

Abstract
In the rodent cerebellum, pharmacological activation of mGluR4 acutely depresses excitatory synaptic transmission at parallel fibre–Purkinje cell synapses. This depression involves the inhibition of presynaptic calcium (Ca2+) influx that ultimately controls glutamate release. In this study, we investigate the molecular basis of mGluR4-mediated inhibition of presynaptic Ca2+ transients. Our results demonstrate that the mGluR4 effect does not depend on selective inhibition of a specific type of presynaptic voltage-gated Ca2+ channel, but rather involves modulation of all classes of Ca2+ channels present in the presynaptic terminals. In addition, this inhibitory effect does not involve the activation of G protein-activated inwardly rectifying potassium channels, TEA-sensitive potassium channels or two-pore-domain potassium channels. Furthermore, this inhibition does not require pertussis toxin-sensitive G proteins, and is independent of any effect on adenylyl cyclases, protein kinase A, mitogen-activated protein kinases or phosphoinositol-3 kinase activity. Interestingly we found that mGluR4 inhibition of presynaptic Ca2+ influx employs a newly defined signalling pathway, notably that involving the activation of phospholipase C and ultimately protein kinase C.
Key points
Glutamate is the major excitatory neurotransmitter of the mammalian brain. It can activate ionotropic receptors underlying fast glutamatergic transmission as well as G protein-coupled metabotropic receptors (mGluR1–8), which are pre- and postsynaptic modulators of this fast excitatory neurotransmission.
In the rodent cerebellum, activation of presynaptic mGluR4 depresses excitatory synaptic transmission at parallel fibre–Purkinje cell synapses. We show that this depression involves the inhibition of presynaptic calcium influx via a newly defined signalling pathway, which notably involves the activation of phospholipase C and ultimately protein kinase C.
The study of the molecular basis of mGluR signalling pathways is an important research topic because these receptors may be implicated in certain neurodegenerative disorders, like Parkinson's or Alzheimer's disease. As such, these receptors are becoming crucial targets for a number of therapeutic agents.
Introduction
In the rodent cerebellum, group III metabotropic glutamate receptors (mGluRs) negatively regulate Ca2+ influx into presynaptic terminals (Daniel & Crepel, 2001; Zhang & Linden, 2009), and thus decrease glutamatergic transmission (Conquet et al. 1994; Pekhletski et al. 1996; Miniaci et al. 2001; Neale et al. 2001; Lorez et al. 2003). We have shown that among the group III mGluRs present at the parallel fibre (PF)–Purkinje cell (PC) synapse, presynaptic mGluR4s are entirely responsible for this regulation (Abitbol et al. 2008).
In PF terminals, Ca2+ influx results from the activation of diverse Ca2+ channels, notably P/Q-, N- and R-type channels (Mintz et al. 1995; Brown et al. 2004; Daniel et al. 2004). At many synapses, group III mGluRs depress neurotransmitter release by inhibiting one or more of these voltage-gated Ca2+ channels (VGCCs). For example, group III mGluR activation inhibits P/Q-type channels in the superior olivary complex (Takahashi et al. 1996), N-type channels in the hippocampus (Capogna, 2004; Rusakov et al. 2004), and both N- and P/Q-type channels in the entorhinal cortex (Woodhall et al. 2007) and in cerebrocortical synaptosomes (Millan et al. 2002). In contrast, group III mGluR-mediated presynaptic inhibition in cultured reticulospinal neurons does not involve selective modulation of any known type of presynaptic VGCC (Krieger et al. 1999).
mGluRs may also have indirect effects on VGCC activity by modulating ionic channels that control presynaptic membrane excitability. Candidate channels include: 4-aminopyridine (4-AP)-sensitive K+ channels (Daniel & Crepel, 2001) and G protein-gated inwardly rectifying K+ channels (GIRKs) (Saugstad et al. 1996; Sharon et al. 1997). In addition, certain subfamilies of two-pore-domain K+ channels (K2P) (Honoré, 2007), including TREK and TASK, are present on cerebellar granule cells (Watkins & Mathie 1996; Talley et al. 2001) and are regulated by G protein-coupled receptors (GPCRs) (Mathie, 2007), including mGluR4 (Cain et al. 2008).
While many group III mGluRs act through pertussis toxin (PTX)-sensitive Gi/o proteins that inhibit adenylyl cyclase (AC) activity and decrease intracellular cAMP levels (Thomsen et al. 1992; Kristensen et al. 1993; Tanabe et al. 1993; Prezeau et al. 1994; Flor et al. 1995; Neil et al. 1996; Conn & Pin, 1997), several studies show alternative signalling pathways for these receptors. For example, they can stimulate AC activity leading to increases in cAMP levels (Lavialle-Defaix et al. 2006) and activation of protein kinase A (PKA) (Evans et al. 2001), probably through Gs proteins. In cultured cerebellar granule cells, native group III mGluRs are functionally coupled to both mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3 kinase (PI3K) pathways (Iacovelli et al. 2002). Finally, within this group, mGluR7 inhibits P/Q-type VGCCs through the phospholipase C (PLC)–protein kinase C (PKC) cascade (Perroy et al. 2000).
We used fluorometric and electrophysiological approaches to investigate mGluR4-mediated inhibition of evoked presynaptic Ca2+ influx. We show that this modulation does not selectively target a specific type of presynaptic VGCC and is independent of any modulation of presynaptic K+ channels, MAPK or PI3K activity, or the Gi/o–AC–PKA signalling pathway. Interestingly we provide evidence suggesting that the activation of PF terminal mGluR4s initiates a non-canonical intracellular cascade that involves PLC and PKC.
Methods
Preparation of cerebellar slices
Animal care and all experimental procedures are in accordance with guidelines from the Centre National de la Recherche Scientifique (CNRS, France). Male Sprague–Dawley rats, 18–34 days old, were stunned and then decapitated. Coronal or sagittal cerebellar slices (200–250 μm thick) were prepared as previously described (Daniel & Crepel, 2001). The slices were kept at room temperature in saline solution gassed with 95% O2–5% CO2 for at least 1 h before recording. This solution contained (in mm): NaCl, 124; KCl, 3; NaHCO3, 24; KH2PO4, 1.15; MgSO4, 1.15; CaCl2, 2; glucose, 10 and registered an osmolarity of 330 mosmol l−1 and a pH of 7.35 at 25°C. The recording chamber was perfused at a rate of 2 ml per minute with this same oxygenated saline solution, supplemented with the GABAA receptor antagonist bicuculline methochloride or methiodide (10 μm, Sigma Aldrich, St Quentin Fallavier, France) to block inhibitory membrane currents mediated by these receptors.
Pharmacological agents
Most pharmacological agents were applied to cerebellar slices by direct addition to the saline solution, with the exception of pertussis toxin (PTX), KT 5720, U-73122, U-73343 and Ro 32-0432 with which the slices were incubated in the saline solution for variable durations before the recording session. In some experiments, slices were pretreated with thapsigargin (10 μm) for 40 min. l-AP4, PTX, KT 5720, Tertiapin Q, U-73122, Ro 32-0432, LY 294002, DHPG, PD98059, WIN55,212-2, NBQX, d-AP5 and phorbol 12 -Myristate 13 - Acetate (PMA) were purchased from Tocris (Illkirch, France). SNX-482, ω-agatoxin TK, ω-conotoxin GVIA and thapsigargin were purchased from Alomone Labs (Israel). 2′,5′-dideoxyadenosine, SQ 22,536, 4-aminopyridine (4-AP), U-73343, ruthenium red, cadmium, TEA and DEA/NO were obtained from Sigma. Fluoxetine was purchased from Ascent Scientific. All drug stocks were prepared in distilled water, except stocks of Fluo-4FF AM (Molecular Probes), 2′,5′-dideoxyadenosine, KT 5720, PD98059, LY 294002, PMA, U-73122, U-73343, SQ 22,536 and Ro 32-0432 which were prepared in dimethylsulfoxide (DMSO). Drug stocks were kept at −20°C until the day of the experiment. Unless otherwise stated, drugs were added to the perfusate at the desired concentration just before application (final concentration of DMSO was 0.1%).
Electrophysiology
Whole-cell patch-clamp recordings of Purkinje cell (PC) somas were performed in sagittal slices with an Axopatch-1D amplifier (Axon Instruments). All recordings were made at 27–28°C. Patch pipettes (3.5–5 MΩ, borosilicate glass) were filled with an internal solution of the following composition (mm): Nacl, 140; KCl, 6; Hepes, 10; EGTA, 0.75; MgCl2, 1; Na-GTP, 0.4; Na2-ATP, 4; pH 7.3 with KOH; 300 milliosmol l−1 As previously reported (Goossens et al. 2001), PCs were clamped at −70 mV (junction potentials corrected) and parallel fibres (PFs) were stimulated once every 6 s through an extracellular glass saline-filled monopolar electrode placed at the surface of the slice, in the lower half of the molecular layer, to evoke PF-mediated excitatory postsynaptic currents (EPSCs). Recorded PF-EPSCs were filtered at 5 kHz, digitized on line at 20 kHz, and analysed on- and off-line with Acquis1 software (Biologic, Grenoble, France). In the cells conserved for analysis, access resistance (usually 5–10 MΩ) was partially compensated (50–70%), according to the procedure described by Llano et al. (1991). Throughout the experiment, PF-EPSCs were elicited on a 10 mV hyperpolarizing voltage step, which allowed monitoring of passive membrane properties of the recorded cells.
In some experiments, PF-EPSCs were evoked with pairs of stimuli of the same intensity applied to the cell with an inter-stimulus interval of 40 ms. Paired-pulse facilitation (PPF) values (Atluri & Regehr, 1996) were calculated on-line as the ratio of the amplitude of the second PF-EPSC over the first one and plotted against time. Corresponding PPF values in individual plots were then averaged for all cells recorded to obtain the plot of mean PPF values before, during and after bath application of the mGluR4 agonist, l-AP4.
The PF volley is an extracellular field potential that was recorded with a saline-filled glass microelectrode placed in the molecular layer 500–800 μm away from the stimulus site. To prevent contamination by excitatory postsynaptic signals, PF volley experiments were performed in the presence of 20 μm NBQX and 50 μm d-AP5, respective antagonists of AMPA–kainate and NMDA receptors.
Calcium-sensitive fluorometric measurements
Using coronal slices, presynaptic PF tracts were labelled by local application of a saline solution containing the low-affinity calcium indicator Fluo-4FF AM (100 μm), as previously described (Daniel & Crepel, 2001). At least 30 min after loading, a confined region of labelled PFs was illuminated at a single excitation wavelength (480 ± 22 nm). Excitation light obtained from a 100 W mercury lamp was gated with an electromechanical shutter (Uniblitz, Rochester, NY, USA). The optical signals were recorded at 27–28°C, through a 20 μm × 50 μm window placed in the molecular layer on the visible narrow band of labelled PFs, approximately 500–700 μm away from the loading site. At this distance, only the loaded fibre tracts were visible in the recording window; no other labelled structures were detectable. PFs located in the recording window were stimulated every 30 s with a single 100 Hz train of five electrical stimuli, through a saline-filled glass microelectrode placed in the molecular layer between the loading site and the recording site. Stimulation-evoked Ca2+-sensitive changes in fluorescence were acquired through a ×60 water-immersion objective of an upright microscope (Zeiss, LePeck France), filtered by a barrier filter at 530 ± 30 nm and converted into an electric signal by a photometer. Fluorometric measurements were analysed on- and off-line using Acquis1 software. The fluorescence data corrected for dye bleaching were expressed as relative fluorescence changes ΔF/F, where F is the baseline fluorescence intensity, and ΔF is the change induced by PF stimulation. When background fluorescence of the tissue in unlabelled regions of the slice was greater than 5% of the resting fluorescence intensity of the indicator, the data were corrected for background fluorescence. Statistical significance was assessed by an unpaired Student's t test, with P < 0.05 (two-tailed) considered as significant. All data are expressed as the mean ± SEM.
Results
The presynaptic molecular events associated with pharmacological activation of mGluR4s were explored in coronal rat cerebellar slices with fluorometric methods, using the low-affinity Ca2+-sensitive dye Fluo-4FF AM, which allows a linear measure of presynaptic Ca2+ influx. As shown in our previous study (Abitbol et al. 2008), a train of five stimulations applied to PFs induced reproducible transient increases in presynaptic fluorescence, which returned to resting levels within a few hundred milliseconds (Fig. 1A, inset). In keeping with the results published by Daniel & Crepel (2001) and Abitbol et al. (2008), 5 min bath application of the broad-spectrum group III mGluR agonist, l-AP4, at a saturating concentration of 100 μm, reversibly decreased the amplitude of presynaptic Ca2+ influxes evoked by PF stimulations by 25.3 ± 2.3% (n= 6, Fig. 1A).
Figure 1. Lack of effect of the K+ channel blocker, TEA on l-AP4-mediated inhibition of presynaptic Ca2+ influx evoked by parallel fibre (PF) stimulation.
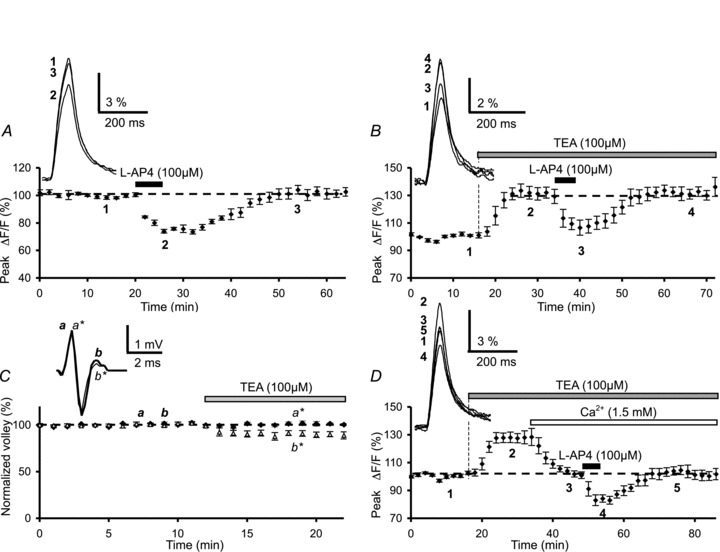
A, effect of l-AP4 on presynaptic Ca2+ influx elicited by PF stimulation. The plot represents the normalized amplitudes of peak Fluo-4FF fluorescence transients (ΔF/F) evoked by 5 PF stimulations (delivered at 100 Hz), plotted as a function of time before (1), during (2) and after (3) bath application of 100 μm l-AP4 (horizontal filled bar). Each point is the mean ± SEM of 6 separate experiments. The inset on the left displays superimposed averaged fluorescence transients in one of these experiments, recorded at the indicated times. B, plot of normalized amplitudes of peak fluorescence transients before, during bath application of TEA (200 μm) and during co-application of l-AP4 (100 μm) (n= 5). The insets are the same as in A. Note that application of TEA in standard extracellular medium (2 mm CaCl2) enhanced the amplitude of the fluorescence transients, but did not prevent their inhibition by l-AP4. C, plot of the normalized amplitude of presynaptic PF volleys recorded as field potentials in the molecular layer, against time before and during bath application of TEA (100 μm) (n= 4). The inset on the left shows an example of averaged PF volleys (25–50 consecutive responses) recorded at the indicated times under control conditions (thick trace) and in the presence of TEA (thin trace). Note that TEA altered the PF volleys: while the amplitude of the first volley was unaffected (a, control versus a*, under TEA, black diamonds), in contrast the amplitude of the second volley was slightly reduced (b, control versus b*, under TEA, open triangles). D, same as in B after lowering the extracellular Ca2+ concentration from 2 to 1.5 mm (n= 5). The insets show averaged fluorescence transients in one of these experiments.
TEA-sensitive, G protein-gated inwardly rectifying (Tertiapin Q-sensitive) or two-pore-domain presynaptic K+ channels do not mediate the depressant effect of mGluR4 activation on presynaptic Ca2+ influx
Axonal voltage-dependent K+ channels play a major role in fibre excitability and as such influence the presynaptic waveform, Ca2+ entry into presynaptic terminals and ultimately neurotransmitter release in mammalian central nervous system synapses (Sabatini & Regehr, 1997). In our previous study (Daniel & Crepel, 2001) we found that application of 1 mm 4-AP, a voltage-sensitive K+ channel blocker, abolished the depressant effect of l-AP4 on PF presynaptic Ca2+ influxes. Since at this concentration 4-AP (i) is fairly unselective (Coetzee et al. 1999) and (ii) strongly alters PF volleys by reducing the positive-going phase of the waveform indicating a slowing of spike repolarisation (Brown et al. 2004), we chose to renew these experiments employing this blocker at a lower concentration (200 μm 4-AP). As described by Brown et al. (2004) for 1 mm 4-AP, we found that bath application of 200 μm 4-AP consistently altered PF volleys by reducing the positive-going phase of the waveform (n= 4, Supplementary Fig. S1A). In addition, bath application of 200 μm 4-AP increased the duration (185%± 10%) and the amplitude (434%± 105%) of the presynaptic fluorescence transients (n= 10, Supplementary Fig. S1B) compared to those recorded in control conditions. As such we abandoned 4-AP and used another K+ channel blocker, tetraethyl ammonium (TEA). In agreement with Brown et al. (2004), bath application of TEA (100 μm) was not entirely without effect on PF volleys (n= 4, Fig. 1C), or Ca2+ transient amplitudes that increased to 131.5 ± 4.5% of control (n= 5, Fig. 1B). Despite the fact that TEA affected presynaptic volleys and Ca2+ influxes, subsequent bath application of l-AP4 (100 μm) reduced the amplitude of presynaptic Ca2+ transients evoked by PF stimulations (Fig. 1B). Indeed, from this TEA-induced plateau (representing increased Ca2+ transients after a 15 min application of 100 μm TEA), co-application of l-AP4 (100 μm) depressed these transients by 19.8 ± 2.3% (n= 5). This value is not significantly different (P > 0.1) to that recorded in control experiments.
In order to evaluate the effect of mGluR4 activation on evoked Ca2+ transients with amplitudes comparable to those recorded in control saline, we reduced the concentration of extracellular Ca2+ from 2 to 1.5 mm (osmolarity was maintained by adjusting the extracellular Mg2+ concentration) (Fig. 1D). Under these conditions, l-AP4 (100 μm) depressed Ca2+ transients by 20.5 ± 1.8% (n= 5), a value not significantly different to that obtained under control conditions (P > 0.1). However, even if the singular effect of TEA alone on evoked presynaptic volleys and presynaptic Ca2+ influxes warrants careful interpretation, these data show that l-AP4-induced depressant effects on PF presynaptic Ca2 influx are independent of TEA-sensitive K+ channels.
We then hypothesized that other K+ channels could be implicated in this mGluR4-mediated effect. Given the importance of two-pore-domain potassium channels (K2P) in the regulation of membrane potential and neuronal excitability, we investigated whether l-AP4 could exert its effects through activating certain K2Ps that are found on cerebellar granule cells like TREK-1 (Talley et al. 2001), TASK-1 (Aller et al. 2005) and TASK-3 (Watkins & Mathie, 1996). To study these TREK channels in the mGluR4-mediated inhibition of evoked presynaptic Ca2+ transients, we determined whether this inhibition was affected by the serotonin re-uptake inhibitor, fluoxetine, a compound that also seems to act as an allosteric blocker of these channels (Kennard et al. 2005; Honoré, 2007). We first determined the effect of fluoxetine on the PF volley. As shown in Fig. 2Aa, 20 min bath applications of 50 μm fluoxetine increased the amplitude of the depolarizing wave of the PF afferent volley, indicating an increased granule cell axon excitability (n= 4). However, using fluorometric measurements, we observed that similar applications of fluoxetine reduced the amplitude of presynaptic Ca2+ transients evoked by PF stimulations by 24.4 ± 7.3% (n= 7, Fig. 2Ab). From this plateau (representing reduced evoked calcium transients after a 20 min application of 50 μm fluoxetine), subsequent co-application of l-AP4 (100 μm) further depressed these calcium transients by 26.1 ± 5.1%, a value not significantly different (P > 0.8) to l-AP4-evoked depression in control experiments.
Figure 2. Lack of effect of the K+ channel blockers fluoxetine, ruthenium red and Tertiapin Q on l-AP4-mediated inhibition of presynaptic Ca2+ influx.
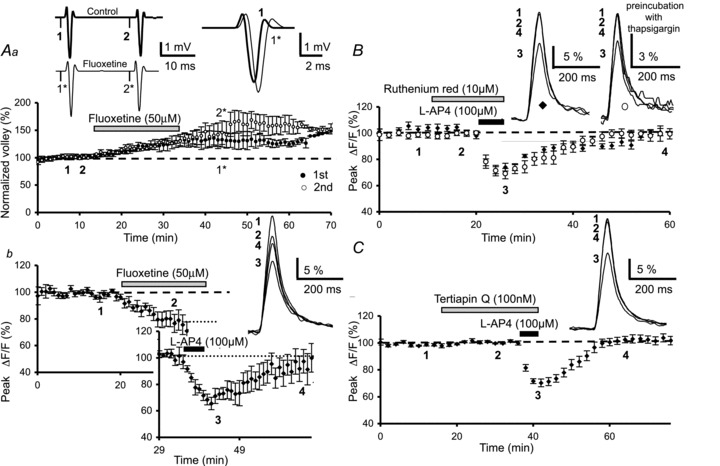
Aa, plot of the normalized amplitude of PF volleys against time before, during and after bath application of fluoxetine (50 μm) (n= 4). The inset on the left shows examples of averaged PF olleys evoked by two stimuli separated by 20 ms and recorded at the indicated times in control conditions (top, thick trace) and in the presence of fluoxetine (bottom, thin trace). Note that fluoxetine increases the amplitude of the first (black circles) and the second (open circles) PF volley. In addition, note the change in the kinetics of the first PF volley (inset on the right). Ab, plot of normalized amplitudes of peak Fluo4-FF fluorescence transients (ΔF/F) recorded as a function of time before, during and after bath application of fluoxetine (50 μm) and co-application of l-AP4 (100 μm) (n= 7). The top inset displays superimposed averaged fluorescence transients in one of these experiments, recorded at the indicated times. The bottom inset (graph) shows the l-AP4-mediated inhibition of the fluorescence transients normalized to the plateau level obtained after application of fluoxetine. Note that application of fluoxetine alone reduced the amplitude of these fluorescence transients. B, the same as in Ab before, during and after bath application of ruthenium red (10 μm) and co-application of l-AP4 (100 μm) in control slices (n= 10, black diamonds) and in slices pre-incubated with thapsigargin (10 μm) (n= 7, open circles). The insets are the same as in Ab recorded in control slices (left) and pre-incubated slices (right). C, the same as in Ab before, during and after bath application of Tertiapin Q (100 nm) and co-application of l-AP4 (100 μm) (n= 10). Top inset as in Ab.
To investigate whether l-AP4 acts by modulating certain TASK channels, we used ruthenium red, a polyvalent cation that has been shown to reduce the open probability of these channels (Musset et al. 2006). A 10 min bath application of 10 μm ruthenium red had no effect on either the PF volley (n= 4, Supplementary Fig. S2A) or the amplitude or duration of evoked presynaptic Ca2+ transients (n= 10, Fig. 2B). When the 10 min application of ruthenium red (10 μm) was followed by 5 min co-application of l-AP4 (100 μm), evoked presynaptic Ca2+ transients were depressed by 25.4 ± 2.7%, a value not significantly different (P > 0.9) to l-AP4-evoked depression in control experiments (n= 4, diamonds, Fig. 2B). Since ruthenium red is also known to block Ca2+-sensitive ryanodine receptors, the activation of which result in the liberation of Ca2+ from intracellular stores (Bezprozvanny et al. 1991), we re-examined the effect of ruthenium red on l-AP4-induced reductions in Ca2+ transients after pre-incubation of slices (40 min) in 10 μm thapsigargin, a molecule that empties intracellular Ca2+ stores (Thastrup et al. 1990). In these slices, after 10 min application of ruthenium red (10 μm), co-application of l-AP4 (5 min, 100 μm) evoked presynaptic Ca2+ transients that were depressed by 25.2 ± 4.5% (n= 7, circles, Fig. 2B), a value not significantly different (P > 0.9) to l-AP4 depression in control experiments. These data suggest that the inhibitory action of l-AP4 on presynaptic Ca2+ transients cannot be attributed to the activation TREK or TASK channels that are sensitive to fluroxetine and ruthenium red, respectively.
Since certain mGluRs are known to couple to G protein-gated inwardly rectifying K+ channels (GIRKs) (see Niswender et al. 2008), we asked whether the mGluR4-mediated inhibition of evoked presynaptic Ca2+ influxes involves GIRK activation. We used Tertiapin Q, which selectively inhibits some subtypes of this K+ channel family with nanomolar affinity (Jin & Lu, 1998). We first determined whether this inhibitor altered the presynaptic waveform. Bath application (20 min) of Tertiapin Q (100 nm) had no effect on the shape of the PF volley (n= 4, Supplementary Fig. S2B), or the amplitude or the duration of presynaptic fluorescence transients evoked by PF stimulations (n= 10, Fig. 2C). When the 20 min application of Tertiapin Q (100 nm) was followed by 5 min co-application of l-AP4 (100 μm), evoked presynaptic Ca2+ transients were depressed by 28.9 ± 2.5% (n= 10, Fig. 2C), a value not significantly different (P > 0.3) to l-AP4-evoked depression in control experiments. Taken together, these data show that the inhibitory action of l-AP4 on presynaptic Ca2+ transients cannot be attributed to the activation of Tertiapin Q-sensitive K+ channels.
mGluR4 activation modulates multiple types of voltage-gated Ca2+ channels
There are at least three pharmacologically distinguishable types of VGCCs that synergistically contribute to neurotransmitter release at PF–PC synapses: the ω-agatoxin TK-sensitive P/Q-type, the ω-conotoxin GVIA-sensitive N-type and the SNX-482-sensitive R-type (Mintz et al. 1995; Brown et al. 2004; Daniel et al. 2004). To test the hypothesis that activation of mGluR4 at these synapses reduces glutamate release by selectively inhibiting one or more of these presynaptic VGCCs, we used specific toxins that target these channels.
First, we blocked P/Q-type Ca2+ channels with ω-agatoxin TK (250 nm) and investigated the effect of l-AP4 on the remaining fraction of presynaptic Ca2+-sensitive fluorescence transients elicited by PF stimulation. Bath application of ω-agatoxin TK (250 nm for 30 min) alone reduced presynaptic Ca2+ transients by 33.6 ± 6.2% (n= 6, Fig. 3A). From this plateau (representing the reduced Ca2+ transients in the presence of ω-agatoxin TK), subsequent application of l-AP4 (100 μm) further decreased presynaptic Ca2+ transients by 25.9 ± 1.5% (n= 6, Fig. 3A). The l-AP4 depression was statistically indistinguishable from that observed with bath application of l-AP4 in control conditions (P > 0.8). We next investigated the l-AP4-mediated inhibition of presynaptic Ca2+ transients after N-type Ca2+ channels were blocked by a 30 min bath application of 250 nmω-conotoxin GVIA. ω-conotoxin GVIA alone reduced presynaptic fluorescence transients by 36.9 ± 6% (n= 5, Fig. 3B). From this plateau (representing the reduced Ca2+ transients in the presence of ω-conotoxin GVIA), subsequent application of l-AP4 (100 μm) further decreased presynaptic Ca2+ transients by 27.6 ± 4.4% (n= 5, Fig. 3B), a value not significantly different (P > 0.8) to that observed with l-AP4 application alone. We next blocked R-type Ca2+ channels with 30 min bath application of 100 nm SNX-482 and examined the effect of l-AP4 on the remaining fraction of presynaptic Ca2+ transients. SNX-482 alone reduced presynaptic fluorescence transients (plateau) by 21.3 ± 3.5% (n= 6, Fig. 3C). Subsequent application of l-AP4 (100 μm) further decreased these transients by 28.9 ± 4% (n= 6, Fig. 3C), a value not significantly different to that observed for l-AP4 in control conditions (P > 0.6).
Figure 3. l-AP4-mediated inhibition of presynaptic Ca2+ influx results from modulation of multiple types of VGCCs.
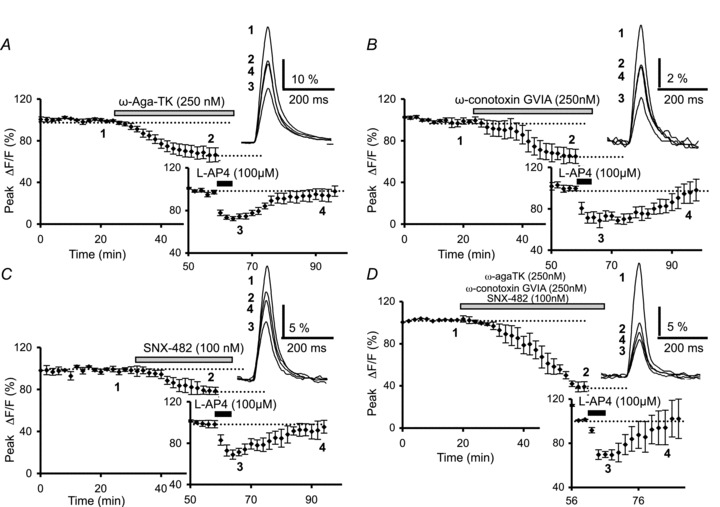
A, the plot represents normalized amplitudes of peak fluorescence transients recorded as a function of time before, during and after sequential bath application of ω-agatoxin TK (250 nm) and l-AP4 (100 μm) (n= 6). The top inset displays superimposed averaged fluorescence transients in one of these experiments recorded at the indicated times. The bottom inset (graph) shows the l-AP4-mediated inhibition of the fluorescence transients normalized to the plateau level obtained after application of ω-agatoxin TK. B, the same as in A with sequential application of ω-conotoxin GVIA (250 nm) and l-AP4 (100 μm) (n= 5). The insets are the same as in A. C, the same as in A with sequential application of SNX-482 (100 nm) and l-AP4 (100 μm) (n= 6). The insets are the same as in A. D, time course of fluorescence transients before, during and after bath application of combined toxin ω-agatoxin TK (250 nm), ω-conotoxin GVIA (250 nm) and SNX-482 (100 nm), followed by application of l-AP4 (100 μm) (n= 4). The insets show averaged fluorescence transients in one of these experiments (top) and the l-AP4-mediated inhibition of these transients normalized to the plateau level obtained after application of combined toxin treatment (bottom).
Finally, to investigate possible cross-interactions between mGluR4 and the different presynaptic VGCCs, we applied ω-agatoxin TK (250 nm), ω-conotoxin GVIA (250 nm) and SNX-482 (100 nm) simultaneously for 30 min. While bath application of cadmium (100 μm) totally abolished Ca2+-sensitive fluorescence transients elicited by PF stimulation (n= 4, Supplementary Fig. S3), the cocktail of Ca2+-blocking toxins decreased fluorescence transients by 60.8 ± 4.7% (n= 4, Fig. 3D). In the presence of this cocktail, subsequent application of l-AP4 (100 μm) further reduced these transients, giving an additional reduction in transient amplitude expressed as a percentage of the transients recorded at the plateau level of 28.1 ± 3.5% (n= 4, Fig. 3D). This value was not significantly different to that observed in control conditions (P > 0.4), or after application of ω-agatoxin TK (P > 0.5), ω-conotoxin GVIA (P > 0.9) or SNX-482 (P > 0.8) alone (see above).
Taken together, these results show that mGluR4 activation does not selectively inhibit P/Q-, N- or SNX-482-sensitive R-type presynaptic VGCCs. Indeed, when a single type of VGCC is selectively blocked, the presynaptic Ca2+ influx resulting from activation of other types of calcium channels, whatever they may be, is still inhibited by pharmacological activation of mGluR4, and to the same degree as mGluR4 activation in control saline. mGluR4 activation by l-AP4 appears to modulate all presynaptic terminal VDCCs to the same degree.
MAPKs or PI3Ks are not implicated in mGluR4-mediated depression of presynaptic Ca2+ influx
Previous studies have demonstrated that in cultured cerebellar granule cells, mGluR4s are coupled to MAPKs and PI3Ks and that these pathways are involved in the neuroprotective role of mGluR4s (Iacovelli et al. 2002). In order to determine if these pathways are also implicated in mGluR4-mediated depression of PF Ca2+ influx, in separate experiments we examined the effect of l-AP4 under conditions in which these kinases were blocked with PD98059 and LY294002, cell-permeable inhibitors of MAPKs and PI3Ks, respectively (Vlahos et al. 1994; Alessi et al. 1995). A 20 min bath application of either PD98059 (20 μm) (n= 8, Fig. 4A) or LY294002 (1 to 50 μm) (n= 8, Fig. 4B) had no effect on fluorescence transients evoked by PF stimulation. When PD98059 was followed by 100 μm l-AP4, Ca2+ transients were reduced by 24.9 ± 1.9%, (n= 8, Fig. 4A), a value not significantly different to that observed with l-AP4 alone (P > 0.4). Similarly, l-AP4 application in the presence of LY294002 reduced presynaptic Ca2+ influx by 27 ± 1.7% on average (n= 8, Fig. 4B), again, a value statistically indistinguishable from that observed in control conditions (P > 0.8). As PD98059 and LY294002 were prepared in DMSO (final concentration 0.1%), we verified that DMSO had no effect under our experimental conditions. After 3.5 h of slice pre-incubation in DMSO (final concentration 0.3%), l-AP4 still depressed fluorescence transients by 22.1 ± 2.8% (n= 5, Fig. 4C), a value not significantly different to that observed in control conditions (no pre-incubation, P > 0.2). Taken together, these data clearly indicate that the mGluR4-mediated depression of PF Ca2+ transients involves neither the MAPK nor the PI3K signalling pathway.
Figure 4. Lack of effect of MAPK and PI3K blockade on l-AP4-mediated inhibition of presynaptic Ca2+ influx.
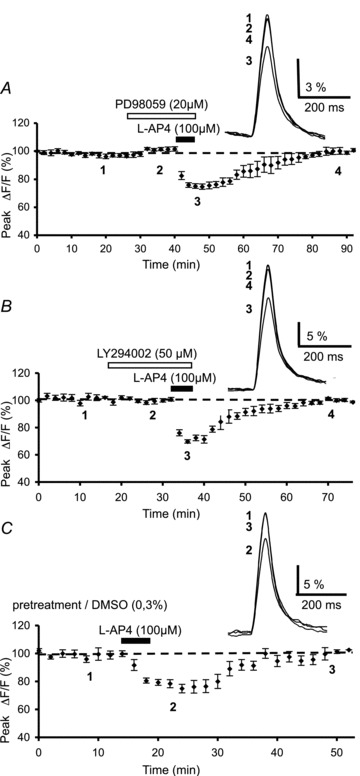
A, plot of normalized amplitudes of peak fluorescence transients before, during and after sequential bath application of the MAPK inhibitor, PD98059 (20 μm) and l-AP4 (100 μm) (n= 8). The inset represents the superimposed averaged fluorescence transients in one of these experiments. B, same as in A with sequential bath application of the PI3K inhibitor, LY294002 (50 μm) and l-AP4 (100 μm) (n= 8). Inset as in A. C, same as in A with bath application of l-AP4 (100 μm) after pre-incubation of slices in DMSO (0.3%) for 3.5 h (n= 5). Insets as in A.
We then verified that in our conditions PD98059 and LY294002 actually inhibit, respectively, MAPK and PI3K activities. In cerebellar culture, it has been demonstrated that phorbol ester-induced long-term depression (LTD) of synaptic transmission between granule cells and PCs is mediated through activation of the MAPK pathway, which may either be a downstream target of PKC or act in parallel with PKC, and that PD98059 completely blocked this form of synaptic plasticity (Endo & Launey, 2003). In cerebellar slices, we performed whole-cell patch-clamp recordings to measure PC excitatory postsynaptic currents (EPSCs) evoked by PF stimulation. A 10 min bath application of phorbol esters (PMA, 200 nm) resulted in a long-term decrease in the amplitude of PF-mediated EPSCs (23.9 ± 2.8%, n= 4, Supplementary Fig. S4A), as previously demonstrated (Endo & Launey, 2003). After 15 min of pretreatment with PD98059 (20 μm), PMA was ineffective in inducing LTD (n= 4, Supplementary Fig. S4B), demonstrating that in our experimental conditions, MAPK activity was effectively inhibited. In the same preparation, we then tested LY294002 since this inhibitor of PI3K has been shown to block the induction of a nitric oxide (NO)-dependent form of plasticity, long-term potentiation (LTP), at PF–PC synapses (Jackson et al. 2010). In agreement with these observations, bath application of the NO donor DEA/NO (10 μm) led to a robust long-term increase in PF-mediated EPSC (40.5 ± 4.4% and 59.6 ± 5.2%, respectively, for the second and first response, n= 4, Supplementary Fig. S4C). As previously demonstrated (Jackson et al. 2010), LY294002 (50 μm) pretreatment for 15 min entirely blocked LTP (n= 4, Supplementary Fig. S4D), showing that this compound effectively blocks PI3K signalling in our experimental conditions.
mGluR4-mediated inhibition of presynaptic Ca2+ influx does not require the Gi/o protein–cAMP–PKA signalling cascade
The classical transduction pathway activated by pharmacological stimulation of mGluR4 is the inhibition of adenylyl cyclase (AC) and protein kinase A (PKA), via Gi/o proteins (Prezeau et al. 1994). In an attempt to verify that the intracellular signals mediating the l-AP4 inhibition of presynaptic Ca2+ influx are part of the Gi/o protein–AC cascade in our model, we examined the effect of l-AP4 on presynaptic fluorescence transients after blocking either Gi/o proteins, AC or PKA activity. Our results are surprising.
Cerebellar slices were incubated with PTX (2–5 μg ml−1) for 12–19 h before the recording session to inhibit Gi/o protein activity. Following PTX pretreatment, the average peak amplitude of the fluorescence transients (8.4 ± 1.9% (ΔF/F), n= 6) was not significantly different from that recorded in control slices (incubated for the same time in the absence of PTX (8.6 ± 2.2% (ΔF/F), n= 8) (P > 0.1). Bath application of 100 μm l-AP4 reduced the amplitude of fluorescence transients by 24.1 ± 2.5% (n= 9, Fig. 5A), which is not significantly different to that observed under control conditions (P > 0.6). In order to confirm the efficacy of the PTX pretreatment, in a separate series of experiments we activated presynaptic type 1 cannabinoid (CB1) receptors with a specific agonist, WIN55,212-2, after pretreatment of slices in PTX. CB1 receptors are known to activate Gi/o proteins and decrease presynaptic Ca2+ influx (Daniel et al. 2004) and synaptic transmission (Kreitzer & Regehr, 2002) at PF–PC synapses. In PTX-pretreated slices, 30 min bath application of WIN55,212-2 (1 μm) had no significant effect on evoked PF Ca2+ transients (n= 9, Fig. 5B). This result concurs with that previously shown by Daniel et al. (2004), and demonstrates unambiguously that PTX was effective in our experimental conditions. In these PTX-pretreated slices, we applied l-AP4 after WIN55,212-2 treatment and observed a reduction in evoked Ca2+ transients of 22.4 ± 2.9%, a value not significantly different from the l-AP4 effect in control conditions (P > 0.8) (n= 9, Fig. 5B). Thus, it appears that the l-AP4-mediated depression of presynaptic Ca2+ influx does not depend on the activation of Gi/o proteins.
Figure 5. Lack of effect of pertussis toxin treatment, adenylyl cyclase inhibition or PKA blockade on l-AP4-mediated inhibition of presynaptic Ca2+ influx.
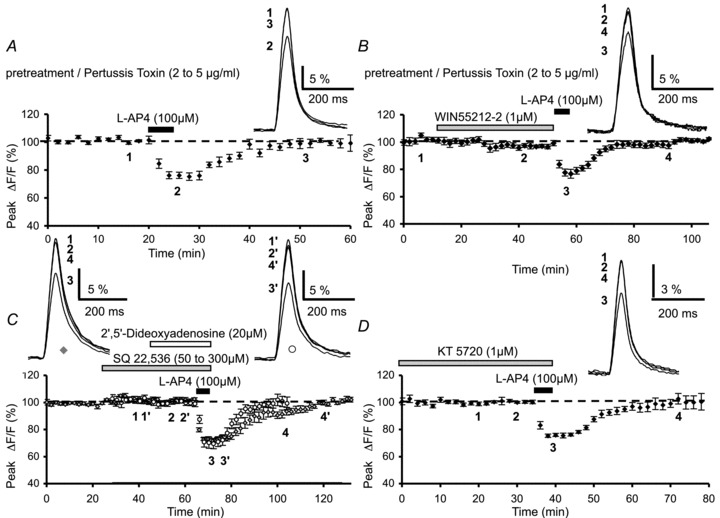
A, plot of normalized amplitudes of peak fluorescence transients before, during and after bath application of l-AP4 (100 μm) (n= 9). Experiments were performed on slices pre-incubated with pertussis toxin (2 to 5 μg ml−1) for 12–19 h. The inset represents the superimposed averaged fluorescence transients in one of these experiments. B, same as in A with sequential bath application of WIN55,212-2 (1 μm) and l-AP4 (100 μm) (n= 9), following similar pre-incubation of slices with pertussis toxin. Inset as in A. Note that after this pre-treatment WIN55,212-2, in contrast to l-AP4, does not inhibit fluorescence transients. C, same as in A with sequential bath application of the adenylyl cyclase inhibitors SQ 22,536 (50 to 300 μm, diamonds, n= 11) or 2′,5′-dideoxyadenosine (20 μm, circles, n= 6), and l-AP4 (100 μm). Insets as in A. D, plot of normalized amplitudes of peak fluorescence transients before, during and after bath sequential application of the PKA inhibitor, KT 5720 (1 μm) and l-AP4 (100 μm) (n= 9). Inset as in A.
Given our results obtained after Gi/o protein inactivation, in separate experiments we tested the involvement of AC in the l-AP4-mediated depression of evoked fluorescence transients. We used two inhibitors of adenylyl cyclase, namely SQ22,536 (Harris et al. 1979) and 2′,5′-dideoxyadenosine (Gille et al. 2004), both of which are membrane-permeable broad-spectrum inhibitors of AC. Bath application of SQ22,536 (50 to 300 μm) for 40 min had no effect on fluorescence transients evoked by PF stimulation (n= 11, diamonds, Fig. 5C). Moreover, this blocker did not prevent the subsequent inhibitory effect of l-AP4, since in the presence of l-AP4, the amplitude of fluorescence transients was reduced by 27.6 ± 1.5% (n= 11, Fig. 5C), a value not significantly different to the l-AP4-mediated inhibition in control conditions (P > 0.7). Similarly, 20 min bath application of 2′,5′-dideoxyadenosine (20 μm) had no effect on fluorescence transients evoked by PF stimulation (n= 6, circles, Fig. 5C). Here again, subsequent application of l-AP4 reduced evoked Ca2+ transients by 29.7 ± 3.8% (n= 6, Fig. 5C), a value statistically indistinguishable to that observed in control conditions (P > 0.7).
As for the PTX experiments described above, we verified that in our conditions SQ22,536 and 2′,5′-dideoxyadenosine inhibit adenylyl cyclase activity. At PF–PC synapses, elevation of cAMP levels by forskolin, an activator of adenylyl cyclase, has been shown to endurably enhance neurotransmitter release (Salin et al. 1996; Chen & Regher, 1997) through a presynaptic mechanism that does not alter resting Ca2+ levels or presynaptic Ca2+ influx, but rather directly increases the probability of vesicular release (Chen & Regher, 1997). Thus, we reasoned that this long-term enhancement of synaptic strength downstream from Ca2+ influx, might provide a selective means to test the efficiency of adenylyl cyclase inhibitors. Again, whole-cell patch-clamp recordings of PCs were performed to record PF-mediated EPSCs. A 10 min bath application of 50 μm forskolin resulted in a large increase in the amplitude of PF-mediated EPSCs (168.2 ± 6.9%, n= 8, Supplementary Fig. S5A). These data are consistent with previous studies (Salin et al. 1996; Chen & Regher, 1997, Daniel et al. 2004). After pretreatment with SQ22,536 (50 μm) for 40 min or 2′,5′-dideoxyadenosine (20 μm) for 20 min, forskolin was ineffective (n= 4, Supplementary Fig. S5B and C), confirming that in our experimental conditions, adenylyl cyclase activity was effectively inhibited. Collectively, these data demonstrate that the mGluR4-mediated depression of presynaptic Ca2+ influx does not involve AC.
As a final step in exploring this signalling pathway, we looked for a role of PKA in the l-AP4-mediated inhibition of presynaptic Ca2+ influx by examining the l-AP4-mediated reduction in Ca2+ transients after inhibition of PKA activity. We employed KT 5720, a specific membrane-permeable inhibitor of PKA that acts with nanomolar affinity (Kase et al. 1987). Bath application of 1 μm KT 5720 for 10 min to 2 h had no effect on evoked fluorescence transients (n= 9, Fig. 5D), nor did this blocker prevent the inhibitory effect of subsequently applied l-AP4. In the presence of KT 5720, l-AP4 depressed PF fluorescence transients by 24.1 ± 1.3% on average (n= 9, Fig. 5D), a value not significantly different to that observed in control conditions (P > 0.9). These data demonstrate that the depressant effect of l-AP4 on presynaptic Ca2+ influx does not require the modulation of PKA activity.
Taken together, our results show that under our experimental conditions, mGluR4-mediated depression of evoked PF Ca2+ influx is independent of the classical Gi/o protein–cAMP–PKA transduction pathway.
mGluR4-mediated inhibition of presynaptic Ca2+ influx probably involves the PLC–PKC signalling cascade
As demonstrated by Perroy et al. (2000) in cultured cerebellar granule cells, mGluR7s, which are part of the group III family, are functionally coupled to a PLC–PKC signalling pathway. In light of our results showing that l-AP4 does not activate the Gi/o protein–AC–PKA transduction pathway, we examined the possibility that PLC and PKC are also intracellular actors in the mechanisms underlying the inhibitory effect of mGluR4 activation on presynaptic Ca2+ influx. We began by blocking the PLC pathway by pre-incubating cerebellar slices for 3.5 h in the membrane-permeable PLC inhibitor, U-73122 (10 μm) (Netzeband et al. 1997). We first tested for putative effects of this compound on fluorescence transients evoked by PF stimulation. U-73122 (10 μm) alone had no effect on these transients (n= 3, not illustrated). Co-application of l-AP4 and U-73122 produced a much smaller depression in evoked Ca2+ influx (9.4 ± 4.2%) than that observed with l-AP4 alone (n= 6, Fig. 6A). This difference was statistically significant (P < 0.001). Furthermore, as expected, pre-incubation of slices for 3.5 h with the inactive analogue U-73343 (10 μm), had no effect per se on fluorescence transients (n= 3, not illustrated), or on the l-AP4-mediated depression of fluorescence transients (22.1 ± 2.5%, n= 5, Fig. 6B), a value not significantly different to that observed in control experiments (P > 0.3).
Figure 6. Effect of phospholipase C blockade on l-AP4-mediated inhibition of presynaptic Ca2+ influx and on DHPG-mediated depression of PF synaptic responses.
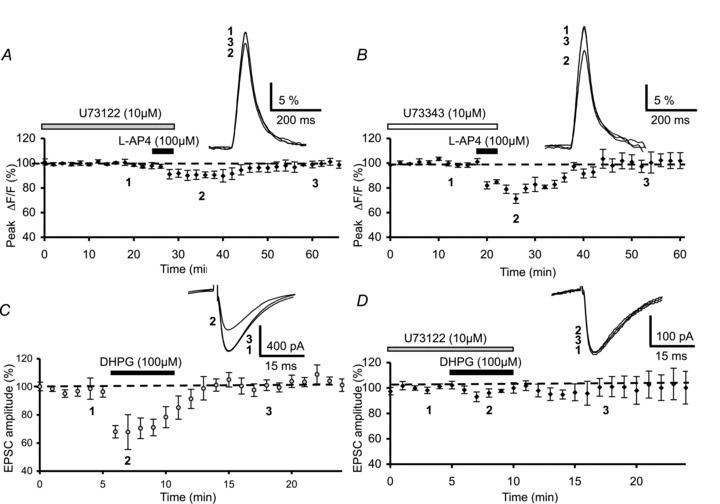
A, plot of normalized amplitudes of peak fluorescence transients before, during and after bath application of l-AP4 (100 μm) in the presence of U-73122 (10 μm) (n= 6). Note that experiments were performed on slices pre-incubated with the PLC inhibitor, U-73122 (10 μm) for 3.5 h. The inset represents superimposed averaged fluorescence transients in one of these experiments. B, same as in A with bath application of l-AP4 (100 μm) in presence of, and after 3.5 h pre-incubation of slices with the inactive analogue of U-73343 (10 μm) (n= 5). Inset as in A. C, plot of normalized amplitudes of PF-mediated excitatory postsynaptic currents (EPSCs) as a function of time before, during and after bath application of 100 μm DHPG in control conditions (circles, n= 4). Inset displays superimposed PF-mediated EPSCs recorded in one of each series of experiments recorded at the indicated time. D, time course of EPSC amplitude before, during and after bath application of 100 μm DHPG and example traces (inset), with slices pre-incubated in U-73122 (10 μm) for 3.5 h (diamonds, n= 4). Note that this pre-incubation prevents the DHPG-mediated depressant effect on PF-mediated EPSC amplitude.
Finally, to ascertain the efficacy of the PLC inhibitor, U-73122 (10 μm), we analysed the effects of this compound on mGluR1-mediated depression of evoked EPSCs, which in cultured Purkinje cell neurons is known to be due, at least in part, to PLC activation (Netzeband et al. 1997). In control experiments, 5 min bath application of DHPG (100 μm), a selective mGluR1 agonist, induced a transient decrease in the amplitude of PF-mediated EPSCs of 31.2 ± 4.9% (n= 4, Fig. 6C), an effect consistent with our previous study (see Fig. 2B in Levenes et al. 2001). This DHPG-mediated inhibitory effect was attenuated by pre-incubation of slices for 3.5 h with U-73122 (10 μm), since the magnitude of the transient decrease in PF-mediated responses was only 4.1 ± 3.1% (n= 5, Fig. 6D), a value significantly smaller than that observed in control experiments (P < 0.001). This finding unambiguously demonstrates that in our experimental conditions, U-73122 (10 μm) blocked PLC activity.
Furthermore, to illustrate the physiological relevance of a PLC-dependent pathway in the l-AP4-mediated modulation of synaptic transmission at PF–PC synapses, we performed a series of electrophysiological experiments to evaluate whether the l-AP4-induced depression of EPSCs and/or the paired-pulse facilitation (PPF) was impaired after inhibiting PLC at these synapses. PPF has been successfully used at PF–PC synapses as an index of a presynaptic site of action of the group III mGluR broad-spectrum agonist l-AP4 (Pekhletski et al. 1996; Miniaci et al. 2001; Lorez et al. 2003). However, taking into account the fact that U-73122 affects both pre- and postsynaptic compartments our results require careful interpretation. In control experiments, 5 min bath application of 100 μm l-AP4 reversibly depressed the amplitude of both the first and the second PF-evoked EPSC elicited by two successive stimuli in PCs recorded in voltage-clamp mode. At its peak, the mean decrease in PF-evoked EPSC amplitude was 74.3 ± 3.5% for the first response (grey triangle) and 65.6 ± 2.8% for the second (grey circle) (n= 10, Fig. 7A top). l-AP4-induced decreases in amplitude were accompanied by a highly significant (P < 0.01) increase in mean PPF, from 2.3 ± 0.1 during the control period to 3.5 ± 0.4 at the peak of l-AP4 depressant effect (Fig. 7A bottom trace, n= 10).
Figure 7. l-AP4-mediated inhibition of synaptic transmission and presynaptic Ca2+ influx involve activation of a PLC–PKC pathway.
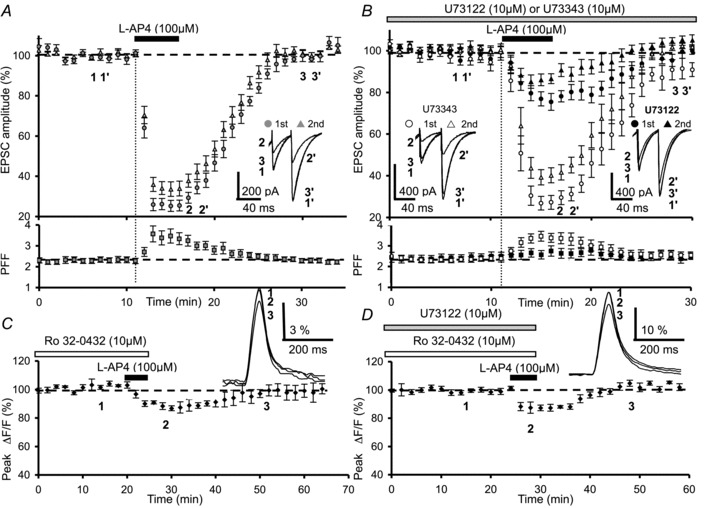
A, time course of normalized amplitudes of PF-mediated EPSCs before, during and after bath application of 100 μm l-AP4 (n= 10). The inset displays superimposed sweeps of representative PF-evoked EPSCs elicited in one Purkinje cell by 2 successive PF stimulations with an inter-stimulus interval of 40 ms, before (1, 1′), during (2, 2′) and after (3, 3′) agonist application. Each trace is an average of 5–15 consecutive trials. The reversible l-AP4 depression of PF-mediated EPSCs is accompanied by a transient increase in paired-pulse facilitation (PPF). B, same as in A, but after pre-incubating slices for 3.5 h in the membrane-permeable PLC inhibitor U-73122 (10 μm, n= 10 black symbols), or its inactive analogue U-73343 (10 μm, n= 10, white symbols). Insets as in A. Note that pre-incubation with the PLC inhibitor (U-73122, 10 μm) partially prevents the l-AP4-mediated depressant effect on PF-evoked EPSCs amplitude and the transient increase in PPF, which is not observed with its inactive analogue. C, plot of normalized amplitudes of peak fluorescence transients before, during and after bath application of l-AP4 (100 μm) in the presence of Ro 32-0432 (10 μm) (n= 6). Experiments were performed on slices pre-incubated with Ro 32-0432 (10 μm) for 1.5 h. The inset represents superimposed averaged fluorescence changes in one of these experiments. D, same as in C with bath application of l-AP4 (100 μm) in presence of Ro 32-0432 (10 μm) and U-73122 (10 μm) (n= 7). Sample traces are shown in the inset. Experiments were performed with slices pre-incubated in Ro 32-0432 and U-73122, for 1.5 h and 3.5 h, respectively.
In marked contrast, for the 10 cells tested after pretreatment with U-73122 (10 μm), the depressant effect on PF-EPSC amplitude following 5 min bath application of 100 μm l-AP4 was 23.8 ± 3.7% and 14.4 ± 3% for the first (black circle) and the second (black triangle) EPSCs, respectively (n= 10, Fig. 7B top). These l-AP4-induced depressions were significantly smaller than those recorded in control experiments for both the first and the second responses (P < 0.0001). PPF remained unchanged (n= 10, Fig. 7B bottom trace, filled squares). In contrast, pretreatment with U-73343 (10 μm), an inactive analogue of the PLC inhibitor, had no effect on the l-AP4-mediated inhibition of PF-EPSC amplitude (71.5 ± 5.6% for the first responses (white triangle) and 58.8 ± 3.9% for the second responses (white circle) (n= 10, Fig. 7B top). These values were not significantly different to those observed for the first and the second PF-EPSCs in control experiments (P > 0.2). In the presence of U-73122, a highly significant (P < 0.001) increase in mean PPF was observed, from 2.2 ± 0.1 during the control period to 3.4 ± 0.2 at the peak depressant effect (n= 10, Fig. 7B bottom trace). The mean PPF at the peak of the l-AP4 depressant effect was not significantly different from that observed in control conditions (P > 0.3).
As a final step we examined the putative role of PKC in the mGluR4-mediated depressant effect of presynaptic Ca2+ influx using fluometric mesurements. To this end, we incubated slices for 1.5 h in 10 μm Ro 32-0432, a cell-permeable inhibitor of PKC (Morreale et al. 1997). While Ro 32-0432 had no effect on presynaptic Ca2+ influx, subsequent co-application of 100 μm l-AP4 and Ro 32-0432 decreased fluorescence transients by only 11.3 ± 2.1% (n= 6, Fig. 7C), a value significantly smaller than that observed with l-AP4 alone (P < 0.001).
In light of our results showing a role for PLC and PKC in the l-AP4-mediated decreases in evoked Ca2+ influx, we finished our study with a set of occlusion experiments to test whether PLC and PKC could interact in mediating the l-AP4-induced effects. After co-incubation of slices in U-73122 (10 μm) and Ro 32-0432 (10 μm) (see above), application of 100 μm l-AP4 decreased fluorescence transients by 12.6 ± 2.7% (n= 7, Fig. 7D), a value significantly smaller than that observed with l-AP4 alone (P < 0.001) but similar to values observed with either U-73122 alone (P > 0.5) or Ro 32-0432 alone (P > 0.8). These results suggest that PLC and PKC are both involved in l-AP4-induced responses, and probably use the same signalling pathway, because the l-AP4 decreases in Ca2+ influx during blockade of both these enzymes are not additive.
Collectively, these data strongly suggest that the mGluR4-mediated depression of presynaptic Ca2+ influx can be attributed to the activation of a PLC–PKC pathway.
Discussion
In this study we investigated the molecular mechanisms underlying mGluR4-mediated depression of presynaptic Ca2+ influx at PF–PC synapses. Our principal finding is that the mGluR4-mediated depression requires neither the ‘classical’ activation of a PTX-sensitive Gi/o protein, nor the AC–PKA signalling pathway. We present evidence that at PF–PC synapses, l-AP4 activation of mGluR4 reduces evoked presynaptic Ca2+ influx by way of a non-canonical signalling pathway, notably one involving PLC and the subsequent activation of PKC.
Presynaptic K+ channels are not involved in mGluR4-induced depression of presynaptic Ca2+ entry
We previously showed that 4-AP (1 mm) dramatically affected the properties of evoked Ca2+ transients and completely abolished the l-AP4-mediated inhibition of these transients (Daniel & Crepel, 2001). However, it has since been demonstrated that 1 mm 4-AP greatly affects the PF afferent volley and prevents spike repolarisation (Brown et al. 2004). As such we re-investigated the mGluR4–K+ channel 4-AP-sensitive link using lower concentrations (200 μm) of this blocker. Even at this concentration, 4-AP profoundly altered both the presynaptic volley and the presynaptic Ca2+ influx, rendering this pharmacological tool inappropriate for studying presynaptic Ca2+ events. Thus, in order to investigate the putative role of K+ channels in the l-AP4-mediated depression of presynaptic Ca2+ influx, we used TEA, another blocker of K+ channels that has less marked effects on Ca2+ influx and only minor effects on the presynaptic waveform (Brown et al. 2004). We conclude that presynaptic TEA-sensitive K+ channels are unlikely to contribute to the mGluR4-mediated inhibition of presynaptic Ca2+ influx. Why in our previous study (Daniel & Crepel, 2001) 1 mm 4-AP hindered l-AP4-induced decreases in presynaptic Ca2+ transient amplitude may lie in the fact that this compound greatly slowed the time course of the Ca2+ signals, even in low extracellular Ca2+ concentrations. In these conditions a residual increase in presynaptic cytosolic Ca2+ levels might contribute to profound alteration of Ca2+ signalling in active zones where mGluR4 is present (Mateos et al. 1999). Thus, these 4-AP-induced variations in presynaptic Ca2+ transients that are not observed either in control conditions or in the presence of low concentrations of TEA, could explain at least in part the puzzling lack of effect of l-AP4 on evoked calcium influx in the presence of 1 mm 4-AP.
Along the same line, we show that Tertiapin Q, at concentrations known to inhibit certain inward rectifying potassium channels including GIRK 1 and 4 (Jin & Lu, 1998), affected neither the shape of the PF volley nor the amplitude or duration of evoked presynaptic Ca2+ transients (Daniel & Crepel, 2001). Since Tertiapin Q had no effect on l-AP4 depression of evoked presynaptic Ca2+ influx, we conclude that GIRK 1 and 4 do not contribute to the l-AP4-induced depression of PF–PC synaptic transmission.
Recent studies have revealed that two-pore-domain K+ channels (K2P) that contribute to the K+ leak current are widely expressed throughout the central nervous system. TREK-1 (Talley et al. 2001), TASK-1 (Aller et al. 2005) and TASK-3 (Watkins & Mathie 1996) channels have been localized to cerebellar granule cells. These channels are tightly regulated by numerous G protein-coupled receptors, including mGluR4 (Cain et al. 2008), and may modulate neuronal excitability through their contribution to background membrane currents (Goldstein et al. 2001; Bayliss et al. 2003; Aller et al. 2005). We show that neither TREK nor TASK are responsible for the mGluR4-mediated inhibition of evoked presynaptic Ca2+ transients in PFs, since inhibiting TREK-1 channels with fluoxetine, or TASK 1 and TASK-3 channels with ruthenium red, did not affect the l-AP4-induced reduction in these transients. However, these results must be interpreted with caution since neither fluoxetine nor ruthenium red were developed as specific K2P channel blockers but have simply been shown to inhibit channel activity.
All identified types of Ca2+ channels are involved in mGluR4 depression of presynaptic Ca2+ entry
Glutamate release at PF–PC synapses is tightly regulated by at least three types of VGCCs that are pharmacologically distinct: the ω-agatoxin TK-sensitive P/Q-type, the ω-conotoxin GVIA-sensitive N-type and the SNX-482-sensitive R-type channels (Mintz et al. 1995; Brown et al. 2004; Daniel et al. 2004). In the CNS, group III mGluR activation depresses transmitter release by directly inhibiting N- or/and P/Q-type VGCCs (Takahashi et al. 1996; Millan et al. 2002; Capogna, 2004; Rusakov et al. 2004; Guo & Ikeda, 2005; Woodhall et al. 2007), but not R-type VGCCs (Woodhall et al. 2007). We studied the role of these VGCCs in the mGluR4-induced depression of evoked presynaptic Ca2+ transients by inhibiting P/Q-, N- and SNX-482-sensitive R-type Ca2+ channels individually and then collectively, using pharmacological compounds selective for each type of VGCC. Our results demonstrate that ω-agatoxin TK, ω-conotoxin GVIA and SNX-482 applied separately or together reduced the peak amplitude of evoked presynaptic fluorescence transients but never entirely eliminated them. It must be noted, however, that evoked calcium transients were entirely blocked by application of cadmium. This suggests that either we used non-saturating concentrations of each toxin or that other Ca2+ channels, not inhibited by these toxins, are functional on PF terminals. In any case, even if these concentrations were non-saturating, at least a portion of each type of Ca2+ channel was inhibited after which l-AP4 reduced remaining presynaptic Ca2+ transients to the same extent as under control conditions. Taken together, these results show that mGluR4 activation by l-AP4 does not selectively inhibit any one kind of Ca2+ channel. Rather, this receptor appears to modulate all types of Ca2+ channel present in the presynaptic terminals to the same degree.
mGluR4-mediated inhibition of presynaptic Ca2+ entry does not involve activation of MAPKs or PI3Ks
There is evidence that group III mGluRs can also activate MAPK and PI3K. In fact, it has been shown that in cultured cerebellar granule cells, mGluR4s are functionally coupled to both MAPKs and PI3Ks (Iacovelli et al. 2002). In our study, selective pharmacological blockade of each of these kinases (MAPK or PI3K) showed that neither of these proteins is involved in the l-AP4-induced depression of evoked presynaptic Ca2+ influx.
mGluR4-mediated inhibition of presynaptic Ca2+ entry does not require activation of the Gi/o protein–AC–PKA cascade
One of the most important finding of our study is that the mGluR4-mediated depression of presynaptic Ca2+ transients is not coupled to the classical group III mGluR transduction pathway described in most other systems. Indeed, these receptors are generally linked to Gi/o proteins that inhibit AC activity resulting in a decrease in intracellular cAMP levels and PKA activity (Prezeau et al. 1994; Neil et al. 1996; Conn & Pin, 1997). It must be mentioned, however, that presynaptic group III mGluRs can be positively coupled to AC and subsequently activate PKA (Evans et al. 2001). In our study, inactivation of Gi/o proteins with PTX had no effect on the inhibition of presynaptic Ca2+ transients following mGluR4 activation. Along the same line, pretreatment with the non-specific AC inhibitors SQ22,536 or 2′,5′-dideoxyadenosine, or with the PKA inhibitor KT 5720, had no effect on the reduction of presynaptic Ca2+ transients following mGluR4 activation. Taken together these results, albeit surprising, suggest that mGluR4-mediated depression of Ca2+ influx at PF terminals does not involve the activation of either Gi/o PTX-sensitive proteins, AC or PKA.
PLC–PKC-dependent signalling pathways are involved in the mGluR4-mediated inhibition of presynaptic Ca2+ entry
Finally, we investigated whether a new signalling pathway involving the activation of PLC and PKC could underlie the mGluR4-mediated depression of evoked presynaptic Ca2+ influx at PF terminals. We show that selective inhibition of either PLC or PKC significantly reduced the magnitude of the l-AP4-mediated depression of presynaptic Ca2+ transients. These findings strongly suggest that mGluR4 depresses presynaptic Ca2+ influx by way of a PLC–PKC intracellular signalling pathway. These results are in line with the observation that in cultured cerebellar granular cells, the activation of another type of the group III mGluR family, mGluR7, inhibits P/Q VGCCs by a PLC and PKC-dependent pathway (Perroy et al. 2000).
In conclusion, we present evidence for a new signalling pathway for PF mGluR4 receptors that involves PLC and/or PKC-dependent signalling cascades responsible for the reduction of evoked presynaptic Ca2+ transients. However, several important questions are pending. For example, what are the intermediate molecular players between mGluR4 and PLC/PKC (for example, Gq proteins) or between PLC/PKC and Ca2+ channels? Does mGluR4 activation also directly affect neurotransmitter exocytosis processes downstream of presynaptic Ca2+ influx, as has been shown in cultured cerebellar granule cells (Chavis et al. 1998)? A recent study has shown that mGluR4 can sequester Munc18-1, a binding partner of syntaxin-1, which is in turn a pivotal constituent of the SNARE complex crucial for exocytosis of synaptic vesicles (Nakajima et al. 2009). Activation of mGluR4 might then reduce vesicular liberation of glutamate through the effects of Munc18-1 sequestration on SNARE activity. It remains to be determined whether an mGluR4 effect on neurotransmitter liberation is direct and/or whether it requires the activation of a specific intracellular signalling pathway, and if so, which pathway is involved.
Acknowledgments
This work was supported by ‘Fondation pour la Recherche Médicale’ (INE20050303431), by ‘Association Nationale pour la Recherche’ (ANR-07-NEURO-047-01) and by ‘Ministère de l’Education Nationale, de la Recherche et de la Technologie’ (scholarships to K.A. and T.B.). The authors wish to thank Professors C. Berrier and F. Crepel, and Drs L. Fagni, M. Galante and A. Ghazi for discussion and comments on the manuscript, and Gérard Sadoc for providing the Acquis1 software employed in these studies.
Glossary
- AC
adenylyl cyclase
- DEA/NO
diethylamine NONOate sodium salt hydrate
- GIRK
G protein-gated inwardly rectifying K+ channels
- GPCR
G protein-coupled receptor
- K2P
two-pore domain K+ channel
- LTD
long-term depression
- LTP
long-term potentiation
- MAPK
mitogen-activated protein kinase
- mGluR
metabotropic glutamate receptor
- PC
Purkinje cell
- PF
parallel fibre
- PLC
phospholipase C
- PPF
paired-pulse facilitation
- PTX
pertussis toxin
- VGCC
voltage-gated calcium channel
Author contributions
Conception and design of the experiments: K.A. and H.D. Collection, analysis and interpretation of data: K.A., H.McL., T.B. and H.D. Drafting and revising the article for important intellectual content: K.A., H.McL. and H.D. All authors approved the final version.
Supplementary material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Movie 5
References
- Abitbol K, Acher F, Daniel H. Depression of excitatory transmission at PF-PC synapse by group III metabotropic glutamate receptors is provided exclusively by mGluR4 in the rodent cerebellar cortex. J Neurochem. 2008;105:2069–2079. doi: 10.1111/j.1471-4159.2008.05290.x. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27 489–27 494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Aller MI, Veale EL, Linden AM, Sandu C, Schwaninger M, Evans LJ, Korpi ER, Mathie A, Wisden W, Brickley SG. Modifying the subunit composition of TASK channels alters the modulation of a leak conductance in cerebellar granule neurons. J Neurosci. 2005;25:11455–11467. doi: 10.1523/JNEUROSCI.3153-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atluri P, Regehr WG. Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J Neurosci. 1996;16:5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss DA, Sirois JE, Talley EM. The TASK family: two-pore domain background K+ channels. Mol Interv. 2003;3:205–219. doi: 10.1124/mi.3.4.205. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Brown SP, Safo PK, Regehr WG. Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci. 2004;24:5623–5631. doi: 10.1523/JNEUROSCI.0918-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain SM, Meadows HJ, Dunlop J, Bushell TJ. mGluR4 potentiation of K2P2.1 is dependant on C-terminal dephosphorylation. Mol Cell Neurosci. 2008;37:32–39. doi: 10.1016/j.mcn.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Capogna M. Distinct properties of presynaptic group II and III metabotropic glutamate receptor-mediated inhibition of perforant pathway-CA1 EPSCs. Eur J Neurosci. 2004;19:2847–2858. doi: 10.1111/j.1460-9568.2004.03378.x. [DOI] [PubMed] [Google Scholar]
- Chavis P, Mollard P, Bockaert J, Manzoni O. Visualization of cyclic AMP-regulated presynaptic activity at cerebellar granule cells. Neuron. 1998;20:773–781. doi: 10.1016/s0896-6273(00)81015-6. [DOI] [PubMed] [Google Scholar]
- Chen C, Regher WG. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B. Molecular diversity of K+ channels. Ann N Y Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Ann Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Condé F, Collingridge L, Crepel F. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- Daniel H, Crepel F. Control of Ca2+ influx by cannabinoid and metabotropic glutamate receptors in rat cerebellar cortex requires K+ channels. J Physiol. 2001;537:793–800. doi: 10.1111/j.1469-7793.2001.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel H, Rancillac A, Crepel F. Mechanisms underlying cannabinoid inhibition of presynaptic Ca2+ influx at parallel fibre synapses of the rat cerebellum. J Physiol. 2004;557:159–174. doi: 10.1113/jphysiol.2004.063263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo S, Thomas Launey T. ERKs regulate PKC-dependent synaptic depression and declustering of glutamate receptors in cerebellar Purkinje cells. Neuropharmacology. 2003;45:863–872. doi: 10.1016/s0028-3908(03)00210-7. [DOI] [PubMed] [Google Scholar]
- Evans DI, Jones RS, Woodhall G. Differential actions of PKA and PKC in the regulation of glutamate release by group III mGluRs in the entorhinal cortex. J Neurophysiol. 2001;85:571–579. doi: 10.1152/jn.2001.85.2.571. [DOI] [PubMed] [Google Scholar]
- Flor PJ, Lukic S, Ruegg D, Leonhardt T, Knopfel T, Kuhn R. Molecular cloning, functional expression and pharmacological characterization of the human metabotropic glutamate receptor type 4. Neuropharmacology. 1995;34:149–155. doi: 10.1016/0028-3908(94)00149-m. [DOI] [PubMed] [Google Scholar]
- Gille A, Lushington GH, Mou TC, Doughty MB, Johnson RA, Seifert R. Differential inhibition of adenylyl cyclase isoforms and soluble guanylyl cyclase by purine and pyrimidine nucleotides. J Biol Chem. 2004;279:19955–19969. doi: 10.1074/jbc.M312560200. [DOI] [PubMed] [Google Scholar]
- Goldstein SN, Bockenhauer D, O’Kelly I, Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci. 2001;2:175–184. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- Goossens J, Daniel H, Rancillac A, van der Steen J, Oberdick J, Crépel F, De Zeeuw CI, Frens MA. Expression of protein kinase C inhibitor blocks cerebellar long-term depression without affecting Purkinje cell excitability in alert mice. J Neurosci. 2001;21:5813–5823. doi: 10.1523/JNEUROSCI.21-15-05813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Ikeda SR. Coupling of metabotropic glutamate receptor 8 to N-type Ca2+ channels in rat sympathetic neurons. Mol Pharmacol. 2005;67:1840–1851. doi: 10.1124/mol.105.010975. [DOI] [PubMed] [Google Scholar]
- Harris DN, Asaad MM, Phillips MB, Goldenberg HJ, Antonaccio MJ. Inhibition of adenylyl cylase in human blood platelets by 9-substituted adenine derivatives. J Cyclic Nucleotide Res. 1979;5:125–134. [PubMed] [Google Scholar]
- Honoré E. The neuronal background K2P channels: focus on TREK1. Nat Neurosci. 2007;8:251–261. doi: 10.1038/nrn2117. [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Bruno V, Salvatore L, Melchiorri D, Gradini R, Caricasole A, Barletta E, De Blasi A, Nicoletti F. Native group-III metabotropic glutamate receptors are coupled to the mitogen-activated protein kinase/phosphatidylinositol-3-kinase pathways. J Neurochem. 2002;82:216–223. doi: 10.1046/j.1471-4159.2002.00929.x. [DOI] [PubMed] [Google Scholar]
- Jackson C, Welch HC, Bellamy TC. Control of cerebellar long-term potentiation by P-Rex-family guanine-nucleotide exchange factors and phosphoinositide 3-kinase. PLoS ONE. 2010;5:e11962. doi: 10.1371/journal.pone.0011962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Lu Z. A novel high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry. 1998;37:13291–13299. doi: 10.1021/bi981178p. [DOI] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun. 1987;142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- Kennard LE, Chumbley JR, Ranatunga KM, Armstrong SJ, Veale EL, Mathie A. Inhibition of the human two-pore domain potassium channel, TREK-1, by fluoxetine and its metabolite norfluoxetine. Br J Pharmacol. 2005;144:821–829. doi: 10.1038/sj.bjp.0706068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde signalling by endocannabinoids. Curr Opin Neurobiol. 2002;12:324–330. doi: 10.1016/s0959-4388(02)00328-8. [DOI] [PubMed] [Google Scholar]
- Krieger P, Buschges A, el Manira A. Calcium channels involved in synaptic transmission from reticulospinal axons in lamprey. J Neurophysiol. 1999;81:1699–1705. doi: 10.1152/jn.1999.81.4.1699. [DOI] [PubMed] [Google Scholar]
- Kristensen P, Suzdak PD, Thomsen C. Expression pattern and pharmacology of the rat type IV metabotropic glutamate receptor. Neurosci Lett. 1993;155:159–162. doi: 10.1016/0304-3940(93)90697-j. [DOI] [PubMed] [Google Scholar]
- Lavialle-Defaix C, Gautier H, Defaix A, Lapied B, Grolleau F. Differential regulation of two distinct voltage-dependent sodium currents by group III metabotropic glutamate receptor activation in insect pacemaker neurons. J Neurophysiol. 2006;96:2437–2450. doi: 10.1152/jn.00588.2006. [DOI] [PubMed] [Google Scholar]
- Levenes C, Daniel H, Crepel F. Retrograde modulation of transmitter release by postsynaptic subtype 1 metabotropic glutamate receptors in the rat cerebellum. J Physiol. 2001;537:125–140. doi: 10.1111/j.1469-7793.2001.0125k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Marty A, Armstrong C, Konnerth A. Synaptic- and agonist-induced excitatory currents of Purkinje cells in rat cerebellar slices. J Physiol. 1991;434:183–213. doi: 10.1113/jphysiol.1991.sp018465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorez M, Humbel U, Pflimlin MC, Kew JN. Group III metabotropic glutamate receptors as autoreceptors in the cerebellar cortex. Br J Pharmacol. 2003;138:614–625. doi: 10.1038/sj.bjp.0705099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos JM, Elezgarai I, Benitez R, Osorio A, Bilbao A, Azkue JJ, Kuhn R, Knöpfel T, Grandes P. Clustering of the group III metabotropic glutamate receptor 4a at parallel fiber synaptic terminals in the rat cerebellar molecular layer. Neurosci Res. 1999;35:71–74. doi: 10.1016/s0168-0102(99)00066-8. [DOI] [PubMed] [Google Scholar]
- Mathie A. Neuronal two-pore-domain potassium channels and their regulation by G protein-coupled receptors. J Physiol. 2007;578:377–385. doi: 10.1113/jphysiol.2006.121582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan C, Lujan R, Shigemoto R, Sanchez-Prieto J. Subtype-specific expression of group III metabotropic glutamate receptors and Ca2+ channels in single nerve terminals. J Biol Chem. 2002;277:47796–47803. doi: 10.1074/jbc.M207531200. [DOI] [PubMed] [Google Scholar]
- Miniaci MC, Bonsi P, Tempia F, Strata P, Pisani A. Presynaptic modulation by group III metabotropic glutamate receptors (mGluRs) of the excitatory postsynaptic potential mediated by mGluR1 in rat cerebellar Purkinje cells. Neurosci Lett. 2001;310:61–65. doi: 10.1016/s0304-3940(01)02082-1. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Morreale A, Mallon B, Beale G, Watson J, Rumsby M. Ro31-8220 inhibits protein kinase C to block the phorbol ester-stimulated release of choline- and ethanolamin-metabolites from CG glioma cells: p70 S6 kinase and MAPKAP kinase-1β do not function downstream of PKC in activating PLD. FEBS Lett. 1997;417:38–42. doi: 10.1016/s0014-5793(97)01252-0. [DOI] [PubMed] [Google Scholar]
- Musset B, Meuth SG, Liu GX, Derst C, Wegner S, Pape HC, Budde T, Preisig-Müller R, Daut J. Effects of divalent cations and spermine on the K+ channel TASK-3 and on the outward current in thalamic neurons. J Physiol. 2006;572:639–657. doi: 10.1113/jphysiol.2006.106898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Mochida S, Okawa K, Nakanishi S. Ca2+-dependent release of Munc18-1 from presynaptic mGluRs in short-term facilitation. Proc Natl Acad Sci U S A. 2009;106:18385–18389. doi: 10.1073/pnas.0910088106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale SA, Garthwaite J, Batchelor AM. Metabotropic glutamate receptor subtypes modulating neurotransmission at parallel fibre-Purkinje cell synapses in rat cerebellum. Neuropharmacology. 2001;41:42–49. doi: 10.1016/s0028-3908(01)00046-6. [DOI] [PubMed] [Google Scholar]
- Neil KE, Kendall DA, Alexander SP. Coupling of metabotropic glutamate receptors to phosphoinositide mobilisation and inhibition of cyclic AMP generation in the guinea-pig cerebellum. Br J Pharmacol. 1996;118:311–316. doi: 10.1111/j.1476-5381.1996.tb15404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzeband JG, Parsons KL, Sweeney DD, Gruol DL. Metabotropic glutamate receptor agonists alter neuronal excitability and Ca2+ levels via the phospholipase C transduction pathway in cultured Purkinje neurons. J Neurophysiol. 1997;78:63–75. doi: 10.1152/jn.1997.78.1.63. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Luo Q, Ayala JE, Kim C, Conn PJ, Weaver CD. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol Pharmacol. 2008;73:1213–1224. doi: 10.1124/mol.107.041053. [DOI] [PubMed] [Google Scholar]
- Pekhletski R, Gerlai R, Overstreet LS, Huang XP, Agopyan N, Slater NT, Abramow-Newerly W, Roder JC, Hampson DR. Impaired cerebellar synaptic plasticity and motor performance in mice lacking the mGluR4 subtype of metabotropic glutamate receptor. J Neurosci. 1996;16:6364–6373. doi: 10.1523/JNEUROSCI.16-20-06364.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perroy J, Prezeau L, De Waard M, Shigemoto R, Bockaert J, Fagni L. Selective blockade of P/Q-type calcium channels by the metabotropic glutamate receptor type 7 involves a phospholipase C pathway in neurons. J Neurosci. 2000;20:7896–7904. doi: 10.1523/JNEUROSCI.20-21-07896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prézeau L, Carrette J, Helpap B, Curry K, Pin JP, Bockaert J. Pharmacological characterization of metabotropic glutamate receptors in several types of brain cells in primary cultures. Mol Pharmacol. 1994;45:570–577. [PubMed] [Google Scholar]
- Rusakov DA, Wuerz A, Kullmann DM. Heterogeneity and specificity of presynaptic Ca2+ current modulation by mGluRs at individual hippocampal synapses. Cereb Cortex. 2004;14:748–758. doi: 10.1093/cercor/bhh035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J Neurosci. 1997;17:3425–3435. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fibre synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- Saugstad JA, Segerson TP, Westbrook GL. Metabotropic glutamate receptors activate G-protein-coupled inwardly rectifying potassium channels in Xenopus oocytes. J Neurosci. 1996;16:5979–5985. doi: 10.1523/JNEUROSCI.16-19-05979.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon D, Vorobiov D, Dascal N. Positive and negative coupling of the metabotropic glutamate receptors to a G protein-activated K+ channel, GIRK, in Xenopus oocytes. J Gen Physiol. 1997;109:477–490. doi: 10.1085/jgp.109.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Forsythe ID, Tsujimoto T, Barnes-Davies M, Onodera K. Presynaptic calcium current modulation by a metabotropic glutamate receptor. Science. 1996;274:594–597. doi: 10.1126/science.274.5287.594. [DOI] [PubMed] [Google Scholar]
- Talley EM, Solorzqno G, Lei Q, Kim D, Bayliss DA. CNS distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci. 2001;21:7491–7505. doi: 10.1523/JNEUROSCI.21-19-07491.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y, Nomura A, Masu M, Shigemoto R, Mizuno N, Nakanishi S. Signal transduction, pharmacological properties, and expression patterns of two rat metabotropic glutamate receptors, mGluR3 and mGluR4. J Neurosci. 1993;13:1372–1378. doi: 10.1523/JNEUROSCI.13-04-01372.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen C, Kristensen P, Mulvihill E, Haldeman B, Suzdak PD. L-2-amino-4-phosphonobutyrate (L-AP4) is an agonist at the type IV metabotropic glutamate receptor which is negatively coupled to adenylyl cyclase. Eur J Pharmacol. 1992;227:361–362. doi: 10.1016/0922-4106(92)90018-q. [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- Watkins CS, Mathie A. A non-inactivating K+ current sensitive to muscarinic receptor activation in rat cultured cerebellar granule neurons. J Physiol. 1996;491:401–412. doi: 10.1113/jphysiol.1996.sp021224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhall GL, Ayman G, Jones RS. Differential control of two forms of glutamate release by group III metabotropic glutamate receptors at rat entorhinal synapses. Neuroscience. 2007;148:7–21. doi: 10.1016/j.neuroscience.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. Neuromodulation at single presynaptic boutons of cerebellar parallel fibres is determined by bouton size and basal action potential-evoked Ca transient amplitude. J Neurosci. 2009;29:15586–15594. doi: 10.1523/JNEUROSCI.3793-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.