol.168.12.5993. [DOI] [PubMed] [Google Scholar]
- 104.Grossmann M., O'Reilly L.A., Gugasyan R., Strasser A., Adams J.M., Gerondakis S. The anti-apoptotic activities of rel and RelA required during B-cell maturation involve the regulation of Bcl-2 expression. EMBO J. 2000;19:6351–6360. doi: 10.1093/emboj/19.23.6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wen R., Chen Y., Xue L., Schuman J., Yang S., Morris S.W., Wang D. Phospholipase Cgamma2 provides survival signals via Bcl2 and A1 in different subpopulations of B cells. J. Biol. Chem. 2003;278:43654–43662. doi: 10.1074/jbc.M307318200. [DOI] [PubMed] [Google Scholar]
- 106.Kuss A.W., Knödel M., Berberich-Siebelt F., Lindemann D., Schimpl A., Berberich I. A1 expression is stimulated by CD40 in B cells and rescues WEHI 231 cells from anti-IgM-induced cell death. Eur. J. Immunol. 1999;29:3077–3088. doi: 10.1002/(SICI)1521-4141(199910)29:10<3077::AID-IMMU3077>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 107.Andre J., Cimaz R., Ranchin B., Galambrun C., Bertrand Y., Bouvier R., Rieux-Laucat F., Trescol-Biemont M.C., Cochat P., Bonnefoy-Berard N. Overexpression of the antiapoptotic gene Bfl-1 in B cells from patients with familial systemic lupus erythematosus. Lupus. 2007;16:95–100. doi: 10.1177/0961203306075382. [DOI] [PubMed] [Google Scholar]
- 108.Hamasaki A., Sendo F., Nakayama K., Ishida N., Negishi I., Nakayama K.-I., Hatakeyama S. Accelerated neutrophil apoptosis in mice lacking A1-a, a subtype of the bcl-2-related A1 gene. J. Exp. Med. 1998;188:1985–1992. doi: 10.1084/jem.188.11.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kausalya S., Somogyi R., Orlofsky A., Prystowsky M.B. Requirement of A1-a for bacillus Calmette-Guerin-mediated protection of macrophages against nitric oxide-induced apoptosis. J. Immunol. 2001;166:4721–4727. doi: 10.4049/jimmunol.166.7.4721. [DOI] [PubMed] [Google Scholar]
- 110.Orlofsky A., Somogyi R.D., Weiss L.M., Prystowsky M.B. The murine antiapoptotic protein A1 is induced in inflammatory macrophages and constitutively expressed in neutrophils. J. Immunol. 1999;163:412–419. [PubMed] [Google Scholar]
- 111.Kathania M., Raje C.I., Raje M., Dutta R.K., Majumdar S. Bfl-1/A1 acts as a negative regulator of autophagy in mycobacteria infected macrophages. Int. J. Biochem. Cell Biol. 2010;43:573–585. doi: 10.1016/j.biocel.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 112.Xiang Z., Ahmed A.A., Moller C., Nakayama K., Hatakeyama S., Nilsson G. Essential role of the prosurvival bcl-2 homologue A1 in mast cell survival after allergic activation. J. Exp. Med. 2001;194:1561–1569. doi: 10.1084/jem.194.11.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xiang Z., Moller C., Nilsson G. IgE-receptor activation induces survival and Bfl-1 expression in human mast cells but not basophils. Allergy. 2006;61:1040–1046. doi: 10.1111/j.1398-9995.2006.01024.x. [DOI] [PubMed] [Google Scholar]
- 114.Ulleras E., Karlberg M., Moller Westerberg C., Alfredsson J., Gerondakis S., Strasser A., Nilsson G. NFAT but not NF-kappaB is critical for transcriptional induction of the prosurvival gene A1 after IgE receptor activation in mast cells. Blood. 2008;111:3081–3089. doi: 10.1182/blood-2006-10-053371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Karsan A., Yee E., Harlan J.M. Endothelial cell death induced by tumor necrosis factor-α is inhibited by the Bcl-2 family member, A1. J. Biol. Chem. 1996;271:27201–27204. doi: 10.1074/jbc.271.44.27201. [DOI] [PubMed] [Google Scholar]
- 116.Sakuma H., Yamamoto M., Okumura M., Kojima T., Maruyama T., Yasuda K. High glucose inhibits apoptosis in human coronary artery smooth muscle cells by increasing bcl-xL and bfl-1/A1. Am. J. Physiol. Cell Physiol. 2002;283:C422–C428. doi: 10.1152/ajpcell.00577.2001. [DOI] [PubMed] [Google Scholar]
- 117.He C.H., Waxman A.B., Lee C.G., Link H., Rabach M.E., Ma B., Chen Q., Zhu Z., Zhong M., Nakayama K., Nakayama K.I., Homer R., Elias J.A. Bcl-2-related protein A1 is an endogenous and cytokine-stimulated mediator of cytoprotection in hyperoxic acute lung injury. J. Clin. Invest. 2005;115:1039–1048. doi: 10.1172/JCI23004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lukiw W.J., Cui J.G., Marcheselli V.L., Bodker M., Botkjaer A., Gotlinger K., Serhan C.N., Bazan N.G. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Invest. 2005;115:2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Capuco A.V., Li M., Long E., Ren S., Hruska K.S., Schorr K., Furth P.A. Concurrent pregnancy retards mammary involution: effects on apoptosis and proliferation of the mammary epithelium after forced weaning of mice. Biol. Reprod. 2002;66:1471–1476. doi: 10.1095/biolreprod66.5.1471. [DOI] [PubMed] [Google Scholar]
- 120.Thangaraju M., Sharan S., Sterneck E. Comparison of mammary gland involution between 129S1 and C57BL/6 inbred mouse strains: differential regulation of Bcl2a1, Trp53, Cebpb, and Cebpd expression. Oncogene. 2004;23:2548–2553. doi: 10.1038/sj.onc.1207363. [DOI] [PubMed] [Google Scholar]
- 121.Brien G., Debaud A.L., Bickle M., Trescol-Biemont M.C., Moncorge O., Colas P., Bonnefoy-Berard N. Characterization of peptide aptamers targeting Bfl-1 anti-apoptotic protein. Biochemistry. 2011;50:5120–5129. doi: 10.1021/bi101839p. [DOI] [PubMed] [Google Scholar]