

Background: Factor V is an inactive procofactor. Removal of its B-domain drives activation through an undefined mechanism.
Results: Basic and acidic regions within the B-domain impair factor Xa binding and cofactor function.
Conclusion: Autoinhibitory B-domain segments maintain the procofactor state and their disruption activates FV.
Significance: The study provides insights into how FV activation unfolds and defines an unexpected sequence-specific function of the B-domain.
Keywords: Coagulation Factors, Hemostasis, Molecular evolution, Mutant, Protein-Protein Interactions, Autoinhibition, Factor V, Factor Va, Procofactor Activation
Abstract
Activation of blood coagulation factor V (FV) is a key reaction of hemostasis. FV circulates in plasma as an inactive procofactor, and proteolytic removal of a large central B-domain converts it to an active cofactor (FVa) for factor Xa (FXa). Here we show that two short evolutionary conserved segments of the B-domain, together termed the procofactor regulatory region, serve an essential autoinhibitory function. This newly identified motif consists of a basic (963–1008) and an acidic (1493–1537) region and defines the minimal sequence requirements to maintain FV as a procofactor. Our data suggest that dismantling this autoinhibitory region via deletion or proteolysis is the driving force to unveil a high affinity binding site(s) for FXa. These findings document an unexpected sequence-specific role for the B-domain by negatively regulating FV function and preventing activity of the procofactor. These new mechanistic insights point to new ways in which the FV procofactor to cofactor transition could be modulated to alter hemostasis.
Introduction
Complex reaction pathways, including blood clotting, often rely on regulated protein activation to elicit a biologic response. Coagulation factors generally circulate in blood as inactive precursors and transition to an active form after limited proteolysis subsequent to vascular damage. For serine protease zymogens involved in clotting, this mechanism is well described (1). Although precise cleavage sites are defined, the mechanism underpinning the conversion of FV to its active form (FVa)3 has remained elusive. FV plays a pivotal role in blood coagulation. It is a large, heavily glycosylated, inactive procofactor protein that is homologous to factor VIII (FVIII) and shares the same domain organization (A1-A2-B-A3-C1-C2) (2, 3). FV cannot participate to any significant degree in the macromolecular enzyme complex prothrombinase. It is converted to the active cofactor state after limited proteolysis within the B-domain. FVa is a nonenzymatic cofactor within the prothrombinase complex that greatly accelerates the ability of factor Xa (FXa) to rapidly convert prothrombin to thrombin (4). FV activation has been studied for more than three decades, yet how bond cleavage within the B-domain facilitates the conversion from an inactive to active state is only starting to become known.
The FV B-domain spans amino acids 710–1545 and is not necessary for procoagulant activity (Fig. 1) (5–8). Thrombin, utilizing both exosites I and II, is considered a key physiologic activator and cleaves three peptide bonds (Arg709, Arg1018, and Arg1545) to generate FVa (5–7, 9–14). Prior studies of FV activation were largely based on correlating bond cleavage with the development of procoagulant activity (for review, see Ref. 3). Broadly, these studies found that variable amounts of cofactor activity are observed depending on which region of the B-domain is cleaved and which assay is used to evaluate activity. A clear consensus is that maximal activity correlates with cleavage at Arg1545 (7, 13, 15–22). Although these studies have advanced the field, such correlative studies have not provided mechanistic insight into how FV activation unfolds.
FIGURE 1.

Recombinant FV derivatives. Top, the human FV B-domain (pale yellow) is defined by residues 710–1545, which are removed after thrombin-mediated proteolysis; cleavage sites are indicated below the B-domain. The blue box represents the BR (963–1008), and the red box represents the AR (1493–1537). Their sequences are provided. Recombinant FV derivative with variable B-domains used in the study are shown schematically (FV-810, FV-B199, FV-B152, and FV-B104). Because FV-B226, FV-B208, and FV-B199 are very similar, only FV-B199 is shown. B-domain residues deleted are provided on the right. The total lengths of the B-domains are as follows: FV-810, 155 residues; FV-B199, 199 residues; FV-B152, 152 residues; FV-B104, 104 residues. The full-length B-domain (710–1545) has 25 potential N-linked carbohydrate sites with a distribution as follows: 709–810, 6 sites; 811–962, 2 sites; 963–1008 (BR), 0 sites; 1009–1491, 16 sites; 1492–1545 (mostly AR), 1 site.
The human FV B-domain (836 residues) is generally not well conserved among vertebrates, varies in size across species, and has no homology to any other known protein, including the FVIII B-domain (3, 9, 23). A longstanding assumption is that the B-domain, due to its size/high carbohydrate content, nonspecifically prevents activity of the procofactor. Initial evidence for this came from the Kane laboratory (17, 24), who generated a FV derivative with a shortened B-domain (FVdes811–1491 or FV-810). Using a transient expression system, they found that this FV derivative had partial but seemingly constitutive activity (17). Our laboratory, using purified FV-810 and a thrombin cleavage site variant, found that FV-810 functions in an identical way to FVa (25). These data imply that proteolysis within the B-domain, although necessary, is incidental to the mechanism by which cofactor function is realized. Instead, activation of FV must eliminate steric and/or conformational constraints contributed by the B-domain. Surprisingly, using a panel of B-domain-truncated variants, we found that specific B-domain sequences appear to play a critical role in stabilizing FV in an inactive procofactor state (26). One of these regions (963–1008) is unusually basic and is one of just a few sections of the B-domain that is conserved across the vertebrate lineage (3). Disruption of these sequences (termed here the basic region (BR)) by mutagenesis or deletion yielded derivatives with cofactor-like properties in the absence of proteolysis (26).
Here we expand on these findings and show that although the BR is necessary, it is not sufficient to suppress cofactor function. Rather, it appears to work in concert with sequences at the C-terminal end of the B-domain (1493–1537, Fig. 1). This region, termed here the acidic region (AR), is similar to the a3 region of FVIII and, like the BR, is conserved across species. Overall the data support the novel idea that these discrete sequences (BR+AR), together defined as the procofactor regulatory region (PRR), are the minimal, autoinhibitory elements required to maintain FV as an inactive procofactor.
EXPERIMENTAL PROCEDURES
Materials
The peptidyl substrate H-d-phenylalanyl-l-pipecolyl-l-arginyl-p-nitroanalide (S2238) was from Diapharma (West Chester, OH). Benzamidine, 4-amidinophenylmethanesulfonyl fluoride hydrochloride (APMSF), and bovine serum albumin (BSA) were from Sigma. Dansylarginine-N-(3-ethyl-1,5-pentanediyl)amide (DAPA) was from Hematologic Technologies (Essex Junction, VT), and Oregon Green488 maleimide (OG488) was from Invitrogen. All tissue culture reagents were from Invitrogen, except insulin-transferrin-sodium selenite Roche Applied Science). Small unilamellar phospholipid vesicles (PCPS) composed of 75% (w/w) hen egg l-α-phosphatidylcholine and 25% (w/w) porcine brain l-α-phosphatidylserine (Avanti Polar Lipids, Alabaster, AL) were prepared and characterized as described (27).
Proteins
Human prothrombin, thrombin, FX, OG488-FXa, and FV were isolated from plasma and/or prepared as described previously (28–31). Recombinant FX (rFX), rFXa, rFVa, plasma-derived FVa, and a partial B-domainless form of human FV (FV-810) were prepared, purified, and characterized as described (25, 32). Molecular weights and extinction coefficients (E0.1%, 280 nm) of the various proteins have been reported previously (26). The molecular weights and extinction coefficients for newly derived rFV variants were: FV-B226, 226,000 and 1.47; FV-B208, 223,000 and 1.43; FV-B199, 222,000 and 1.49; FV-B152, 215,000 and 1.51; FV-B104, 210,000 and 1.57; FV-B8-BR1, 247,000 and 1.51; FV-B8-BR2, 247,000 and 1.51; FV-B8-BR3, 247,000 and 1.51. All functional assays were performed at 25 °C in 20 mm Hepes, 0.15 m NaCl, 5 mm CaCl2, 0.1% polyethylene glycol 8000, pH 7.5 (assay buffer).
Construction of rFV Variants
Construction of FV-810 and FV-1033 has previously been described (25, 26). Specific oligonucleotides used to generate FV-B226 were as follows: forward primer A, 5′-GAAGAGGTGGGAATACTTC-3′, encoding for amino acid residues 320–325; reverse primer B, 5′-GTGGCCACTCTGCTTTCCAGGATCCTCTATAGGGTCTTCAG-3′, in which the first 21 bases correspond to full-length FV cDNA, encoding for residues 964–970 and the last 19 bases to residues 805–810; forward primer C, 5′-CTGAAGACCCTATAGAGGATCCTGGAAAGCAGAGTGGCCAC-3′, in which the first 19 bases correspond to residues 805–810 and the last 21 bases correspond to full-length FV cDNA residues 964–970; reverse primer D, 5′-TCTGTCCATGATAAGAAATGG-3′, corresponding to the full-length FV cDNA sequence encoding for residues 1871–1877. The resulting DNA fragment was TOPO-cloned (Invitrogen), digested with Bsu36I and SnaBI, gel-purified, and subcloned into pED-FV digested with the same enzymes (25). To ensure the absence of polymerase-induced errors, the entire modified cDNA was sequenced. The remaining FV constructs outlined in the manuscript were prepared in the same type of way using appropriately modified primers. The FV cDNA sequence of Gallus gallus (chicken) was derived from the Ensembl genome data base (Version 35). The base pairs encoding the chicken B-domain were synthesized (GenScript) and flanked by HpaI and SnaBI restriction sites to facilitate cloning.
Expression and Purification of rFV
Transfection of the plasmids encoding FV constructs into baby hamster kidney cells, selection of stable clones, and expression/purification of rFV derivatives were performed as described (25, 33). Protein purity was assessed by SDS-PAGE using precast 4–12% gradient gels (Invitrogen) under reducing conditions (50 mm dithiothreitol) using the MOPS buffer system followed by staining with Coomassie Brilliant Blue R-250.
FV-specific Prothrombin Time (PT)-based Clotting Assay
FV (600 nm) derivatives were prepared in assay buffer. For experiments in which pretreatment with thrombin was intended, FV derivatives were incubated at 37 °C for 15 min with 10–60 nm thrombin. All thrombin-dependent activation reactions were quenched by the addition of a 1.2-fold molar excess of hirudin. Samples were diluted to less than 3 nm in assay buffer with 0.1% BSA, and the specific clotting activity using FV-deficient plasma (George King Biomedical, Overland Park, KS) and TriniClot PT Excel (Tcoag, Wicklow, Ireland) was assayed as described (21). The data are presented as the mean values ± S.D.
Prothrombin Activation
Steady state initial velocities of prothrombin cleavage were determined discontinuously at 25 °C as described (32). Progress curves of prothrombin activation were performed using the following reaction conditions. PCPS (50 μm), DAPA (3 μm), and prothrombin (1.4 μm) were incubated with the various rFV(a) derivatives (0.1 nm) in assay buffer, and the reaction was initiated with FXa (1.0 nm). At various time points, aliquots of the reaction mixture were quenched, and thrombin generation was determined using the chromogenic substrate S2238 as described (32).
Fluorescence Measurements
Steady state fluorescence intensity and/or anisotropy was measured at 25 °C in a PTI QuantaMaster fluorescence spectrophotometer (Photon Technology International) using λex = 480 nm and λem = 520 nm with long pass filters (KV500, CVI Melles Griot) in the emission beam. Reaction mixtures (2.5 ml) in a 1 × 1-cm2 quartz cuvette contained 20 or 30 nm OG488-FXa and 50 μm PCPS in assay buffer to which increasing concentrations of FV/V(a) derivative (0.5–100 nm) were added. Fluorescence anisotropy or intensity measurements, including controls, were performed as described (29, 34).
Data Analysis
Data were analyzed according to the indicated equations by nonlinear least squares regression analysis using the Marquardt Algorithm (35), and the quality of each fit was assessed by the criteria described (36). Dissociation constants (Kd), stoichiometries (n), and the maximum increase in anisotropy at saturating rFV were obtained for the membrane-dependent interaction between FXa and rFV from the dependence of the anisotropy with increasing concentrations of rFV(a), which was corrected for the overall change in fluorescence intensity (37, 38).
RESULTS
Role of the BR in Preserving the Procofactor State
We have previously shown that removal of FV-B-domain residues 902–1033 and more specifically 963–1008, termed here the basic region or BR, drives the expression of FVa procoagulant function (26). The BR appears to stabilize the inactive FV procofactor state through a mechanism that is not well understood. To examine if the BR is sufficient to keep FV inactive, we produced a new panel of recombinant FV variants. Using a constitutively active FV derivative, FV-810 (residues 811–1491 deleted), as a scaffold (25), different extensions of the BR were inserted after residue 811, and the functional properties of the new variants (FV-B226, FV-B208, and FV-B199) were evaluated. Because these new FV variants are very similar, only FV-B199 is shown in Fig. 1. These variants have residues 964–1033 (FV-B226), 964–1015 (FV-B208), and 964–1007 (FV-B199) added back to FV-810. Each variant migrated on SDS-PAGE with the expected mobility either before or after treatment with thrombin (Fig. 2).
FIGURE 2.
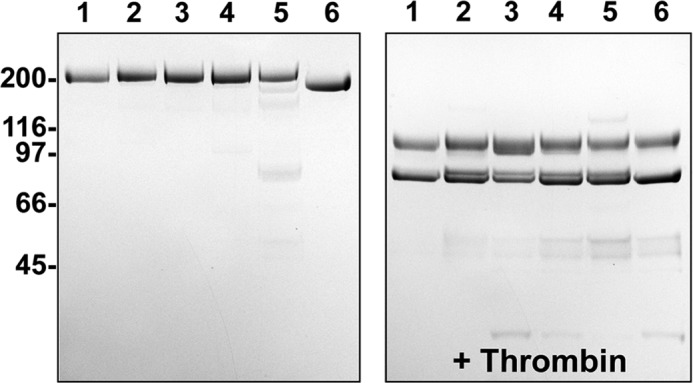
SDS-PAGE analysis. Purified proteins (5 μg/lane) before (left) and after treatment with thrombin (right) were subjected to SDS-PAGE under reducing conditions and visualized by staining with Coomassie Brilliant Blue R-250. Lane 1, FV-810; lane 2, FV-B226; lane 3, FV-B208; lane 4, FV-B199; lane 5, FV-B152; lane 6, FV-B104. The apparent molecular weights of the standards are indicated.
Consistent with previous studies (25, 26), FV-810-like FVa exhibits high specific activity before or after treatment with thrombin in a one-stage PT-based clotting assay using FV-deficient plasma (Fig. 3A and Table 1). In contrast, FV-B226, FV-B208, or FV-B199 with variable BRs inserted reduced the specific activity to a level comparable with PD-FV. Treatment of these procofactor-like derivatives or PD-FV with thrombin resulted in a substantial increase in specific activity, with levels comparable with FV-810 (Fig. 3A and Table 1). Additionally, progress curves of prothrombin conversion to thrombin using FXa-FV(a) yielded results consistent with the clotting assay. Unlike FV-810, the procofactor-like derivatives and PD-FV had very little activity and displayed progress curves of thrombin generation characterized by a substantial lag (Fig. 3B). Conversion of the FV variants to FVa by thrombin resulted in maximal cofactor activity and yielded rates of prothrombin activation comparable with FV-810 and FVa (Fig. 3C).
FIGURE 3.

Characterization of rFV+BR derivatives. Panel A, specific clotting activities of recombinant FV/V(a) derivatives before (white bars) or after treatment with thrombin (black bars) were determined by a FV-specific PT-based clotting assay as described under “Experimental Procedures.” The data are the mean ± S.D. of at least three similar experiments. Panels B and C. Reaction mixtures containing 50 μm PCPS, 3.0 μm DAPA, 1.4 μm prothrombin, and 0.1 nm FV/V(a) derivatives before (panel B) or after treatment with thrombin (panel C) were incubated for 5 min at 25 °C. The reaction was initiated with 1.0 nm FXa, and thrombin generation was monitored as described under “Experimental Procedures.” The symbols represent the following: ▴, FV-810; ◊, FV-B226; ×, FV-B208; ○, FV-B199; ●, FV-B152; △, FV-B104; □, PD-FV; ■, rFVa. The lines represent a linear fit of the data, except for FV-B226, FV-B208, FV-B199, FV-B104, and PD-FV in panel B, which result from a polynomial fit. The data are the means ± S.D. of 2–4 similar experiments.
TABLE 1.
Characterization of B-domain FV derivatives
ND, not determined; NA, not able to determine an accurate value.
| Cofactor species | Specific activitya | Specific activityb (+thrombin) | Kdc |
|---|---|---|---|
| Units/nmol | Units/nmol | nm | |
| FV-810 | 265 ± 49 | 308 ± 41 | 1.1 ± 0.3 |
| rFVa | 264 ± 30 | ND | 1.8 ± 0.2 |
| PD-FV | 27 ± 2 | 214 ± 9 | NA |
| FV-B226 | 29 ± 2 | 382 ± 29 | NA |
| FV-B208 | 4 ± 1 | 271 ± 20 | NA |
| FV-B199 | 9 ± 1 | 339 ± 74 | NA |
| FV-B152 | 223 ± 11 | 286 ± 43 | 2.6 ± 0.4 |
| FV-B104 | 13 ± 2 | 311 ± 33 | NA |
| FV-B-Ptex | 212 ± 49 | 308 ± 41 | 1.5 ± 0.2 |
| FV-B-Fish | 223 ± 39 | 245 ± 43 | 1.8 ± 0.8 |
| FV-B-Ch | 192 ± 31 | 327 ± 62 | 1.9 ± 0.3 |
| FV-B-Ch+hBR | 60 ± 13 | 273 ± 49 | NA |
| FV-B8-BR1 | 260 ± 16 | 293 ± 18 | ND |
| FV-B8-BR2 | 152 ± 15 | 268 ± 39 | ND |
| FV-B8-BR3 | 76 ± 13 | 232 ± 11 | ND |
a The specific clotting activity was determined as described under “Experimental Procedures.” The mean values ± S.D. of at least three determinations are presented.
b FV variants were incubated with thrombin before analysis.
c Data for binding studies can be found in Fig. 4 and supplemental Figs. S1 and S3.
Although activity measurements are informative, they can be misleading, as feedback reactions during the assay could compromise data interpretation. To address this, we evaluated the ability of each of the FV variants to bind membrane-bound FXa using fluorescence measurements. Consistent with previous observations (25, 26), FVa and FV-810 bound with high affinity to active site-blocked, membrane-bound FXa (Fig. 4, A and B, Table 1). In contrast, the interaction of FV-B226, FV-B208, FV-B199, or PD-FV with membrane bound-FXa was dramatically reduced compared with FV-810 as no saturable binding was observed (Fig. 4A, Table 1). Binding parameters for these proteins could not accurately be assessed; however, a lower limit estimate of the affinities indicates that they are at least >50-fold reduced compared with FV-810. Treatment of FV-B226, FV-B208, or FV-B199 with thrombin followed by assessment of direct binding yielded dissociation constants similar to thrombin-treated FV-810 and FVa (Fig. 4B), indicating that once activated each of the variants can assemble within prothrombinase. The binding data and functional measurements demonstrate that the BR is a necessary segment of the B-domain and plays a crucial role in keeping FV in an inactive, procofactor-like state. It appears to do so by obscuring FXa binding either directly or allosterically despite >75% of the B-domain being absent. Remarkably, the insertion of as little as 44 BR residues (e.g. FV-B199) had a major influence on FXa binding, shifting the functional status from cofactor-like to procofactor-like.
FIGURE 4.

Direct binding measurements. Reaction mixtures containing 20 nm OG488-FXa and 50 μm PCPS were titrated with increasing concentrations of FV-810 (▴), FV-B226 (◊), FV-B208 (×), FV-B199 (○), PD-FV (□), and rFVa (▾; only shown in panel B) at 25 °C without prior treatment (panel A) or with proteins treated with thrombin before the experiment (panel B). The change in fluorescence anisotropy (Δr) was measured and analyzed as described in “Experimental Procedures.” The lines are drawn after analysis to independent, non-interacting sites. The data are representative of 2–4 similar experiments. The dissociation constants (Kd) for FV-810 in panel A was 1.1 ± 0.3 nm. We were not able to accurately determine Kd values for FV-B226, FV-B208, FV-B199, and PD-FV; they are estimated to be >50 nm. In panel B, the dissociation constants for the variants after thrombin treatment are as follows: FV-810, 1.3 ± 0.3 nm; FV-B226, 1.8 ± 0.5 nm; FV-B208, 2.1 ± 0.7 nm; FV-B199, 1.3 ± 0.3 nm; plasma-derived FVa, 1.5 ± 0.3 nm; rFVa, 1.8 ± 0.2 nm.
Contribution of Additional B-domain Sequences to Maintaining FV as a Procofactor
The findings suggest that a small B-domain harboring the BR (e.g. FV-B199) contains the necessary structural features to maintain the FV procofactor state. However, residual B-domain sequences apart from the BR could also play an important stabilizing role. Using FV-B199 as a scaffold, we removed sequences on either the C-terminal side (residues 1492–1538; FV-B152) or N-terminal side (residues 716–810; FV-B104) of the B-domain (see Fig. 1). Residues flanking Arg709 and Arg1545 were left intact to retain efficient processing by thrombin. FV-B152 or FV-B104 migrated on SDS-PAGE as expected and were cleaved by thrombin to yield FVa (Fig. 2).
Elimination of ∼100 residues (716–810) from FV-B199 on the N-terminal side of the B-domain (FV-B104; see Fig. 1) was without functional consequence as FV-B104 remained a procofactor as assessed by clotting, purified component assays, and direct binding measurements to FXa (Fig. 3, A and B, supplemental Fig. S1 and Table 1). After treatment with thrombin, FV-B104 exhibited functional properties similar to FV-810 (Fig. 3, A and C). In contrast, removing sequences C-terminal to the BR (residues 1492–1538; FV-B152, see Fig. 1) converted an otherwise inactive FV derivative (e.g. FV-B199) to an active cofactor even in the presence of the BR. The overall activity of FV-B152 was similar to FV-810, and it bound FXa with high affinity (Fig. 3, A–C, supplemental Fig. S1 and Table 1). These findings indicate that in addition to the BR, C-terminal B-domain sequences 1493–1537 also contribute to stabilizing FV in an inactive procofactor state. Inspection of this region of the B-domain reveals that 1493–1537 is highly acidic (termed herein the acidic region), is generally conserved across the vertebrate lineage (see supplemental Fig. S2), and is similar to the a3 region of FVIII at the beginning of its light chain. Overall, these data suggest that the BR works in concert with the AR to suppress cofactor function. Thus the BR and AR, consisting of ∼100 residues and together defined here as the procofactor regulatory region, are the minimal, autoinhibitory elements required to maintain FV as a procofactor. Elimination of either the BR (e.g. FV-810) or AR (e.g. FV-B152) removes critical structural motifs that otherwise keep FV functionally inert. Furthermore, the juxtaposition in the primary sequence of the BR and AR in FV-B199 and FV-B104 (see Fig. 1) suggest that these segments act as independent motifs and do not appear to be influenced in a major way by the rest of the B-domain.
Specificity of the BR and Fine Mapping
Considering the functional importance of the PRR, it is not surprising that most, if not all, of available FV sequences across the vertebrate lineage have a conserved BR and AR, albeit with weaker conservation in lower vertebrates (see supplemental Fig. S2). An exception to this is FV derived from the venom of certain Australian snakes (39–41). Venom FV from Pseudonaja textilis (ptex-FV) has a B-domain of just 46 residues that lacks the BR but contains a region similar to AR. Consistent with the importance of an intact PRR in keeping FV inactive, we have shown that ptex-FV is constitutively active and does not require processing/removal of its B-domain (42, 43).
To test specificity and functionality, we examined whether B-domains from select vertebrates, which have divergent PRRs, preserve the human FV procofactor state. As expected, exchanging only the human FV B-domain (836 residues) with that of ptex-FV (46 residues, no BR; FV-B-Ptex) yielded a FV derivative that was constitutively active, as determined by clotting assay, and it exhibited high affinity binding to FXa (Table 1 and supplemental Fig. S3). Surprisingly, however, B-domains from lower vertebrates, which harbor a PRR, did not suppress cofactor activity. For example, a human FV derivative harboring only the chicken B-domain (FV-B-Ch) or pufferfish B-domain (FV-B-Fish) displayed a high specific clotting activity and high affinity FXa binding in the absence of thrombin activation (Table 1 and supplemental Figs. S3 and S4). These data imply that the mechanism for maintaining the procofactor state of FV in lower vertebrates is fundamentally different, or alternatively, a high level of specificity mediates the autoinhibitory function of the PRR. Lending some support to the latter, when we placed only 44 residues of the human BR within the chicken B-domain (FV-B-Ch+hBR), the procofactor state was reestablished (supplemental Figs. S3 and S4). This procofactor-like species could be fully processed and activated by thrombin to FVa (supplemental Fig. S4). These data suggest that either other factors may serve to inhibit the function of FV in these lower vertebrates or that the mode of inhibition of the PRR is highly specific and a major contributor to this specificity is the BR.
We next mapped the human BR further to define the extent to which specific portions stabilize FV. Using a previously described procofactor-like variant (FV-1033, Fig. 5A) as a scaffold (26), short segments of the BR were retained, and the remaining sequence was replaced with unrelated FVIII B-domain sequence. Three variants were expressed and purified (FV-B8-BR1, FV-B8-BR2, and FV-B8-BR3; Fig. 5B), and they are schematically shown in Fig. 5A with the original FV BR residues highlighted in blue and FVIII B-domain residues shown in green. This approach allowed us to focus on the individual role of stretches of 10–12 BR residues.
FIGURE 5.
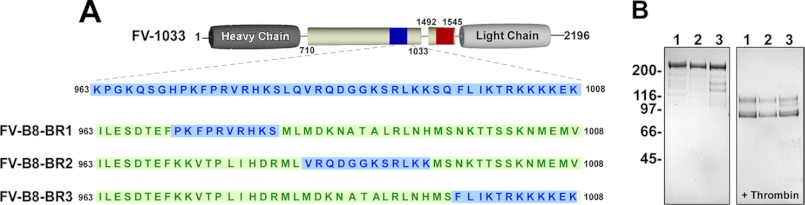
Schematic representation and SDS-PAGE analysis of FV-B8-BR variants. Panel A, FV-1033 is a procofactor-like FV derivative that harbors both a BR (blue box; sequence shown) and AR (red box) and has B-domain residues 1034–1491 deleted (26). With FV-1033 as a scaffold, portions of the BR were retained, and the remaining sequence was replaced with non-homologous segments of FVIII B-domain (green). The BR region retained for the different variants is as follows: 971–980 (FV-B8-BR1), 983–994 (FV-B8-BR2), and 997–1008 (FV-B8-BR3). Panel B, purified proteins (5 μg/lane) before (left) and after treatment with thrombin (right) were subjected to SDS-PAGE under reducing conditions and visualized by staining with Coomassie Brilliant Blue R-250. Lane 1, FV-B8-BR1; lane 2, FV-B8-BR2; lane 3, FV-B8-BR3. The apparent molecular weights of the standards are indicated.
Previous analysis of a FV variant in which the entire basic sequence was exchanged for FVIII B-domain residues (FV-B8–46) demonstrated that this derivative has cofactor-like properties similar to FV-810 (26). Assessment of the specific clotting activities of the newly generated FV-B8-BR variants revealed a gradual decline in cofactor activity depending on which BR sequence was preserved. Maintaining residues 971–980 (BR1), while exchanging the rest of the BR for FVIII B-domain sequence resulted in a cofactor-like FV variant (FV-B8-BR1; Fig. 6A). However, preserving the second (BR2; residues 983–994) or third (BR3; residues 997–1008) subset of the BR reduced the specific activity, thereby generating an intermediate (FV-B8-BR2) or more procofactor-like FV variant (FV-B8-BR3). These findings were confirmed in a purified system where we monitored the conversion of prothrombin to thrombin (Fig. 6B). Upon treatment with thrombin, the FV variants displayed a high specific activity by clotting assay (Fig. 6A). Furthermore, in the purified component assay, FV-B8-BR3 displayed a high level of activity, whereas there was only a very modest change in activity of FV-B8-BR2 after thrombin treatment (Fig. 6C). Based on these observations, we conclude that the C-terminal portion (residues 997–1008) of the BR has the most influence in suppressing cofactor function, thereby maintaining the procofactor state.
FIGURE 6.

Characterization of FV-B8-BR variants. Panel A, specific clotting activities of FV/FV(a) derivatives before (white bars) or after treatment with thrombin (black bars) were determined by a FV-specific PT-based clotting assay as described under “Experimental Procedures.” The data are the means ± S.D. of at least three experiments. FV-810 and PD-FV are for reference and are from Fig. 3A. Panels B and C, reaction mixtures containing 50 μm PCPS, 3.0 μm DAPA, 1.4 μm prothrombin, and 0.1 nm FV derivatives were incubated for 5 min at 25 °C. The reaction was initiated with 1.0 nm FXa, and thrombin generation was monitored as described under “Experimental procedures.” The symbols represent the following: FV-810 (▴), FV-B8-BR1 (▾), FV-B8-BR2 (*), FVB8-BR3 (⎔), PD-FV (□). The lines represent a linear fit of the data, except for FV-B8-BR3 and PD-FV in panel B, which results from a polynomial fit. The data are the means ± S.D. of 2–4 similar experiments.
DISCUSSION
It has been recognized for more than 30 years that the large central B-domain of FV is responsible for the lack of activity in the precursor (3). Due to its size/high carbohydrate content, it has been generally assumed that the B-domain nonspecifically prevents activity of the procofactor. Here we show that the B-domain harbors an autoinhibitory motif that is essential for the regulation of FV function. Termed the procofactor regulatory region, the motif consists of two evolutionary conserved segments of the B-domain, one which is highly basic (BR) and one that has a concentration of ARs. These regions work together to obscure a high affinity binding site for FXa. The unraveling of the autoinhibitory PRR via B-domain proteolysis is the central driving force converting the inactive procofactor (FV) to the active cofactor state (FVa).
Keeping FV as an inactive procofactor undoubtedly plays a critical regulatory role rooted in the tight regulation of thrombin generation. Having a potent regulatory motif such as the PRR that provides “on-site” repression of cofactor activity is thus of central importance for maintaining normal hemostasis. Due to its significance, it is not surprising that the ∼100 amino acids that make up the PRR are generally conserved across vertebrate evolution (supplemental Fig. S2) even though there is weak homology among B-domains from mammals (<50% sequence identity) and lower vertebrates (<20%) (3, 44). Surprisingly, even the length of the B-domain varies across the vertebrate lineage, from 836 residues in humans to 471 in pufferfish. However, common threads of these B-domains include high carbohydrate content, large size, and the presence of the PRR. Results of the current study suggest that the large size of the B-domain likely has little, if any, impact in keeping FV inactive. For example, we found that >85% of the B-domain is dispensable with respect to the inhibition of FV cofactor activity. Functional measurements revealed that FV-B104, consisting of just 104 amino acids and essentially only harbors the PRR, is a procofactor. Although further work would need to be done, it is possible that other factors apart from the PRR especially in lower vertebrates may serve to inhibit the function of FV.
Within the PRR, both the BR and AR play critical roles in keeping FV inactive as elimination of either shifts the functional status to the cofactor-like state. The BR and AR are separated in the primary sequence of human FV by ∼480 amino acids. Interestingly, approximately half of this region has 31 copies of 9-amino acid tandem repeats (9, 45, 46). However, lower vertebrates such as lizard, chicken, and pufferfish do not have any repeat segments, and the length separating the BR and AR is very short at <55 residues in these species. In this study we essentially recapitulated this type of orientation with human FV as juxtaposition of the BR and AR (e.g. FV-B199 and FV-B104, see Fig. 1) had no effect on PRR function. This suggests that the BR and AR are likely specialized motifs that function together independent of the vast majority of the B-domain. Interestingly, the region separating the BR and AR has increased in size during the course of evolution; however, this part of the B-domain has no known function. This region in mammals is heavily glycosylated and could play a role in intracellular trafficking or influence the circulating half-life of FV (47, 48).
We speculate that the PRR functions as an autoinhibitory motif within the large B-domain and is a modular effector of cofactor function. Throughout biology, a prevalent mechanism employed for negatively regulating otherwise constitutively active proteins is the incorporation of cis-acting inhibitory sequences (49, 50). A hallmark of this autoinhibition mechanism is the identification of the protein fragment(s) that inhibits or represses the mature, active protein. For FV, our data support the idea that the PRR solely serves this function (see Fig. 7). The autoinhibitory effect of the PRR could be mediated by direct masking of a functional region or could occur by an indirect/allosteric mechanism. In either scenario, the underpinnings of this inhibition likely derive from altering the interaction with FXa. Notably, FXa binding is significantly inhibited or essentially undetectable in all FV derivatives that harbor an intact PRR. The FXa binding site is thought to reside on the A2 domain with contributions from the A3. Largely driven by mutagenesis studies, it appears that several key residues in the A2 contribute in a major way to the binding energy (51). This is consistent with reports suggesting that a region surrounding the activated protein C cleavage site at Arg506 is involved in FXa binding (52–54). In principle, relief of the autoinhibitory interaction could occur by several mechanisms; however for FV, proteolysis within the B-domain is the driving force. Cleavage at Arg709, Arg1018, and Arg1545 ultimately dismantles the PRR consequently exposing the FXa binding site (Fig. 7). At present, it is not clear which of the individual cleavage sites has the most impact on destabilizing the PRR. However, cleavage at Arg1545, which is adjacent to the AR, must have a significant impact. Support for this comes from the longstanding observation in the field that cleavage of FV by a protease in Russell's viper venom at Arg1545 results in full activation (7, 13, 15–20).
FIGURE 7.

Schematic of FV activation and functional landmarks. After thrombin processing to FVa, the B-domain (black lines) was released as two large fragments thereby exposing a FXa binding site. For FV, the BR (dark blue) and AR (dark red) are shown as cylinders; the putative FXa binding site is shown as a white sphere; green circles represent the end of the A2 and beginning of A3 domains.
Due to the nature of how the PRR inhibits cofactor function, it is likely that there are multiple ways in which FV could be activated. As long as the PRR is sufficiently disrupted or disengaged from the core heavy/light region, the protein should bind FXa and function in prothrombinase. Clear examples of this from our work are FV-810 (lacks AR) and FV-B152 (lacks BR), which have altered PRRs and are constitutively active. Furthermore, numerous proteases have been identified that process FV to varying functional states in a manner different from thrombin (for review, see Ref. 3). The use of sequence-specific B-domain regions to suppress cofactor activity is in stark contrast to the mechanism underlying FVIII activation. For example, B-domainless FVIII remains a procofactor, and processing between the A1 and A2 domain at Arg372 is absolutely required for the development of FVIIIa activity (3, 23). Furthermore, FVIII association with von Willebrand factor also contributes to maintaining the procofactor state as it obscures functional membrane binding sites (55). The clear differences in the mechanism by which FVIII and FV are kept inactive are also evident from naturally occurring mutations. For FVIII, missense mutations have been identified (e.g. Arg372 and Arg1689) that alter the rate and/or extent of FVIII activation and cause hemophilia A. Although FV deficiency is rarer, there are no known missense mutations for FV that alter its activation (56). We speculate that because there are multiple routes to disrupt the PRR, a single amino acid change would unlikely be so unfavorable as to substantially impact FV activation. The observation that FV and FVIII employ differential mechanisms to maintain the procofactor state is surprising considering the clear evolutionary link between the two proteins (57, 58). However, it is possible that there were different selective pressures on FV and FVIII that necessitated a diverse and/or stringent mechanism of control of cofactor activity development. The observation that FVIII must be cleaved at very specific sites and dissociate from von Willebrand factor (unlike FV) to express activity suggests that this could be the case (59).
The ability of the PRR to regulate the FV procofactor to cofactor transition through an autoinhibitory mechanism provides an unexpected twist to the problem of FV activation. Structural information would provide much needed insight into how the PRR maintains FV as an inactive procofactor. However, apart from electron microscope images of FV that show the B-domain as an appendage stemming from a globular core, presumed to be the heavy/light chain (60–65), there is no structural information. Although the structure of inactivated bovine FVa lacking the A2 domain is available, little can be gleaned in terms of FV activation (66). Due to its large size and high carbohydrate content, detailed structural information of the FV B-domain may not be possible. However, information obtained in this study identifying the PRR as the core functional unit of the B-domain possibly makes structural studies of this region within grasp. High resolution structural information may reveal whether the PRR is keeping FV inactive directly or allosterically. This information may also shed light on where FXa engages the heavy/light chain region, as the PRR appears to obscure its binding. Furthermore, this information could also be used to develop therapeutic probes that target the autoinhibitory PRR or even mimic the PRR to suppress cofactor function. The identification of the sequence-specific function of the PRR makes the development of these types of stabilizing or destabilizing probes to modulate FV/FVa function possible.
In summary, work in this study unexpectedly shows that there are well conserved, discrete segments of the FV B-domain, termed the procofactor regulatory region, that serves an essential autoinhibitory function. The PRR is a cis-acting motif that through a direct or allosteric mechanism plays a key role in keeping FV inactive. Mechanisms that counteract the autoinhibitory function of the PRR drive FV activation. The predominant way this occurs after the initiation of coagulation is via discrete proteolysis within the B-domain. Cleavage at these sites must destabilize or facilitate the disengagement of the PRR to enable FXa to bind to the heavy/light chain region and thus allow for prothrombinase assembly. These insights are unique and could not have been uncovered by correlating B-domain cleavage with activity development. The findings detailed here also provide the starting point for unraveling new mechanistic details of FV activation and point to novel strategies for modulation of FV/FVa function for therapeutic purposes.
Supplementary Material
Acknowledgments
We are grateful to Dr. Sriram Krishnaswamy for assistance with fluorescence measurements, useful suggestions, and critical review of the manuscript. We also thank Drs. Raffaella Toso, Matthew Bunce, and Steven Orcutt for critical review of the manuscript. The technical assistance of Michael Boltz is greatly appreciated. Takifugu rubripes (pufferfish) FV cDNA was generously provided by Dr. John H. McVey (Thrombosis Research Institute, London, UK).
This work was supported, in whole or in part, by National Institutes of Health Grants R01 HL88010 and P01 HL74124, Project 2 (to R. M. C.). This work was also supported by the National Hemophilia Foundation (Judith Graham Pool Postdoctoral Research Fellowship; to M. H. A. B.).

This article contains supplemental Figs. 1–4.
- FVa
- activated factor V (FV)
- FX
- factor X
- FXa
- activated FX
- FVIII
- factor VIII
- rFV
- recombinant FV
- DAPA
- dansylarginine-N-(3-ethyl-1,5-pentanediyl)amide
- PCPS
- small unilamellar phospholipid vesicles composed of 75% (w/w) hen egg l-α-phosphatidylcholine and 25% (w/w) porcine brain l-α-phosphatidylserine
- OG488-FXa
- human FXa modified with Oregon Green488 maleimide
- PD-FV
- plasma-derived FV
- PT
- prothrombin time
- BR
- basic region
- AR
- acidic region
- PRR
- procofactor regulatory region.
REFERENCES
- 1. Khan A. R., James M. N. (1998) Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 7, 815–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mann K. G., Kalafatis M. (2003) Factor V. A combination of Dr. Jekyll and Mr. Hyde. Blood 101, 20–30 [DOI] [PubMed] [Google Scholar]
- 3. Camire R. M., Bos M. H. (2009) The molecular basis of factor V and VIII procofactor activation. J. Thromb. Haemost. 7, 1951–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mann K. G., Nesheim M. E., Church W. R., Haley P., Krishnaswamy S. (1990) Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood 76, 1–16 [PubMed] [Google Scholar]
- 5. Nesheim M. E., Mann K. G. (1979) Thrombin-catalyzed activation of single chain bovine factor V. J. Biol. Chem. 254, 1326–1334 [PubMed] [Google Scholar]
- 6. Esmon C. T. (1979) The subunit structure of thrombin-activated factor V. Isolation of activated factor V, separation of subunits, and reconstitution of biological activity. J. Biol. Chem. 254, 964–973 [PubMed] [Google Scholar]
- 7. Suzuki K., Dahlbäck B., Stenflo J. (1982) Thrombin-catalyzed activation of human coagulation factor V. J. Biol. Chem. 257, 6556–6564 [PubMed] [Google Scholar]
- 8. Nesheim M. E., Foster W. B., Hewick R., Mann K. G. (1984) Characterization of factor V activation intermediates. J. Biol. Chem. 259, 3187–3196 [PubMed] [Google Scholar]
- 9. Jenny R. J., Pittman D. D., Toole J. J., Kriz R. W., Aldape R. A., Hewick R. M., Kaufman R. J., Mann K. G. (1987) Complete cDNA and derived amino acid sequence of human factor V. Proc. Natl. Acad. Sci. U.S.A. 84, 4846–4850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Esmon C. T., Lollar P. (1996) Involvement of thrombin anion binding exosites 1 and 2 in the activation of factor V and factor VIII. J. Biol. Chem. 271, 13882–13887 [DOI] [PubMed] [Google Scholar]
- 11. Dharmawardana K. R., Bock P. E. (1998) Demonstration of exosite I-dependent interactions of thrombin with human factor V and factor Va involving the factor Va heavy chain. Analysis by affinity chromatography employing a novel method for active-site selective immobilization of serine proteinases. Biochemistry 37, 13143–13152 [DOI] [PubMed] [Google Scholar]
- 12. Dharmawardana K. R., Olson S. T., Bock P. E. (1999) Role of regulatory exosite I in binding of thrombin to human factor V, factor Va, factor Va subunits, and activation fragments. J. Biol. Chem. 274, 18635–18643 [DOI] [PubMed] [Google Scholar]
- 13. Segers K., Dahlbäck B., Bock P. E., Tans G., Rosing J., Nicolaes G. A. (2007) The role of thrombin exosites I and II in the activation of human coagulation factor V. J. Biol. Chem. 282, 33915–33924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Corral-Rodríguez M. Á., Bock P. E., Hernández-Carvajal E., Gutiérrez-Gallego R., Fuentes-Prior P. (2011) Structural basis of thrombin-mediated factor V activation. The Glu-666–Glu-672 sequence is critical for processing at the heavy chain-B domain junction. Blood 117, 7164–7173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kane W. H., Majerus P. W. (1981) Purification and characterization of human coagulation factor V. J. Biol. Chem. 256, 1002–1007 [PubMed] [Google Scholar]
- 16. Siigur J., Aaspõllu A., Tõnismägi K., Trummal K., Samel M., Vija H., Subbi J., Siigur E. (2001) Proteases from Vipera lebetina venom affecting coagulation and fibrinolysis. Haemostasis 31, 123–132 [DOI] [PubMed] [Google Scholar]
- 17. Keller F. G., Ortel T. L., Quinn-Allen M. A., Kane W. H. (1995) Thrombin-catalyzed activation of recombinant human factor V. Biochemistry 34, 4118–4124 [DOI] [PubMed] [Google Scholar]
- 18. Thorelli E., Kaufman R. J., Dahlbäck B. (1997) Cleavage requirements for activation of factor V by factor Xa. Eur. J. Biochem. 247, 12–20 [DOI] [PubMed] [Google Scholar]
- 19. Steen M., Dahlbäck B. (2002) Thrombin-mediated proteolysis of factor V resulting in gradual B-domain release and exposure of the factor Xa-binding site. J. Biol. Chem. 277, 38424–38430 [DOI] [PubMed] [Google Scholar]
- 20. Segers K., Rosing J., Nicolaes G. A. (2006) Structural models of the snake venom factor V activators from Daboia russelli and Daboia lebetina. Proteins 64, 968–984 [DOI] [PubMed] [Google Scholar]
- 21. Camire R. M., Kalafatis M., Tracy P. B. (1998) Proteolysis of factor V by cathepsin G and elastase indicates that cleavage at Arg1545 optimizes cofactor function by facilitating factor Xa binding. Biochemistry 37, 11896–11906 [DOI] [PubMed] [Google Scholar]
- 22. Kalafatis M., Beck D. O., Mann K. G. (2003) Structural requirements for expression of factor Va activity. J. Biol. Chem. 278, 33550–33561 [DOI] [PubMed] [Google Scholar]
- 23. Fay P. J. (2006) Factor VIII structure and function. Int. J. Hematol. 83, 103–108 [DOI] [PubMed] [Google Scholar]
- 24. Kane W. H., Devore-Carter D., Ortel T. L. (1990) Expression and characterization of recombinant human factor V and a mutant lacking a major portion of the connecting region. Biochemistry 29, 6762–6768 [DOI] [PubMed] [Google Scholar]
- 25. Toso R., Camire R. M. (2004) Removal of B-domain sequences from factor V rather than specific proteolysis underlies the mechanism by which cofactor function is realized. J. Biol. Chem. 279, 21643–21650 [DOI] [PubMed] [Google Scholar]
- 26. Zhu H., Toso R., Camire R. M. (2007) Inhibitory sequences within the B-domain stabilize circulating factor V in an inactive state. J. Biol. Chem. 282, 15033–15039 [DOI] [PubMed] [Google Scholar]
- 27. Higgins D. L., Mann K. G. (1983) The Interaction of bovine factor V and factor V-derived peptides with phospholipid vesicles. J. Biol. Chem. 258, 6503–6508 [PubMed] [Google Scholar]
- 28. Baugh R. J., Krishnaswamy S. (1996) Role of the activation peptide domain in human factor X activation by the extrinsic Xase complex. J. Biol. Chem. 271, 16126–16134 [DOI] [PubMed] [Google Scholar]
- 29. Buddai S. K., Toulokhonova L., Bergum P. W., Vlasuk G. P., Krishnaswamy S. (2002) Nematode anticoagulant protein c2 reveals a site on factor Xa that is important for macromolecular substrate binding to human prothrombinase. J. Biol. Chem. 277, 26689–26698 [DOI] [PubMed] [Google Scholar]
- 30. Katzmann J. A., Nesheim M. E., Hibbard L. S., Mann K. G. (1981) Isolation of functional human coagulation Factor V by using a hybridoma antibody. Proc. Natl. Acad. Sci. U.S.A. 78, 162–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mann K. G. (1976) Methods Enzymol. 45, 123–156 [DOI] [PubMed] [Google Scholar]
- 32. Camire R. M. (2002) Prothrombinase assembly and S1 site occupation restore the catalytic activity of FXa impaired by mutation at the sodium-binding site. J. Biol. Chem. 277, 37863–37870 [DOI] [PubMed] [Google Scholar]
- 33. Bradford H. N., Micucci J. A., Krishnaswamy S. (2010) Regulated cleavage of prothrombin by prothrombinase. Repositioning a cleavage site reveals the unique kinetic behavior of the action of prothrombinase on its compound substrate. J. Biol. Chem. 285, 328–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Betz A., Krishnaswamy S. (1998) Regions remote from the site of cleavage determine macromolecular substrate recognition by the prothrombinase complex. J. Biol. Chem. 273, 10709–10718 [DOI] [PubMed] [Google Scholar]
- 35. Bevington P. R., Robinson K. D. (1992) Data Reduction and Error Analysis for the Physical Sciences, McGraw-Hill Inc., New York [Google Scholar]
- 36. Straume M., Johnson M. L. (1992) Analysis of residuals. Criteria for determining goodness-of-fit. Methods Enzymol. 210, 87–105 [DOI] [PubMed] [Google Scholar]
- 37. Buddai S. K., Layzer J. M., Lu G., Rusconi C. P., Sullenger B. A., Monroe D. M., Krishnaswamy S. (2010) An anticoagulant RNA aptamer that inhibits proteinase-cofactor interactions within prothrombinase. J. Biol. Chem. 285, 5212–5223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krishnaswamy S. (1990) Prothrombinase complex assembly. Contributions of protein-protein and protein-membrane interactions toward complex formation. J. Biol. Chem. 265, 3708–3718 [PubMed] [Google Scholar]
- 39. Rao V. S., Swarup S., Kini R. M. (2003) The nonenzymatic subunit of pseutarin C, a prothrombin activator from eastern brown snake (Pseudonaja textilis) venom, shows structural similarity to mammalian coagulation factor V. Blood 102, 1347–1354 [DOI] [PubMed] [Google Scholar]
- 40. St Pierre L., Masci P. P., Filippovich I., Sorokina N., Marsh N., Miller D. J., Lavin M. F. (2005) Comparative analysis of prothrombin activators from the venom of Australian elapids. Mol. Biol. Evol. 22, 1853–1864 [DOI] [PubMed] [Google Scholar]
- 41. Welton R. E., Burnell J. N. (2005) Full-length nucleotide sequence of a factor V-like subunit of oscutarin from Oxyuranus scutellatus scutellatus (coastal Taipan). Toxicon 46, 328–336 [DOI] [PubMed] [Google Scholar]
- 42. Bos M. H., Boltz M., St Pierre L., Masci P. P., de Jersey J., Lavin M. F., Camire R. M. (2009) Venom factor V from the common brown snake escapes hemostatic regulation through procoagulant adaptations. Blood 114, 686–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bos M. H., Camire R. M. (2010) Procoagulant adaptation of a blood coagulation prothrombinase-like enzyme complex in Australian elapid venom. Toxins 2, 1554–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vos H. L., van Wijngaarden A. (2009) Variation and conservation of the B-domain of factor V. J. Thromb. Haemost. 7, 368 [Google Scholar]
- 45. Kane W. H., Davie E. W. (1986) Cloning of a cDNA coding for human factor V, a blood coagulation factor homologous to factor VIII and ceruloplasmin. Proc. Natl. Acad. Sci. U.S.A. 83, 6800–6804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cripe L. D., Moore K. D., Kane W. H. (1992) Structure of the gene for human coagulation factor V. Biochemistry 31, 3777–3785 [DOI] [PubMed] [Google Scholar]
- 47. Zhang B. (2009) Recent developments in the understanding of the combined deficiency of FV and FVIII. Br. J. Haematol. 145, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rand M. D., Hanson S. R., Mann K. G. (1995) Factor V turnover in a primate model. Blood 86, 2616–2623 [PubMed] [Google Scholar]
- 49. Pufall M. A., Graves B. J. (2002) Autoinhibitory domains. Modular effectors of cellular regulation. Annu. Rev. Cell Dev. Biol. 18, 421–462 [DOI] [PubMed] [Google Scholar]
- 50. Peterson J. R., Golemis E. A. (2004) Autoinhibited proteins as promising drug targets. J. Cell. Biochem. 93, 68–73 [DOI] [PubMed] [Google Scholar]
- 51. Steen M., Tran S., Autin L., Villoutreix B. O., Tholander A. L., Dahlbäck B. (2008) Mapping of the factor Xa binding site on factor Va by site-directed mutagenesis. J. Biol. Chem. 283, 20805–20812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heeb M. J., Kojima Y., Hackeng T. M., Griffin J. H. (1996) Binding sites for blood coagulation factor Xa and protein S involving residues 493–506 in factor Va. Protein Sci. 5, 1883–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Steen M., Villoutreix B. O., Norstrøm E. A., Yamazaki T., Dahlbäck B. (2002) Defining the factor Xa-binding site on factor Va by site-directed glycosylation. J. Biol. Chem. 277, 50022–50029 [DOI] [PubMed] [Google Scholar]
- 54. Gale A. J., Yegneswaran S., Xu X., Pellequer J. L., Griffin J. H. (2007) Characterization of a factor Xa binding site on factor Va near the Arg-506-activated protein C cleavage site. J. Biol. Chem. 282, 21848–21855 [DOI] [PubMed] [Google Scholar]
- 55. Fay P. J. (2004) Activation of factor VIII and mechanisms of cofactor action. Blood Rev. 18, 1–15 [DOI] [PubMed] [Google Scholar]
- 56. Duckers C., Simioni P., Rosing J., Castoldi E. (2009) Advances in understanding the bleeding diathesis in factor V deficiency. Br. J. Haematol. 146, 17–26 [DOI] [PubMed] [Google Scholar]
- 57. Jiang Y., Doolittle R. F. (2003) The evolution of vertebrate blood coagulation as viewed from a comparison of puffer fish and sea squirt genomes. Proc. Natl. Acad. Sci. U.S.A. 100, 7527–7532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Davidson C. J., Tuddenham E. G., McVey J. H. (2003) 450 million years of hemostasis. J. Thromb. Haemost. 1, 1487–1494 [DOI] [PubMed] [Google Scholar]
- 59. Hill-Eubanks D. C., Parker C. G., Lollar P. (1989) Differential proteolytic activation of factor VIII-von Willebrand factor complex by thrombin. Proc. Natl. Acad. Sci. U.S.A. 86, 6508–6512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nesheim M. E., Myrmel K. H., Hibbard L., Mann K. G. (1979) Isolation and characterization of single chain bovine factor V. J. Biol. Chem. 254, 508–517 [PubMed] [Google Scholar]
- 61. Laue T. M., Johnson A. E., Esmon C. T., Yphantis D. A. (1984) Structure of bovine blood coagulation factor Va. Determination of the subunit associations, molecular weights, and asymmetries by analytical ultracentrifugation. Biochemistry 23, 1339–1348 [DOI] [PubMed] [Google Scholar]
- 62. Lampe P. D., Pusey M. L., Wei G. J., Nelsestuen G. L. (1984) Electron microscopy and hydrodynamic properties of blood clotting factor V and activation fragments of factor V with phospholipid vesicles. J. Biol. Chem. 259, 9959–9964 [PubMed] [Google Scholar]
- 63. Mosesson M. W., Nesheim M. E., DiOrio J., Hainfeld J. F., Wall J. S., Mann K. G. (1985) Studies on the structure of bovine factor V by scanning transmission electron microscopy. Blood 65, 1158–1162 [PubMed] [Google Scholar]
- 64. Fowler W. E., Fay P. J., Arvan D. S., Marder V. J. (1990) Electron microscopy of human factor V and factor VIII. Correlation of morphology with domain structure and localization of factor V activation fragments. Proc. Natl. Acad. Sci. U.S.A. 87, 7648–7652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mosesson M. W., Church W. R., DiOrio J. P., Krishnaswamy S., Mann K. G., Hainfeld J. F., Wall J. S. (1990) Structural model of factors V and Va based on scanning transmission electron microscope images and mass analysis. J. Biol. Chem. 265, 8863–8868 [PubMed] [Google Scholar]
- 66. Adams T. E., Hockin M. F., Mann K. G., Everse S. J. (2004) The crystal structure of activated protein C-inactivated bovine factor Va. Implications for cofactor function. Proc. Natl. Acad. Sci. U.S.A. 101, 8918–8923 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.