

Abstract
The industrially important organism Corynebacterium glutamicum has been characterized in recent years for its robust ability to assimilate aromatic compounds. In this study, C. glutamicum strain AS 1.542 was investigated for its ability to catabolize phenylacetic acid (PAA). The paa genes were identified; they are organized as a continuous paa gene cluster. The type strain of C. glutamicum, ATCC 13032, is not able to catabolize PAA, but the recombinant strain ATCC 13032/pEC-K18mob2::paa gained the ability to grow on PAA. The paaR gene, encoding a TetR family transcription regulator, was studied in detail. Disruption of paaR in strain AS 1.542 resulted in transcriptional increases of all paa genes. Transcription start sites and putative promoter regions were determined. An imperfect palindromic motif (5′-ACTNACCGNNCGNNCGGTNAGT-3′; 22 bp) was identified in the upstream regions of paa genes. Electrophoretic mobility shift assays (EMSA) demonstrated specific binding of PaaR to this motif, and phenylacetyl coenzyme A (PA-CoA) blocked binding. It was concluded that PaaR is the negative regulator of PAA degradation and that PA-CoA is the PaaR effector. In addition, GlxR binding sites were found, and binding to GlxR was confirmed. Therefore, PAA catabolism in C. glutamicum is regulated by the pathway-specific repressor PaaR, and also likely by the global transcription regulator GlxR. By comparative genomic analysis, we reconstructed orthologous PaaR regulons in 57 species, including species of Actinobacteria, Proteobacteria, and Flavobacteria, that carry PAA utilization genes and operate by conserved binding motifs, suggesting that PaaR-like regulation might commonly exist in these bacteria.
INTRODUCTION
Phenylacetic acid (PAA) is an important intermediate during the microbial degradation of aromatic compounds such as styrene, ethylbenzene, 2-phenylethylamine, tropic acid, phenylacetaldehyde, phenylacetyl esters, and amides (26), and PAA degradation pathways have been detected in a wide range of bacterial species (7, 10, 27, 30, 31). The initial reaction of PAA degradation is the activation of PAA to phenylacetyl coenzyme A (PA-CoA). Subsequently, PA-CoA is degraded via the function of a PA-CoA catabolon core (26) (see Fig. 1). Recently, it was discovered that PA-CoA is epoxidized and isomerized to form a seven-membered C-O-heterocyclic enol ether (oxepin-CoA), followed by hydrolytic ring cleavage and β-oxidation-like steps yielding tricarboxylic acid cycle intermediates (45, 46).
Fig 1.

(A) Schematic diagram of the phenylacetic acid (PAA) catabolic pathway (adapted from reference 45 with permission). (B) Genetic organization of the paa gene cluster in C. glutamicum strain AS 1.542, C. glutamicum strain R, C. efficiens YS-314, and E. coli K-12 MG1655. Homologous genes are presented in the same color, and the proposed enzyme names are listed at the bottom. The amino acid sequence similarities (expressed as percentages) of paa genes in Corynebacterium strains with those in E. coli K-12 are shown. N/A, not applicable; the paaT and paaR genes do not exist in E. coli K-12.
Genes encoding the PAA degradation pathways are diversely organized in bacteria (26). In Pseudomonas putida strain U, 15 paa genes are organized in five contiguous operons and are grouped in five functional units, including PAA transport, PAA activation, ring hydroxylation and cleavage, β-oxidation-like catalysis, and regulation (26, 28). In Escherichia coli, 14 paa genes are organized in three transcription units that function in PAA activation, ring hydroxylation and cleavage, β-oxidation, and regulation (10, 26). The regulation of the paa genes has been investigated in several bacterial species. In E. coli and Pseudomonas species, a GntR family transcriptional regulator named PaaX represses the transcription of the paa genes (7, 9). In Burkholderia cenocepacia K56-2 and Thermus thermophilus HB8, a transcriptional regulator of the TetR family, named PaaR, has been reported as a repressor of the PAA degradation pathway (13, 38). Instead of PAA, PA-CoA acts as the effector for all the regulatory proteins identified so far (9, 11, 38, 48).
C. glutamicum is a well-studied, high-GC-content, Gram-positive soil bacterium of industrial importance. The ability of C. glutamicum to metabolize a variety of aromatic compounds has been studied for its type strain, ATCC 13032 (5, 15, 41–43, 49). However, no degradation of PAA by C. glutamicum was reported. In this study, we investigated the abilities of various strains of C. glutamicum to degrade PAA. The results showed that strain ATCC 13032 is not able to utilize PAA but that a serine-auxotrophic strain, AS 1.542 (formerly Corynebacterium crenatum), isolated from winery soil in southeast China (6), is able to grow on PAA. The paa gene cluster in strain AS 1.542 was functionally characterized, and its regulation by a TetR-type PaaR protein was studied.
MATERIALS AND METHODS
Bacterial strains, growth conditions, primers, and plasmids.
The bacterial strains, primers, and plasmids used in this study are listed in Table S1 in the supplemental material. C. glutamicum strains were grown at 30°C in minimal medium (20) with 5 mM glucose or PAA as the sole carbon source. For strain AS 1.542, minimal medium was supplemented with 1 mM serine. E. coli was grown at 37°C in Luria-Bertani (LB) broth or on LB agar plates (39). When needed, antibiotics were added at the following concentrations: 50 μg/ml kanamycin or 200 μg/ml ampicillin for E. coli and 25 μg/ml kanamycin for C. glutamicum. Bacterial growth was monitored by measuring the optical density at 600 nm (OD600) or by using a Nephelostar Galaxy instrument (BMG Laboratories) (37).
DNA extraction and manipulation.
C. glutamicum chromosomal DNA was isolated with the GenElute kit (Sigma-Aldrich). E. coli plasmid isolation and transformation, DNA manipulation, ligation, and agarose gel electrophoresis were carried out using standard protocols (39). Transformation of C. glutamicum with plasmids was performed according to the method of Tauch et al. (44).
Identification and annotation of the paa gene cluster.
The genome sequence of strain AS 1.542 was determined with the GS FLX system (454 Life Sciences), and the paa gene cluster was identified after the initial sequence assembly. Annotation and functional assignment were carried out with GenDB software (29) and according to similarity to the paa genes of E. coli K-12 (45) using the NCBI BLAST interface (17).
Disruption of paaR.
paaR was disrupted by the gene splicing by overlap extension (GeneSOEing) method (14), removing a DNA fragment overlapping the putative helix-turn-helix (HTH) domain (amino acid residues 8 to 102) of PaaR. The primers used for gene SOE are listed in Table S1 in the supplemental material. The disrupted paaR gene (paaRΔΗΤΗ) was ligated into EcoRI/BamHI-treated pK18mobsacB, generating pK18mobsacB::paaRΔΗΤΗ. This plasmid was used to perform an allelic exchange of paaR in the chromosome of strain AS 1.542, according to the work of Schäfer et al. (40). An AS 1.542/paaRΔΗΤΗ mutant was obtained, and the disruption of paaR was confirmed by PCR amplification.
Introduction of the paa gene cluster into C. glutamicum strain ATCC 13032.
One-step isothermal DNA assembly (12) was used for the construction of plasmid pEC-K18mob2::paa. Primers (see Table S1 in the supplemental material) were designed to allow for a 40- or 41-bp overlap between the ends of the pEC-K18mob2 plasmid (22) and the amplified paa gene cluster. PCR products from the plasmid and the paa gene cluster were first purified and then treated with a DNA-blunting enzyme (CloneJET PCR cloning kit; Fermentas). After one-step isothermal DNA assembly, the assembled pEC-K18mob2::paa plasmid was isolated and introduced into C. glutamicum ATCC 13032.
RT-qPCR measurements of transcription levels.
For the preparation of total RNA, bacterial cultures were grown in minimal medium with either 5 mM glucose or PAA as the sole carbon source, and cells from a 10-ml culture at exponential phase (OD600, 1.0) were harvested by centrifugation at 14,000 rpm for 1 min. The supernatant was discarded, and the pellet was immediately frozen in liquid nitrogen. Subsequent total-RNA isolation and purification were performed using the RNeasy Mini kit (Qiagen) along with the DNase I kit (Roche) and the RNase-free DNase set (Qiagen) (16). Reverse transcription-quantitative PCR (RT-qPCR) was carried out using the SensiMix SYBR one-step kit (Quantace) and the Opticon Monitor system (Bio-Rad). The specificity of RT-qPCR products was verified by gel electrophoresis and melting curve analysis. For all conditions, measurements were taken for a total amount of 300 ng RNA with two biological duplicates and two technical replicates for each sample. The difference in the cycle threshold between each strain and the wild-type strain AS 1.542 grown in 5 mM glucose was expressed as a ratio calculated by 2−ΔCP, where ΔCP is the difference between the measured crossing points (CP).
Identification of transcription start sites (TSS) and promoter regions by RNAseq.
RNA was isolated as described for the RT-qPCR experiments. In total, 5 μg of RNA was pooled from equal amounts of RNAs from strain AS 1.542 grown on glucose, PAA, and LB broth.
A 5′-end-enriched RNAseq library was constructed according to the following procedures. (i) Depletion of stable rRNA and enrichment of mRNA molecules were performed using the Ribo-Zero rRNA removal kit for Gram-positive bacteria (Epicentre Biotechnologies). (ii) The enriched mRNA was fragmented by magnesium potassium acetate (MgKOAc) hydrolysis. Four volumes of RNA solution was mixed with 1 volume of MgKOAc solution (100 mM KOAc and 30 mM MgOAc in 200 mM Tris-HCl [pH 8.1]), and the mixture was incubated for 2.5 min at 94°C. The reaction was stopped by adding an equal volume of 1× TE (10 mM Tris, 1 mM EDTA [pH 8]) and chilling on ice for 5 min. (iii) The fragmented RNA was precipitated by the addition of 3 volumes 0.3 M NaAc in ethanol with 2 μl glycogen and overnight incubation at −20°C. (iv) The precipitated RNA fragments were dissolved in water, and the 5′-end RNA fragments were enriched by using Terminator 5′-phosphate-dependent exonuclease (Epicentre Biotechnologies). (v) After RNA precipitation (see step iii above), the triphosphates were removed using RNA 5′ polyphosphatase (Epicentre Biotechnologies). (vi) After RNA precipitation (see step iii above), the 5′-enriched, monophosphorylated RNA fragments were used to construct a cDNA library by using the Small RNA Sample prep kit (Illumina).
The fragmentation of RNA molecules (fragment sizes, 200 to 500 bp) and the RNA concentration were monitored using the RNA 6000 Pico assay on an Agilent 2100 bioanalyzer (Agilent).
The cDNA library was sequenced on the GA IIx platform (Illumina). The resulting reads were aligned to the paa cluster genomic sequence by using the mapping software SARUMAN (4). By combining published information about promoter regions in C. glutamicum (34) with 5′-end-enriched RNAseq data, probable TSS and promoter regions were deduced for the paa gene cluster in C. glutamicum AS 1.542.
Bioinformatic analysis of PaaR binding sites and regulons.
For the prediction of PaaR binding sites, we searched for a common motif in the upstream sequences of all genes in the paa cluster using the de novo motif extraction program MEME (1). Based on the distribution of known regulator binding sites in C. glutamicum (2), upstream regions were defined as noncoding sequence ranging from position +20 relative to the translation start codon up to the next coding sequence (CDS), with a maximum size of 620 bp and a minimum size of 40 bp. CDSs smaller than 150 bp were ignored in determining the maximum length of the upstream sequences (23). Most TetR family transcription regulators reportedly bind with 15-bp DNA sequences (35), so parameters for MEME were set to detect 15- to 22-bp palindromic motifs. Genomewide detection of putative binding sites in genomic upstream regions was carried out using PoSSumSearch (3). A positional weight matrix (PWM)-based model of the respective binding motif was generated from known binding sites for both PaaR (this study) and GlxR (18, 23, 24).
Comparative genomic identification of PaaR binding sites and reconstruction of PaaR regulons in a reference set of 57 bacterial species were performed according to the approach established previously (36), implemented in the RegPredict Web server tool (regpredict.lbl.gov) (33). The PaaR regulons were reconstructed separately in each taxonomic group containing a PaaR ortholog. In each group of genomes, a PaaR binding motif was identified, and a PWM was constructed using the motif discovery program in the RegPredict server. Then the taxonomically specific PWMs were used to scan all reference genomes in the respective groups and to predict novel genes in the PaaR regulon. Scores of candidate sites were defined as the sum of positional nucleotide weights. The details of the reconstructed PaaR regulons are captured and displayed in the RegPrecise database of bacterial regulons inferred by the comparative genomics approach (regprecise.lbl.gov) (32).
PaaR purification and EMSA.
E. coli strain ER2566 carrying the expression plasmid pTXB1::paaR was grown in LB broth at 37°C with 200 μg/ml ampicillin. Isopropyl-β-d-thiogalactopyranoside (IPTG) was added at a final concentration of 0.4 mM when the cell density reached an OD600 of 0.7, and the temperature was adjusted to 18°C, followed by overnight incubation. PaaR was purified using the IMPACT system (New England BioLabs). For electrophoretic mobility shift assays (EMSA) with PaaR, Cy3-labeled DNA probes of genomic upstream regions were obtained by PCR and column-based purification. The 20-μl reaction mixtures contained 20 mM Na2HPO4, 75 mM KCl, 0.1 mM EDTA, 5% glycerin, 25 μg/ml salmon sperm blocking DNA, 0.1 pmol DNA probe, and purified PaaR at specified concentrations. In case of competition EMSA with PaaR, an unlabeled 40-bp fragment in a gradient of 0, 0.1, 0.5, 1, 5, and 10 pmol was also added to the reaction mixture. Reaction mixtures were incubated at 30°C for 20 min before being loaded onto a cooled 1.5% (wt/vol) agarose gel. The gel buffer and running buffer contained 20 mM Na2HPO4. Hexahistidyl-tagged GlxR was purified, and EMSA were performed, as described previously (23, 25). Gel electrophoresis was performed for 40 min at a voltage of 90 V, and the resulting band shifts were visualized using a Typhoon 8600 imager (Amersham Bioscience).
RESULTS
Bioinformatic and functional identification of a paa gene cluster from C. glutamicum AS 1.542.
Genome sequencing of C. glutamicum AS 1.542 revealed a gene cluster of 15 genes (EBI-EMBL accession no. HE649965) that were deduced to be responsible for PAA transport, metabolism, and regulation (Fig. 1). Except for the putative regulator (paaR) and transporter (paaT) genes, these genes show significant amino acid sequence similarities (32 to 66% [Fig. 1]) to the PAA degradation genes of E. coli (45). The paaT gene, encoding a putative transporter, is located at the beginning of the paa gene cluster in strain AS 1.542 but is absent in E. coli K-12. In contrast to the GntR-type regulatory gene paaX in E. coli, the paa cluster in strain AS 1.542 contains a putative TetR-type regulatory gene, paaR. The genetic organization of the paa gene cluster in C. glutamicum is apparently different in general from that in E. coli (Fig. 1), although a coherent paaABCDE cassette was observed in E. coli and in C. glutamicum, as well as in other bacteria (27).
paa gene clusters were also identified in the genomes of C. glutamicum strain R and Corynebacterium efficiens YS-314, with high sequence similarity and identical genetic organization to the paa gene cluster of strain AS 1.542 (Fig. 1). But no such paa gene cluster was found in C. glutamicum strain ATCC 13032 (19). The paa gene cluster of AS 1.542 was cloned into pEC-K18mob2, and the plasmid constructed, pEC-K18mob2::paa, was successfully mobilized into strain ATCC 13032. The transformant, ATCC 13032/pEC-K18mob2::paa, obtained the ability to grow on PAA as the sole carbon source (see Fig. S1 in the supplemental material), indicating that the paa gene cluster of AS 1.542 is sufficient for PAA degradation.
Transcriptional organization of the paa gene cluster in strain AS 1.542.
A 5′-end-enriched RNAseq library was constructed, and 6 probable TSS and promoter regions were identified by mapping sequence reads to the paa gene cluster of strain AS 1.542 (Table 1; see also Fig. S2 in the supplemental material). The identified TSS and promoter regions were manually checked and were adjusted according to the nucleotide distribution within the C. glutamicum promoter sequences (34). These results suggest that the paa gene cluster of strain AS 1.542 is organized into six transcription units: paaTK, paaABCDEGJFH, paaR, paaI, paaY, and paaZ.
Table 1.
Transcriptional organization of the paa gene cluster in C. glutamicum strain AS 1.542
| Operon | Strand | TSS positiona | TSS base | Sequence |
Match coverageb |
||
|---|---|---|---|---|---|---|---|
| −10 | −35 | Perfect | Best | ||||
| paaTK | − | −213 | A | TAGCGT | TGGACA | 336 | 911 |
| paaABCDEGJFH | + | −44 | G | TATCCT | TTGACC | 27 | 209 |
| paaR | − | −76 | A | TACAGT | TTCCCG | 1 | 7 |
| paaI | + | −60 | A | TAGACT | GTGAAA | 22 | 59 |
| paaY | − | −54 | C | TAATAT | TACACT | 0 | 35 |
| paaZ | + | −57 | A | TATACT | TTCACC | 21 | 170 |
Relative to the translation start codon.
Perfect match coverage and best match coverage are defined according to Blom et al. (4).
paaR encodes a transcription regulator repressing the transcription of paa genes in strain AS 1.542.
The paaR gene was annotated as a putative TetR family transcription regulator. The growth of strain AS 1.542 and its mutant AS 1.542/paaRΔΗΤΗ was observed on 5 mM PAA as a carbon source. The mutant AS 1.542/paaRΔΗΤΗ exhibited significantly faster adaptation to PAA as a carbon source than did AS 1.542. Furthermore, the induction of the paa genes and the influence of paaR disruption on the transcriptional control of the paa genes were determined by RT-qPCR (Fig. 2). The results showed that the mRNA levels of all paa genes of strain AS 1.542 increased when this strain was grown on PAA rather than glucose as a carbon source. The levels of the six transcription units, paaABCDEGJFH, paaTK, paaR, paaI, paaY, and paaZ, increased 178- to 896-, 36- to 64-, 13-, 7-, 59-, and 49-fold, respectively. When the mutant AS 1.542/paaRΔΗΤΗ was grown on glucose, the mRNA levels of all paa genes were comparable to those in AS 1.542/paaRΔΗΤΗ grown on PAA. These results clearly demonstrate that PaaR negatively regulates the transcription of paa genes in AS 1.542.
Fig 2.

Transcriptional expression of paa genes in AS 1.542 (wild type) and the AS 1.542/paaRΔΗΤΗ mutant. Cells were cultivated in mineral medium with 5 mM glucose or 5 mM PAA as the sole carbon source. The ratios of mRNA levels were calculated relative to strain AS 1.542 grown on glucose and are presented on a logarithmic scale. Gene names are indicated at the bottom. The betB gene is located outside the cluster and was used as a negative control.
PaaR binds to the upstream regions of the paa gene cluster, and phenylacetyl-CoA (PA-CoA) is the effector.
EMSA of the upstream regions of paaK (271 bp), paaA (258 bp), paaYZ (203 bp), and paaRI (247 bp) were performed with Cy3-labeled DNA probes. The addition of 5 pmol of PaaR (a 50-fold excess over the DNA probe) resulted in clear shifts with the DNA probes of paaK, paaA, and paaYZ but not with the DNA probe of paaRI (Fig. 3A). A distinct shift of the paaRI DNA probe was observed only at a higher concentration (60 pmol) of PaaR, and a blurred smear was observed for the negative-control hemH probe (114 bp) under these conditions (Fig. 3B).
Fig 3.

Electrophoretic mobility shift assays of PaaR binding to the upstream regions of the paaK, paaA, paaYZ, and paaRI genes. The upstream region of hemH from C. glutamicum AS 1.542 was tested in parallel as a negative reference (A and B). Cy3-labeled DNA probes were added at 0.1 pmol, and PaaR protein was added at 5 pmol (A and D), at 60 pmol (C), or as indicated. The effector PA-CoA was added at 250 μM (A) or as indicated (C). For competition EMSA experiments with the paaRI DNA probe (C) or with the paaK, paaA, or paaYZ DNA probe (D), unlabeled 40-bp DNA fragments carrying the corresponding putative binding sites were added. As a negative control, an unlabeled fragment (40 bp) of the argC gene was added to an EMSA mixture with the Cy3-labeled paaA probe (D).
The effects of PAA, acetyl-CoA, and PA-CoA at a concentration of 0, 10, 25, 50, 100, 250, or 500 μM on the binding between PaaR and the paaA DNA probe were determined. The results showed that PAA and acetyl-CoA did not affect the binding between PaaR and the DNA probe of paaA (see Fig. S3 in the supplemental material). It was observed that 250 μM PA-CoA abolished the binding of PaaR to the probes for paaK, paaA, and paaYZ (Fig. 3A). For the upstream region of paaRI, which required 60 pmol PaaR to introduce an observable shift, the effector identified above, PA-CoA, did not result in a clear abortion of PaaR binding at 250 μM or even at 3 mM PA-CoA. Instead, smeared bands were observed under these conditions (Fig. 3C).
Determination of the PaaR binding sites.
The upstream regions of the paa genes were searched with the MEME software for a putative PaaR binding motif, and a 22-bp palindromic motif (ACTnACCGnnCGnnCGGTnAGT) was identified at each of the upstream regions of paaK, paaA, and paaYZ. A putative PaaR binding site at the upstream region of paaRI (Fig. 4) was observed, and the sequence of this site was less similar to the 22-bp palindromic motif identified. Forty-base-pair fragments, each carrying the 22-bp motif and additional 9-bp flanks at both sides (thus totaling 40 bp), were synthesized and were used for competition EMSA. The results showed that 0.5 pmol (i.e., a 5-fold excess) of the respective 40-bp fragment competed effectively for PaaR binding with the Cy3-labeled DNA probes of the upstream regions of paaK, paaA, and paaYZ (Fig. 3D). But 0.5 pmol (i.e., a 5-fold excess) or even 6 pmol (i.e., a 60-fold excess) of the 40-bp paaRI competition DNA fragment resulted in only partial competition for PaaR binding with the upstream DNA probe of paaRI (Fig. 3C).
Fig 4.

Regulatory regions of the paa gene cluster in strain AS 1.542. Upstream DNA sequences of the paaK (I), paaA (II), paaYZ (III), and paaRI (IV) genes are shown in 5′ → 3′ orientation relative to the translation start codons of the respective genes. The determined transcription start site is in boldface, and bent arrows indicate the direction of transcription; the derived −10 and −35 regions are underlined. PaaR binding sites are indicated by boxes with solid borders, and GlxR binding sites are indicated by dotted boxes with dotted borders.
GlxR binds to the upstream regions of the paa gene cluster.
It was found that the paa gene transcription levels in AS 1.542/paaRΔΗΤΗ were lower when this mutant was grown on glucose than when it was grown on PAA (Fig. 2). This indicated that a second transcriptional regulator was involved in paa gene regulation in AS 1.542. It was reported that GlxR acts as a global regulator for aromatic-compound degradation in C. glutamicum ATCC 13032, and the consensus sequence TGTGAnnTAnnTCACA was identified (23). By application of the PoSSuMsearch tool, possible GlxR binding sites were searched for in the upstream regions of the paa genes from C. glutamicum strain AS 1.542 and strain R, as well as from C. efficiens. GlxR binding sites were found in the upstream regions of the paaK, paaA, and paaYZ genes of all three Corynebacterium strains. In C. glutamicum strain AS 1.542 and strain R, less-conserved GlxR binding sites were also found in the upstream region of paaRI. They were located around the putative promoter regions of the paa genes and upstream of the PaaR binding sites (Fig. 4). EMSA were performed to confirm the putative GlxR binding sites in strain AS 1.542. The results showed that the 40-bp fragments of the upstream regions of paaK, paaA, and paaYZ containing the previously identified GlxR binding motif (23) specifically bound with GlxR in the presence of cyclic AMP (cAMP) (Fig. 5).
Fig 5.
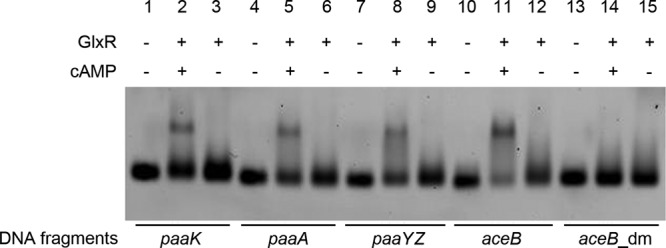
Electrophoretic mobility shift assays for identification of the GlxR protein binding sites from the upstream regions of paaK, paaA, and paaYZ in strain AS 1.542. Cy3-labeled 40-bp DNA fragments carrying the GlxR binding sites (TGTGAnnTAnnTCACA) with an additional 12 bp at each side were used. A fragment with the GlxR binding site of aceB was used as a positive control, and the same fragment with the double-mutated binding sites (aceB_dm) was used as a negative control (23). GlxR protein was added at 60 pmol; the effector cAMP was added at 0.2 mM.
Comparative-genomics reconstruction of orthologous PaaR regulons in bacteria.
In addition to the PaaR protein from C. glutamicum, two PaaR proteins from T. thermophilus and B. cenocepacia had been identified previously (13, 38). By using these functionally characterized PaaR proteins, PaaR orthologs were identified by BLAST searches against the nonredundant set of sequenced bacterial genomes and were used to analyze the phylogenetic relationships between these proteins (Fig. 6). As a result, a single copy of the paaR gene was detected in the reference set of 57 bacterial genomes from the following taxonomic groups: Actinobacteria, Alpha- and Betaproteobacteria, Deinococcus-Thermus, and Flavobacteria. In most of these genomes, the paaR gene is located in the vicinity of the paa genes; the only exception is 3 Deinococcus spp. For inference of PaaR regulons in these genomes, we applied the comparative-genomics approach implemented in the RegPredict Web server (33, 36).
Fig 6.

Phylogenetic tree of a selection of PaaR orthologs marked with taxonomic groups and identified PaaR-binding DNA motifs (generated by http://weblogo.berkeley.edu) for each group. The tree was constructed using the neighbor-joining method with 1,000 bootstraps. For the identities of the PaaR orthologs shown here, please see Table S2 in the supplemental material.
Phylogenetic analysis of the PaaR-like proteins detected (Fig. 6) provided a basis for transcription factor binding site (TFBS) identification among different phyla. First, we collected training sets of prospective target genes for each of 10 defined taxonomic groups possessing PaaR orthologs. These training sets were defined by the analysis of chromosomal colocalizations of paaR genes and information on the known PaaR binding sites from strain AS 1.542 from this study, as well as from T. thermophilus and B. cenocepacia (13, 38). Using the motif recognition program applied to each training set of upstream gene regions, we identified conserved PaaR binding motifs, constructed a positional weight matrix for each motif identified, and searched for additional PaaR-binding sites in the genomes analyzed. Except for three Deinococcus species, most of the PaaR binding sites detected were located adjacent to their target genes (see the “TFBS” sheet of Table S2 in the supplemental material; also see Discussion). The resulting PaaR binding motifs are well conserved between the Actinobacteria and Proteobacteria phyla, whereas the PaaR binding motifs in the Deinococcus-Thermus group and Flavobacteria have several substituted nucleotides in their consensus sequences (Fig. 6). The reconstructed PaaR regulons are summarized in Table S2 in the supplemental material, and the detailed information on the predicted PaaR-binding sites and downstream regulated genes are available in an updated version of the RegPrecise database (32).
DISCUSSION
C. glutamicum strain AS 1.542 possesses a complete paa gene cluster, conferring a PAA degradation pathway and its regulation. This paa gene cluster is subjected to regulation by the TetR-type regulator PaaR. In contrast to those in P. putida (11), E. coli (9, 10), B. cenocepacia (13), and T. thermophilus (38), the paa gene cluster in C. glutamicum consists of 6 transcription units. The differences in the transcription levels of the paa genes suggest that PaaR strongly regulates the transcription of paaABCDEGJFH, paaY, paaZ, and paaTK, while it weakly regulates the transcription of paaR and paaI. EMSA results from this study indicated that a 12-times-higher concentration of PaaR was needed to retard the probe of the paaIR upstream region than to retard the other probes. Therefore, it is proposed that PaaR exerts a weak self-repression and a strong repression of the other genes in the cluster in C. glutamicum.
The PAA catabolic pathway is widespread in the bacterial domain (45). Bacteria have evolved two different systems to regulate this PAA catabolism, namely, the TetR-type regulation represented by the PaaR proteins of C. glutamicum (this study), T. thermophilus (38), and B. cenocepacia (13) and the GntR-type regulation represented by PaaX of E. coli (9), P. putida, and other Pseudomonas species (7, 11). The binding consensus sequence of GntR-type regulator PaaX was identified as TGATTC(N27)GAATCA in E. coli (21), and a similar inverted repeat sequence was found in Pseudomonas strain Y2 (7). The binding sequences of the TetR-type regulators from the PaaR proteins of T. thermophilus HB8 (38) and B. cenocepacia K56-2 (13) show little similarity to the binding consensus sequences of GntR-type regulators from E. coli (21) and Pseudomonas species (7). In this study, the identified binding sequence of the TetR-type PaaR protein from C. glutamicum strain AS 1.542 shows some similarity to the binding sequences of PaaR from B. cenocepacia K56-2 (13). Moreover, analysis of the respective paa gene clusters from C. glutamicum strain R and C. efficiens reveals a binding consensus sequence of the TetR-type PaaR protein in Corynebacterium species, which is characterized by an imperfect palindromic sequence (ACTnACCGnnCGnnCGGTnAGT; 22 bp).
Based on these findings and by integration of a comparative-genomics approach, we were able to define PaaR regulons in 57 distinct bacterial species from the Actinobacteria, Alphaproteobacteria and Betaproteobacteria, Flavobacteria, and Deinococcus-Thermus phyla. PaaR regulation was restricted to paa catabolic and transport genes in a chromosomal cluster, and the sizes of predicted PaaR regulons range from one target operon (e.g., in Flavobacteria) to nine target operons (as in Deinococcus). Phylogenetic analysis of PaaR proteins and the observed conservation of the PaaR binding motifs within each taxonomic group (Fig. 6) indicate that the PaaR regulators and motifs coevolved via presumably vertical evolution in Bacteria. Interestingly, in Deinococcus species, the deduced PaaR binding motifs are not associated with PAA utilization genes, and there are no PaaR binding motifs adjacent to paa genes. Therefore, how the paa gene cluster is regulated in the Deinococcus species is still not clear.
A recent study has identified in vivo GlxR binding sites at the corresponding paaK, paaA, and paaYZ promoter regions in C. glutamicum strain R (47); of these, the paa gene cluster is almost identical to that of strain AS 1.542. This is in agreement with our EMSA results for GlxR in strain AS 1.542. As proposed in this study and previous reports (23, 24), GlxR regulates PAA degradation, although the mechanism of this regulation needs more investigation. It is known that GlxR is a cAMP-binding global regulator (18, 23, 24). The cAMP receptor protein (CRP), a homolog to GlxR in C. glutamicum, senses the intracellular cAMP levels and superimposed the PaaX-mediated repression in E. coli (9). Thus, it would be interesting to determine if phenylacetic acid catabolism is related to the intracellular cAMP level. The involvement of the global regulator GlxR and a pathway-specific regulator, PaaR, of the PAA catabolic pathway in strain AS 1.542 exemplifies the proposal of two levels of transcriptional regulation for aromatic degradation (8). It would be of interest to investigate exactly how GlxR and PaaR coordinate in the regulation of aromatic-compound degradation in C. glutamicum and other bacteria.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (30725001) and the Ministry of Science and Technology (973 program 2012CB721104). X.C. acknowledges support from the German Academic Exchange Service (DAAD) and the Graduate Cluster Industrial Biotechnology (CLIB2021) at Bielefeld University, Bielefeld, Germany. The Graduate Cluster is supported by a grant from the Federal Ministry of Innovation, Science, Research and Technology (MIWFT) of the federal state North Rhine-Westphalia, Germany. C.R. and J.K. acknowledge funding from a research grant awarded by the MIWFT within the BIO.NRW initiative (grant 280371902). D.A.R. was supported by contract DE-SC0004999 from the U.S. Department of Energy, Office of Science (Biological and Environmental Research), as part of the Genomic Science Program.
Footnotes
Published ahead of print 8 June 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Bailey TL, et al. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37:W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baumbach J. 2007. CoryneRegNet 4.0—a reference database for corynebacterial gene regulatory networks. BMC Bioinformatics 8:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beckstette M, Homann R, Giegerich R, Kurtz S. 2006. Fast index based algorithms and software for matching position specific scoring matrices. BMC Bioinformatics 7:389 doi:10.1186/1471-2105-7-389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blom J, et al. 2011. Exact and complete short-read alignment to microbial genomes using Graphics Processing Unit programming. Bioinformatics 27:1351–1358 [DOI] [PubMed] [Google Scholar]
- 5. Brinkrolf K, Brune I, Tauch A. 2006. Transcriptional regulation of catabolic pathways for aromatic compounds in Corynebacterium glutamicum. Genet. Mol. Res. 5:773–789 [PubMed] [Google Scholar]
- 6. Chen Q, Li L-G. 1975. The identification of a l-glutamate-producing bacterium Corynebacterium crenatum AS 1.542. Acta Microbiol. Sin. (Wei Sheng Wu Xue Bao) 15:119–124 (In Chinese.) [Google Scholar]
- 7. del Peso-Santos T, et al. 2006. Coregulation by phenylacetyl-coenzyme A-responsive PaaX integrates control of the upper and lower pathways for catabolism of styrene by Pseudomonas sp. strain Y2. J. Bacteriol. 188:4812–4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Díaz E, Prieto MA. 2000. Bacterial promoters triggering biodegradation of aromatic pollutants. Curr. Opin. Biotechnol. 11:467–475 [DOI] [PubMed] [Google Scholar]
- 9. Ferrández A, García JL, Díaz E. 2000. Transcriptional regulation of the divergent paa catabolic operons for phenylacetic acid degradation in Escherichia coli. J. Biol. Chem. 275:12214–12222 [DOI] [PubMed] [Google Scholar]
- 10. Ferrández A, et al. 1998. Catabolism of phenylacetic acid in Escherichia coli. Characterization of a new aerobic hybrid pathway. J. Biol. Chem. 273:25974–25986 [DOI] [PubMed] [Google Scholar]
- 11. García B, et al. 2000. Phenylacetyl-coenzyme A is the true inducer of the phenylacetic acid catabolism pathway in Pseudomonas putida U. Appl. Environ. Microbiol. 66:4575–4578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gibson DG, et al. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6:343–345 [DOI] [PubMed] [Google Scholar]
- 13. Hamlin JNR, Bloodworth RAM, Cardona ST. 2009. Regulation of phenylacetic acid degradation genes of Burkholderia cenocepacia K56-2. BMC Microbiol. 9:222 doi:10.1186/1471-2180-9-222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68 [DOI] [PubMed] [Google Scholar]
- 15. Huang Y, et al. 2006. Genetic characterization of the resorcinol catabolic pathway in Corynebacterium glutamicum. Appl. Environ. Microbiol. 72:7238–7245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hüser AT, et al. 2003. Development of a Corynebacterium glutamicum DNA microarray and validation by genome-wide expression profiling during growth with propionate as carbon source. J. Biotechnol. 106:269–286 [DOI] [PubMed] [Google Scholar]
- 17. Johnson M, et al. 2008. NCBI BLAST: a better web interface. Nucleic Acids Res. 36:W5–W9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jungwirth B, et al. 2008. Triple transcriptional control of the resuscitation promoting factor 2 (rpf2) gene of Corynebacterium glutamicum by the regulators of acetate metabolism RamA and RamB and the cAMP-dependent regulator GlxR. FEMS Microbiol. Lett. 281:190–197 [DOI] [PubMed] [Google Scholar]
- 19. Kalinowski J, et al. 2003. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of l-aspartate-derived amino acids and vitamins. J. Biotechnol. 104:5–25 [DOI] [PubMed] [Google Scholar]
- 20. Katsumata R, Ozaki A, Oka T, Furuya A. 1984. Protoplast transformation of glutamate-producing bacteria with plasmid DNA. J. Bacteriol. 159:306–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim HS, Kang TS, Hyun JS, Kang HS. 2004. Regulation of penicillin G acylase gene expression in Escherichia coli by repressor PaaX and the cAMP-cAMP receptor protein complex. J. Biol. Chem. 279:33253–33262 [DOI] [PubMed] [Google Scholar]
- 22. Kirchner O, Tauch A. 2003. Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J. Biotechnol. 104:287–299 [DOI] [PubMed] [Google Scholar]
- 23. Kohl TA, Baumbach J, Jungwirth B, Pühler A, Tauch A. 2008. The GlxR regulon of the amino acid producer Corynebacterium glutamicum: in silico and in vitro detection of DNA binding sites of a global transcription regulator. J. Biotechnol. 135:340–350 [DOI] [PubMed] [Google Scholar]
- 24. Kohl TA, Tauch A. 2009. The GlxR regulon of the amino acid producer Corynebacterium glutamicum: detection of the corynebacterial core regulon and integration into the transcriptional regulatory network model. J. Biotechnol. 143:239–246 [DOI] [PubMed] [Google Scholar]
- 25. Letek M, et al. 2006. Characterization and use of catabolite-repressed promoters from gluconate genes in Corynebacterium glutamicum. J. Bacteriol. 188:409–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luengo JM, García JL, Olivera ER. 2001. The phenylacetyl-CoA catabolon: a complex catabolic unit with broad biotechnological applications. Mol. Microbiol. 39:1434–1442 [DOI] [PubMed] [Google Scholar]
- 27. Martin FJ, McInerney JO. 2009. Recurring cluster and operon assembly for phenylacetate degradation genes. BMC Evol. Biol. 9:36 doi:10.1186/1471-2148-9-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martínez-Blanco H, Reglero A, Rodriguez-Aparicio LB, Luengo JM. 1990. Purification and biochemical characterization of phenylacetyl-CoA ligase from Pseudomonas putida. A specific enzyme for the catabolism of phenylacetic acid. J. Biol. Chem. 265:7084–7090 [PubMed] [Google Scholar]
- 29. Meyer F, et al. 2003. GenDB—an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 31:2187–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mohamed ME-S, Ismail W, Heider J, Fuchs G. 2002. Aerobic metabolism of phenylacetic acids in Azoarcus evansii. Arch. Microbiol. 178:180–192 [DOI] [PubMed] [Google Scholar]
- 31. Navarro-Llorens JM, et al. 2005. Phenylacetate catabolism in Rhodococcus sp. strain RHA1: a central pathway for degradation of aromatic compounds. J. Bacteriol. 187:4497–4504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Novichkov PS, et al. 2010. RegPrecise: a database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. Nucleic Acids Res. 38:D111–D118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Novichkov PS, et al. 2010. RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 38:W299–W307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pátek M, Nešvera J. 2011. Sigma factors and promoters in Corynebacterium glutamicum. J. Biotechnol. 154:101–113 [DOI] [PubMed] [Google Scholar]
- 35. Ramos JL, et al. 2005. The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69:326–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rodionov DA. 2007. Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem. Rev. 107:3467–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rückert C, et al. 2005. Functional genomics and expression analysis of the Corynebacterium glutamicum fpr2-cysIXHDNYZ gene cluster involved in assimilatory sulphate reduction. BMC Genomics 6:121 doi:10.1186/1471-2164-6-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sakamoto K, Agari Y, Kuramitsu S, Shinkai A. 2011. Phenylacetyl-coenzyme A is an effector molecule of TetR family transcriptional repressor PaaR from Thermus thermophilus HB8. J. Bacteriol. 193:4388–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 40. Schäfer A, et al. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73 [DOI] [PubMed] [Google Scholar]
- 41. Shen X-H, Huang Y, Liu S-J. 2005. Genomic analysis and identification of catabolic pathways for aromatic compounds in Corynebacterium glutamicum. Microb. Environ. 20:160–167 [Google Scholar]
- 42. Shen X-H, Jiang C-Y, Huang Y, Liu Z-P, Liu S-J. 2005. Functional identification of novel genes involved in the glutathione-independent gentisate pathway in Corynebacterium glutamicum. Appl. Environ. Microbiol. 71:3442–3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shen X-H, Liu S-J. 2005. Key enzymes of the protocatechuate branch of the beta-ketoadipate pathway for aromatic degradation in Corynebacterium glutamicum. Sci. China 48:241–249 [DOI] [PubMed] [Google Scholar]
- 44. Tauch A, et al. 2002. Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr. Microbiol. 45:362–367 [DOI] [PubMed] [Google Scholar]
- 45. Teufel R, et al. 2010. Bacterial phenylalanine and phenylacetate catabolic pathway revealed. Proc. Natl. Acad. Sci. U. S. A. 107:14390–14395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Teufel R, et al. 2011. Studies on the mechanism of ring hydrolysis in phenylacetate degradation: a metabolic branching point. J. Biol. Chem. 286:11021–11034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Toyoda K, Teramoto H, Inui M, Yukawa H. 2011. Genome-wide identification of in vivo binding sites of GlxR, a cyclic AMP receptor protein-type regulator in Corynebacterium glutamicum. J. Bacteriol. 193:4123–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yudistira H, et al. 2011. Phenylalanine induces Burkholderia cenocepacia phenylacetic acid catabolism through degradation to phenylacetyl-CoA in synthetic cystic fibrosis sputum medium. Microb. Pathog. 51:186–193 [DOI] [PubMed] [Google Scholar]
- 49. Zhao K-X, Huang Y, Chen X, Wang N-X, Liu S-J. 2010. PcaO positively regulates pcaHG of the beta-ketoadipate pathway in Corynebacterium glutamicum. J. Bacteriol. 192:1565–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.