

Abstract
BAC arrays were used to evaluate genomic instability along the colon of patients with ulcerative colitis (UC). Genomic instability increases with disease progression and biopsies more proximal to dysplasia showed increased instability. Pan-colonic field copy number gain or loss involving small (< 1 Mb) regions were detected in most patients and were particularly apparent in the UC progressor patients who had dysplasia or cancer. Chromosomal copy gains or losses affecting large regions were mainly restricted to dysplastic biopsies. Areas of significant chromosomal losses were detected in the UC progressors on chromosomes 2q36, 3q25, 3p21, 4q34, 4p16.2, 15q22, and 16p13 (p-value ≤ 0.04). These results extend our understanding of the dynamic nature of pan-colonic genomic instability in this disease.
Keywords: ulcerative colitis, genomic instability, BAC array
1. Introduction
Ulcerative colitis (UC) is an inflammatory bowel disease (IBD) associated with an increased risk of developing colorectal cancer. Two clear clinical risk factors for UC-associated colon cancer have been identified: the duration of disease (> 8 years) and the concomitant diagnosis of primary sclerosing cholangitis (PSC), a disease of the biliary tract [1–3]. The standard of care for management of colon cancer risk in UC is currently colonoscopy (with 8 – 40 random biopsies) with follow up every 1 – 3 years [4;5]. This approach is time-consuming and expensive. With the incidence of IBD increasing, it is becoming even more important to develop molecular techniques for improved surveillance and identification of those patients with the greatest cancer risk (i.e., those who currently have pre-cancer or dysplasia). Therefore, we hope to identify DNA biomarkers which were significantly altered in both non-dysplastic and dysplastic biopsies from UC progressor patients (patients with high-grade dysplasia or cancer), but absent or minimally altered in control groups, including non-UC controls and UC non-progressors. Towards that end, we took the approach of a mapping study, in which several biopsies per individual were analyzed by BAC array-CGH (comparative genome hybridization) and then tested for conserved alterations within patient samples and between patients. Multiple biopsies (ranging from 2 to 9) were analyzed per patient in UC and UC/PSC progressors. Included in these sets were sites of varying histologic grades of dysplasia (negative for dysplasia, low grade dysplasia (LGD) or high grade dysplasia (HGD)) sampled along the full length of the colon and biopsy samples that were taken at a range of distances from sites of cancer or high grade dysplasia. The tissue samples for the progressors and non-progressors were graded and matched for severity of inflammation (grade 0–3+).
Cancer development with UC occurs through a multi-stage process involving genomic instability and clonal outgrowth which eventually leads to dysplasia. Progression of ulcerative colitis to cancer is facilitated by an environment in which chronic inflammation and damage from bile acid results in repeated cycles of injury and repair. We have shown premature telomere length shortening and increased DNA damage resulting from chronic inflammation in ulcerative colitis patients compared to age-matched normal control individuals [6]. Propagation of mutations resulting from this damage leads to a mutator phenotype [7] and subsequently a field defect, in which localized regions within the colon have acquired the same mutation(s) or other genomic alterations [7–10]. We and others have also demonstrated that pan-colonic chromosomal instability is detectable early in disease progression and even in non-dysplastic biopsies, by FISH [11;12] and by arbitrarily primed PCR (AP-PCR) or inter–simple sequence repeat PCR (ISSR-PCR)[11;13;14]. While these assays demonstrate clearly that instability is present and highly associated with UC neoplasia, none of these assays are amenable to the high-throughput that would be required of a clinical biomarker.
In this retrospective study, we addressed the following: (1) whether pan-colonic genomic changes could be detected by a relatively inexpensive and high through-put technology such as BAC array-CGH; (2) whether specific genomic changes found in colonic biopsies of a single patient are conserved regardless of differences in the histological grade and/or spatial location of the biopsy within the colon; and (3) whether genomic instability could serve as a biomarker that could distinguish UC non-progressors from progressors. Since field defects are typically acquired during the early stages of neoplastic progression, we hypothesized that this approach could elucidate clinically relevant biomarkers of cancer risk which precede dysplasia, thereby making them applicable for cancer surveillance.
2. Materials and Methods
2.1 Patient information
Patient samples were collected at either the University of Washington Medical Center or the Cleveland Clinic Foundation with the approval of the Institutional Human Subjects Review Board at each institute. Patient data is summarized in Table 1; biopsy samples were collected from surgically resected colons or colonoscopy from three groups of patients: 10 UC progressors (patients with the diagnosis of high-grade dysplasia or cancer), 10 UC non-progressors (patients without any dysplasia or cancer), and 8 non-UC colons (diagnoses included diverticulitis, hyperplastic polyps, appendicitis, tentative endometrial cancer, and meningomyelocele). Five patients with UC also had concomitant PSC. None of the patients had a known family history of colorectal carcinoma. For most patients, no medication data was available. However, patient C was on asacol, patient E was on azulfidin and transplant meds, patient H was on sulfasalazine, and patient I was on FK506, asacol, & URSO (letters correspond to those shown in Figure 1). For one UC progressor patient, disease duration was at least 8 years; however, the exact age of disease onset was not available. Two of the 10 UC non-progressor patients subsequently developed colon cancer and underwent colectomy 7 and 8 years later after the biopsies in this study were taken. The data for these patients is included in the non-progressor subset because that is the initial classification of the patient. None of the other UC non-progressor patients developed high-grade dysplasia or cancer.
Table 1.
Patient information.
| Group | Number of Patients | Total number of biopsies tested(range per patient) | Age (Mean ± SD) | Duration of disease (Mean ± SD) | Age of Onset (Mean ± SD) |
|---|---|---|---|---|---|
| Non-UC3 | 8 | 8(1) | 41.9 ± 7.6 | ||
| UC Non-Progressors | 10 | 17 (1–4) | 48.5 ± 14.3 | 19.6 ± 7.1 | 28.2 ± 9.9 |
| UC Progressors | 10 | 58 (2–9) | 41.2 ± 11.2 | 14.3 ± 7.2 * | 24.9 ± 9.0 * |
Shown are the number of patients per group with the number of biopsies studied per patient, the average age of patients, duration of disease in years (±standard deviation), and average ages of onset (±standard deviation).
denotes that information was unavailable for one patient
Figure 1. Instability along the colon of UC progressor patients.

Shown are whole genome plots illustrating the copy number instability in biopsies sampled taken along the colons of ten UC progressor patients (A–J). Left, maps of the respective colons showing relative spatial locations and histological diagnoses. Blue, negative for dysplasia; green, indefinite for dysplasia; yellow, low grade dysplasia; pink, high grade dysplasia; red, cancer; blank, no histological diagnosis available. Right, whole genome plots showing normalized log2 ratios for chromosomes 1–22 and X (ordered from left to right). Roman numerals correspond to the biopsy position on the respective colon maps. The horizontal line indicates log2 ratio of zero, or normal copy number of 2. Spots above the line indicate BACs with copy number gain; spots below the line indicate BACs with copy number loss.
Epithelial cells were isolated from frozen colon biopsies as described previously [12]. Genomic DNA was isolated from purified epithelium using Qiagen DNA extraction kits as per manufacturer’s instructions. DNA was adjusted to a concentration of 50 ng/μl after quantification with Nanodrop-1000. For BAC arrays, one hundred ng of DNA was used for labeling and hybridization at the Fred Hutchinson Cancer Research Center Microarray Facility as described previously [15]. Fluorescence intensity values were entered into the BAC-arrayer software for block normalization and filtering of the data to generate normalized log2 ratio intensities.
2.2 Analysis by array
Arrays were also analyzed using a variety of algorithms including Wavelet (levels=3; as described by Hsu et al., 2005)[16], circular binary segmentation (CBS), and GLAD with threshold set to 0.2 and processed with the following website: http://compbio.med.harvard.edu/CGHweb [17]). Regions of copy gain or loss were visualized using Ideogram Browser with no pre-filtering [18]. MAD (median absolute deviation), and statistical calculations for all data points were generated in StatView version 5.0.1 (SAS Institute Inc).
2.3 Analysis by group
Significant regions of conserved copy number gains and losses as determined by wavelet were recorded in a binary format for 1 Mb windows (based on start and end positions) per chromosome arm and analyzed with Statistical Analysis of array-CGH (STAC) version 2.0.1 software [20]. Confidence cutoff was set at 0.95; five hundred permutations of the data were performed. A footprint p-value cutoff of 0.05 was used to select significant regions of copy gain or loss. Cytoband locations and genes were obtained with NCBI MapViewer Build 36.3.
3. Results
In this retrospective study, we examined colon biopsies taken from UC progressor and UC non-progressor patients to explore the extent and conservation of genomic alterations. UC progressor patients were defined as individuals who had at least one biopsy diagnosed as high-grade dysplasia or cancer. UC patients included in this study had a long duration of disease (UC non-progressors, mean 19.6 years; UC progressors, mean 14.3 years). In total, 88 arrays were analyzed including samples from 8 non-UC colons (1 site per patient), 10 UC non-progressor patients (2 patients, 4 sites each; 9 patients, 1 site; 17 samples total), and 10 UC progressor patients (average = 5.7 (range 2 to 9) biopsies per patient; for a total of 57 samples). For the UC progressor patients, DNA was analyzed from biopsies diagnosed as negative for dysplasia (n=44), low grade dysplasia (LGD; n=9), low/high grade dysplasia (LGD/HGD; n=2), or high grade dysplasia (HGD; n=3).
3.1 Instability affected by environment
Using this mapping approach, we were able to evaluate genomic instability along the length of the colon for individual UC progressor patients. Figure 1 shows copy number plots for our UC progressor patients. As expected, we observed more extensive and increasing numbers of copy number alterations in dysplastic samples compared to samples which were non-dysplastic. However, non-dysplastic biopsies immediately adjacent to dysplasia typically showed numerous common regions of chromosomal losses and gains in the DNA, irrespective of the degree of dysplasia (high or low grade or indefinite for dysplasia) (Figure 1A). While the field defect of DNA instability in the normal-appearing mucosa surrounding neoplasia was as expected, there were several findings that were surprising. First, copy number alterations could vary greatly even between adjacent biopsies (spatial distances < 2 cm apart such as biopsies III and IV, Figure 1A). Moreover, biopsies adjacent to regions of high-grade dysplasia or cancer (Figure 1A, plots I and II) could have relatively normal copy numbers. Finally, as shown in Figure 1J, we observed that some non-dysplastic biopsies from the progressors demonstrated substantial genomic instability (sometimes involving entire chromosome arms) while other non-dysplastic biopsies from the same colon did not. For example, we observed much more instability on chromosomes 7, 18, and 19 in a non-dysplastic biopsies in one region of a progressor colon (Figure 1E, plot I) compared with another negative biopsy from the same patient (Figure 1E, plot V). We also observed that a biopsy graded indefinite for dysplasia (Figure 1E, Biopsy VI) showed intermediate levels of instability whereas much greater instability was detected in the low-grade dysplasia biopsy from the same patient (Figure 1E, IV).
3.2 Instability increases with proximity to dysplasia
Several algorithms were used to detect BACs with copy number alterations (see Methods); we report here the results using the wavelet algorithm [16]. Consistent with our previous findings [21], the total number of BACs showing copy alterations (copy number gain or copy number loss) in non-dysplastic biopsies increased with proximity to dysplasia in the progressor patients, with a sharp increase in copy number alterations for biopsies located within 10 cm of dysplasia (Figure 2). There was no trend observed in a similar comparison of altered BACs relative to physical location of the biopsy sample in the colon (plotted as distance to rectum in Figure 2B). This suggests that there was no left or right colon bias in these patients.
Figure 2. BAC alterations affected by distance to dysplasia but not distance to rectum.

Shown are scatterplots of number of BACs with copy number gain or loss as detected by wavelet. A, Alterations plotted as a function of distance to dysplasia (cm). B, Alterations plotted relative to distance to rectum (cm). Negative for dysplasia, open blue triangles; dysplasia, open red squares. This data suggests that a single non-dysplastic biopsy from the rectum could detect genomic instability.
3.3 Absolute number of Copy number gains, but not losses, distinguishes UC progressors from UC non-progressors and controls
Having observed an increase in the size and number of alterations with histological grade progression, we next asked whether samples grouped by histological grade would show a difference in extent of genomic instability. For this analysis, we focused on chromosomes 1–22 since individuals could have one or two copies of chromosome X. We observed a significant increase in both the number and maximum size of regions of copy gains (Kruskal Wallis p-values = 0.0007 and 0.0002, respectively) with neoplastic progression (Figure 3A, 3C). Although trends of increasing size and number of regions of copy number loss were also observed, these failed to reach statistical significance (Kruskal Wallis test). The copy number losses observed in the non-UC controls and UC non-progressor patients suggest that small fields of highly unstable clones can be well tolerated without developing into cancer.
Figure 3. The number and max size of copy number alterations increases with UC progression.

A & C, The number of regions of copy number gain (A) and loss (C) increase with UC disease progression. Shown is the average number of regions of copy gain +/− SEM for non-UC, UC non-progressors, UC progressors at sites negative for dysplasia (located > 10 cm or < 10 cm from a site of dysplasia) and UC progressors at dysplastic sites. Asterisks indicate significant difference for comparisons to UC NP using Mann Whitney * p<0.05; ** p-value <0.01. B & D, The maximum size of regions of copy number gain (B) or loss (D) increase with UC disease progression. Shown is the maximum size (Mb) of regions of copy gain +/− SEM for non-UC, UC non-progressors, UC progressors at sites negative for dysplasia (located > 10 cm or < 10 cm from a site of dysplasia) and UC progressors at dysplastic sites. Asterisks indicate significant difference for comparisons to UC NP using Mann Whitney * p<0.05; ** p-value <0.01.
3.4 Field effect of copy number alterations observed along the colon
By comparing array data from multiple biopsies sampled from the same patient, we identified alterations specific to individual UC progressor patients (see Supplemental Table 1). Analyses of 9 UC progressor patients (each with four or more biopsies per patient) showed that the majority (average 84.8 ± 4.8%) of copy number alterations involving two or more consecutive BACs were detected in only a single biopsy whereas a small fraction, average 2.4 ± 1.5%, of all copy number gains and losses were conserved between all biopsies sampled from an individual patient. All of the UC progressors had widespread individual-specific genomic alterations consistent with a field effect (field size ranged from 49 to 161 cm). In general, there were more conserved copy losses than copy gains. Chromosome arms which showed the greatest frequency of copy number loss were 1p, 3p, 9p, and 15q. Three of the nine patients showed loss at the PTPRD (protein-tyrosine phosphatase receptor-type delta) locus on chromosome arm 9p in all biopsies sampled. However, there was relatively low concordance between patients in terms of actual regions of copy number gain or loss, supporting the mutator phenotype [22].
To determine whether there were conserved alterations within the colons of UC progressor patients, —we examined whether a specific chromosomal gain or loss could be detected in two or more biopsies (regardless of histological diagnosis)--amongst the colons of the UC progressor patients. While there were no regions of copy number gain which reached statistical significance, we identified 26 regions of copy number loss which were detectable in multiple biopsies from individual UC progressor patients as well as in multiple patients (Supplemental Table 1). These regions were located on chromosomes 1 through 6, 9, 10, 11, 13, 15, 16, and 17.
Having shown that there is conservation in copy number alterations in colonic biopsies, we next asked whether BAC arrays would have utility to develop a clinical biomarker.
3.5 An overall metric of instability increases with UC progression
To examine whether the overall levels of genomic instability detected by BAC arrays could distinguish patients grouped by disease progression we used median absolute deviation (MAD) values as a measure of the variability of copy number estimates. As expected, we observed a general increase in copy number variability from the minimal changes in colonic biopsies of non-UC controls (range 0.036 to 0.057, median 0.0505), compared to UC non-progressors (0.05 to 0.067; median, 0.054), and to UC progressors (range 0.039 to 0.15; median, 0.063) (Figure 4) with statistically significant differences between groups (ANOVA, p-value <0.01). A wide scatter was observed in the UC progressor samples; greater instability was observed in non-dysplastic samples located within a 10cm radius surrounding sites of dysplasia (data not shown). While the non-UC controls were statistically distinct from UC groups (t-test, p=0.015–0.03) and the UC non-progressors could be distinguished from UC progressors as a whole (t-test, p=0.027), the overlap between these the MAD value in the progressors and non-progressor groups decreases the clinically utility of using the MAD value to distinguish UC patients with dysplasia from those that are dysplasia-free. For the purposes of a clinical biomarker in these setting, the MAD values would need to be mutually exclusive.
Figure 4. MAD (median absolute deviation) values of global genomic instability increase with neoplastic progression.
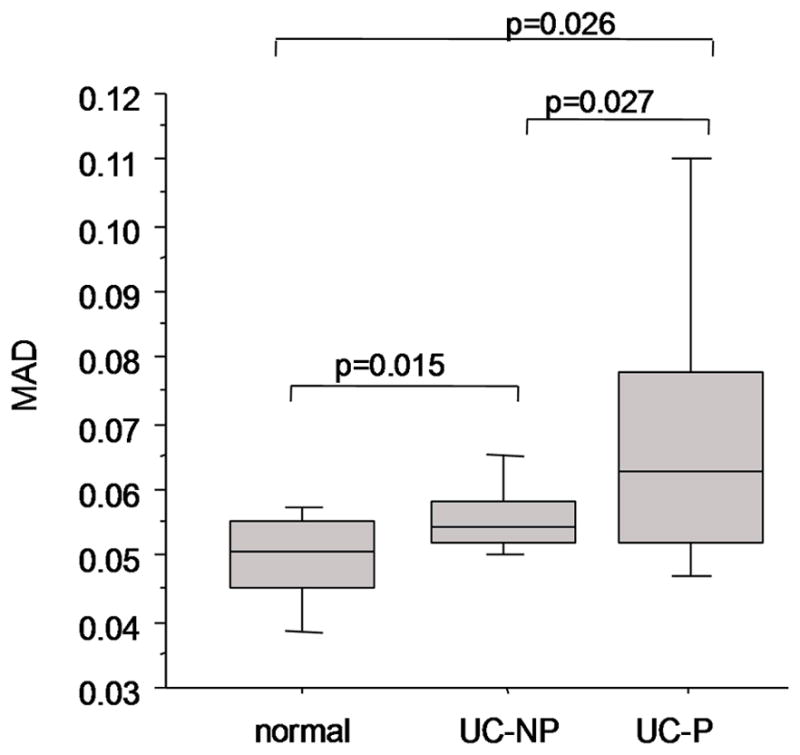
Shown are boxplots of the MAD values for the BAC arrays. The lines indicate the median values and the boxes denote the 75% and 25% for the arrays within each group. The error bars indicate the 90% and 10% for the arrays within each group. The MAD values for genomic instability sufficiently overlap between UC non-progressors (UC-NP) and UC progressors (UC-P), prevent the use of this measure as a clinically meaningful biomarker of neoplasia.
3.6 Common copy number alterations detectable in negative rectal biopsies
Ideally, a clinically useful biomarker test for cancer or dysplasia in UC could use a single colonic biopsy that is easily obtained (i.e. located in the rectum) and the biomarker should accurately distinguish UC progressors from non-progressors regardless of the histologic diagnosis of the biopsy used (i.e. the biomarker should be detectable even in non-dysplastic biopsies of UC progressors). Two biopsies were selected per UC progressor patient, including the most distal (distance to rectum ranged from 2.6 cm to 32 cm; mean, 14.6 ± 9.8 cm) non-dysplastic biopsy for UC non-progressors and one dysplastic biopsy (9 LGD sites and 2 LGD/HGD biopsies). The latter provides an internal positive control for the UC progressors. Alterations detected in non-UC controls (n = 8), and UC non-progressors (n = 10) were compared to UC progressors (n=10). The STAC algorithm [20] was applied to regions of copy number gain or loss identified using the wavelet analysis to detect conserved copy number alterations at 1 Mb intervals.
We postulated that alterations would fall into one of a number of groups, and results for regions of copy number gain or loss, frequency, and footprint p-value are summarized in Table 2. No significant alterations were detected in the samples from normal control patients. Some alterations might only be detectable in UC non-progressors (group 1) and others found in all UC samples tested (group 5). Moreover, some alterations might be detectable only in UC progressors, but exclusively in negative for dysplasia biopsies (group 2), or exclusively in dysplastic biopsies (group 3). The most interesting alterations would be those detectable in both negative for dysplasia and dysplastic biopsies from UC progressors, but not observed in UC non-progressors (group 4).
Table 2.
Significant regions of gain or loss in BAC arrays.
| NP | P neg | P dys | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr | Location | Cytoband | Gain/Loss | freq | foot pvalue | freq | foot pvalue | freq | foot pvalue | Genes of Interest | ||
| 1 | NP only | 4 | 28Mb | 4p15.1 | gain | 25.0% | 0.002 | |||||
| 7 | 9Mb | 7p21 | loss | 25.0% | 0.034 | NXPH1 | ||||||
| 9 | 91Mb | 9q22 | loss | 25.0% | 0.010 | SPIN1, SHC3 | ||||||
| 2 | P neg only | 11 | 64Mb | 11q13 | gain | 20% | 0.002 | COX8A, BAD, NRXN2, MAP4K2 | ||||
| 15 | 19Mb | 15q11.2 | gain | 20% | 0.010 | |||||||
| 2 | 25Mb | 2p23 | loss | 20% | 0.010 | CENPO, ADCY3 | ||||||
| 3 | P dys | 6 | 20Mb | 6p22 | gain | 20% | 0.008 | E2F3 | ||||
| 10 | 24Mb | 10p12 | gain | 20% | 0.002 | ARHGAP21 | ||||||
| 5 | 133Mb | 5q31 | loss | 20% | 0.048 | SKP1, VDAC1, PPP2CA | ||||||
| 7 | 129Mb | 7q32 | loss | 20% | 0.044 | MIR183 | ||||||
| 4 | P neg & dys | 2 | 230–231Mb | 2q36 | loss | 30% | 0.020 | 30% | 0.004 | FBXO36 | ||
| 3 | 171Mb | 3q26 | loss | 30% | 0.010 | 30% | 0.004 | TNIK, PLD1, FNDC3B | ||||
| 3 | 44Mb | 3p21 | loss | 40% | 0.020 | 30% | 0.020 | KIF15 | ||||
| 4 | 173Mb | 4q34 | loss | 40% | 0.020 | 50% | 0.006 | GALNTL6 | ||||
| 4 | 4–5Mb | 4p16.2 | loss | 20% | 0.010 | 40% | 0.004 | STX18 | ||||
| 15 | 69–70Mb | 15q22 | loss | 50% | 0.020 | 60% | 0.004 | MIR629 | ||||
| 16 | 6Mb | 16p13 | loss | 30% | 0.010 | 20% | 0.124 | A2BP1 | ||||
| 5 | NP & P | 1 | 103–104Mb | 1p21 | loss | 37.5% | 0.004 | 40% | 0.010 | 50% | 0.004 | COL11A1 |
| 2 | 233Mb | 2q37 | loss | 25.0% | 0.002 | 20% | 0.090 | 30% | 0.004 | ALPI | ||
| 3 | 8Mb | 3p26 | loss | 37.5% | 0.002 | 40% | 0.020 | 30% | 0.032 | RAD18 | ||
| 6 | 16–18Mb | 6p22 | loss | 37.5% | 0.004 | 40% | 0.010 | 30% | 0.002 | ATXN1 | ||
| 9 | 9Mb | 9p23 | loss | 25.0% | 0.054 | 30% | 0.050 | 50% | 0.004 | PTPRD | ||
Frequency of regions of copy gain and copy loss as detected by STAC analysis for UC non-progressors (n=9; left), and UC progressors (n=11) at non-dysplastic (middle columns) or dysplastic (right columns) sites as indicated by cytoband and genomic location (NCBI Build 36.3). Number of samples and relative percentage (in parentheses) are shown for each alteration per group. Data is grouped to reflect sites specific to one or more of the sample sets (non-progressor, NP; progressor, P; progressor (dysplasia), P dys). NS = p-value > 0.05; not significant.
For the UC non-progressors, one region of significant gain was detected on chromosome 4p15.1 (Table 2). For the UC progressors, regions of gain on chromosomes 11q13, and 15p11.2 were detected in the negative/rectal biopsies but not in the dysplastic samples. However, 2 regions of copy number gain at chromosome arm 6p22 and 10p12 were identified only in UC progressor dysplastic samples, as well as two regions of copy number loss at 5q31 and 7q32. By comparison, seven regions of copy number loss were detected using this analysis, including ten sites in which changes were significant in both the negative and dysplastic biopsies. Significant losses were specifically in the UC progressors at sites on chromosomes 2q36, 3q25, 3p21, 4q34, 4p16.2, 15q22, and 16p13 (footprint p-values = 0.0002 to 0.04). It looks like chromosomal gains, but not losses, could potentially be used to distinguish UC progressors regardless of the histological diagnosis of the biopsies.
4. Discussion
UC patients are at increased risk for colon cancer and efforts are underway to find biomarkers that could help define which of these patients are undergoing neoplastic progression and need close colonoscopic surveillance versus those who do not. We and others have previously described evidence of DNA changes in the epithelium of UC progressors; our goal in this study was to determine whether an assay of genomic instability, that is amenable to high-throughput and objective assessment (eg. the BAC array), could be used as a clinical tool for measuring genomic instability as a neoplastic biomarker in UC. The mapping studies described herein were designed 1) to measure the extent of genomic instability, 2) to determine if BAC-array measured instability could act as a biomarker that can distinguish UC progressors from UC non-progressors, and 3) whether the genomic instability alters as biopsies become increasingly more dysplastic.
4.1 Instability occurs as a field defect
Our data supports the hypothesis that a field effect, stemming from chronic inflammation and clonal outgrowth, is present in the colons of patients with ulcerative colitis [23]. The inflammatory response prompts stem cells with damaged DNA to expand causing the clonal copy number alterations, methylation changes, and nucleotide mutations to spread through whole crypts, crypt clusters and eventually larger colon patches through a process of crypt fission[9;13;24–26]. Patches of mutations can reflect the unequal division of mutated vs. wildtype clones, thereby mutated crypts can give rise to wildtype, mixed wildtype and mutated crypts, or exclusively mutated crypts. Clonal expansion of these genomic mutations within epithelial sheets has been described [23;25]. Evidence from FISH studies have shown that chromosomal instability increases dramatically from low grade to high grade dysplasia, leading to gain (or loss) of whole chromosome arms as demonstrated in high grade dysplasia and cancer samples [12]. Our data supports a study by Greaves et al. in which the mean patch size of cytochrome c oxidase deficient crypts increases with age [26]. We have previously reported that UC is a disease of premature aging due to telomere length shortening and increased DNA damage [6]. Thus the large field defects that we have observed in UC may be due to not only to the chronic inflammation and mutator phenotypes, but also due to the premature aging of the UC colon whereby the colon of a 30 year old UC patient is more analogous to the colon of a 60 to 70 year old without UC.
4.2 BAC array-measured genomic instability as a biomarker to distinguish UC Progressors from C Non-Progressors
The BAC arrays could objectively detect small interstitial changes in DNA copy number in both the non-dysplastic and dysplastic colonic epithelium UC progressors; however instability could also be measured in the UC non-progressors. Thus, very small segments of DNA (< 1Mb) were lost and/or gained in the normal appearing mucosa in all of the UC patients regardless of whether they were graded as histologically dysplastic or not. In order for a neoplastic biomarker to be clinically relevant in UC, it must detect all of the patients who have dysplasia (achieve 100% sensitivity) and have at least some discrimination for the neoplasia-free patients to make it worth using. As seen in Figure 4, when using total genomic instability as the potential biomarker--there is too much overlap of the amounts of global instability found non-progressors and progressors to clinically separate the 2 cohorts. However, if we examine a panel of specific DNA loci, in its capability to distinguish progressors from non-progressors, we find the data is more promising. ROC curves of copy number gains or copy number losses detected by these BAC arrays have at least a moderate sensitivity (AUC=0.716 and AUC=0.813 for copy number gains and losses, respectively) and specificity for distinguishing between UC non-progressors and progressors (data not shown). This data will require futher validation with a larger patient set before determining whether a subset of BACs could be as a discriminating biomarker for UC neoplasia.
4.3 Potential markers identified in this study
Many of the loci shown in Table 2 have not previously been detected in studies of ulcerative colitis or inflammatory bowel disease. Although we did not detect alterations in any of the BACs reported in a recent study by our group [27], we believe that this is due to the small sample sizes and the increased proximity to dysplasia in samples used in the earlier study. However, loss at 5q31 agrees with proteomic studies showing downregulation of the cellular stress response protein, VDAC1 [28]; in addition, 5q31 has been described as a susceptibility locus for IBD, although the association is much stronger for Crohn’s Disease than for UC [29]. MicroRNA 629 (MIR629) at chromosome arm 15q22 was expressed at a level several-fold lower in active UC compared to normal tissues [30]. Downregulation of alkaline phosphatase expression has also been suggested to contribute to chronic inflammation; oral administration of intestinal alkaline phosphatase tablets resulted in marked decrease in intestinal inflammation and improved intestinal wall morphology in a rat model of colitis [31]. The gain at chromosome arm 6p22.1 in UC progressors negative for dysplasia is interesting since superoxide dismutase (SOD2) functions to dissipate oxidative stress in chronically inflamed UC colons [32]. Experiments to validate alterations which would distinguish UC progressors from non-progressors are currently underway.
4.4 Dysplasia to Cancer defect gradient
Tiny DNA alterations were found throughout the entire colon of all UC patients. In order for these changes to be detectable by BAC array, they have to be clonal— e.g. involving at least 15–20% of the cells within a biopsy to be distinguishable from the background. The copy number alterations became much more extensive in the genomic DNA from the dysplastic mucosa: not only are there more incidents of copy changes throughout the DNA but in addition, the originally small defects become much larger in size and encompass a greater proportion of the biopsy (i.e. greater deviation of log2 ratios from the baseline). This progression of instability is undoubtedly associated with the other simultaneous events that are unraveling in the transition to dysplasia—which include breakage of chromosomes associated with shortened telomeres, development of aneuploidy, impairment of DNA repair function caused by reactive oxygen and reactive nitrogen species, and loss of p53 function to just name a few [9]. The fundamental underlying event for the DNA changes is likely caused by chronic inflammation. While the role of inflammation has long been accepted in UC tumorigenesis, we found it quite interesting to note that interstitial, small scale genomic alterations could be detected at a low level even in the colons of control individuals and at an intermediate level in patients with diverticulitis (data not shown). The hypothesis that acute inflammation in the general population could be linked to colon tumorigenesis is supported by the association of recurrent diverticulitis with an elevated risk of sporadic colon cancer [33;34]. Whether patients who have apparently sporadic cancer might actually have occult or asymptomatic bouts of acute colonic inflammation remains an area of future interest.
4.5 Genomic instability in UC patients with and without Primary Sclerosing Cholangitis (PSC)
Three of the main clinical risks for development of UC-associated neoplasia are the duration of disease, the extent of disease, and the concomitant presence of the biliary tract disease: PSC. In the assessment of BAC-measured genomic instability as a biomarker for neoplasia, we studied UC progressors with and without PSC (n=5 cases and n=6 cases, respectively). As noted above, there were many more conserved alterations in the patients that progressed to cancer than those that progressed to high grade dysplasia, however the set of UC only patients, as compared to individuals with UC and PSC, showed a substantial greater number DNA alterations (average 11 to 3.6 respectively). This may seem odd, as the reverse might be expected since PSC confers an approximately 5-fold increased risk of developing colorectal cancer in the setting of UC [1;5]. These UC and UC/PSC patients had comparable disease duration (average 13 vs. 11, respectively); the UC only patients had slightly earlier ages of onset (average 22.4 vs. 30 years respectively). Yet the clonal expansion seemed less robust in the PSC patients. These findings suggest that tumorigenesis might be driven through different etiologies in UC patients with and without PSC or may be affected by the treatment regimen. The smaller number of DNA alterations may be attributable to the milder pancolitis typically observed in UC patients with PSC, however the abnormal bile conjugates present in UC/PSC patients may augment the inflammatory induced genomic instability pathway with an alternative/supplemental pathway of tumorigenesis [35]. Altered bile acids, such as would be present in PSC patients, have been shown to stimulate colon cancer production through a blockage of apoptosis, trans-activation of epidermal growth factor receptors, and increased proliferation [36;37]. Moreover, some bile conjugates can directly interact with DNA causing adducts and base substitutions. Changes such as these would not be detected by BAC-array copy number analysis. This information suggests that even in the setting of mild inflammation, the addition of abnormal bile conjugates can provide a substantial increase in cancer formation. The somewhat differing pathogenesis of colonic tumorigenesis in UC patients with and without PSC could be pertinent to future considerations of chemoprevention in these patients.
4.6 Summary
In conclusion, these findings of widespread genomic instability support that there is a tolerated degree of genomic damage in the colons of ulcerative colitis patients, irrespective of whether they progress to high-grade dysplasia or cancer. These DNA changes are likely clonal in nature and have been identified consistently in samples taken along the full length of the colon from the same patients. The defects in DNA increase in frequency and size as the epithelium becomes increasingly more dysplastic. While BAC-measured global measures of instability could not distinguish between UC progressors and non-progressors in a clinically meaningful way, it is possible that a panel of selected BAC targets might be useful as a functional neoplastic biomarker. Future work will be required to determine whether the use of arrays with greater resolution or the use of specific array targets can be used to distinguish UC progressors from non-progressors
Supplementary Material
Acknowledgments
The authors would like to thank Jasmine Gallaher and Cassie Sather for technical assistance, and Allyn Stevens, Yasuko Tamura, Jeanne Stanton, and Mallory Smith for research biopsy coordination. This study was supported by NIH P20 CA103728, R01 CA068124, P30 AG13280, and the Crohn’s and Colitis Foundation of America.
Footnotes
Conflict of Interest Statement
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brentnall TA, Haggitt RC, Rabinovitch PS, Kimmey MB, Bronner MP, Levine DS, Kowdley KV, Stevens AC, Crispin DA, Emond M, Rubin CE. Risk and natural history of colonic neoplasia in patients with primacy sclerosing cholangitis and ulcerative colitis. Gastroenterology. 1996;110:331–8. doi: 10.1053/gast.1996.v110.pm8566577. [DOI] [PubMed] [Google Scholar]
- 2.Brentnall TA. Molecular underpinnings of cancer in ulcerative colitis. Curr Opin Gastro. 2003;19:64–8. doi: 10.1097/00001574-200301000-00011. [DOI] [PubMed] [Google Scholar]
- 3.Itzkowitz SH, Harpaz N. Diagnosis and management of dysplasia in patients with inflammatory bowel diseases. Gastro. 2004;126:1634–48. doi: 10.1053/j.gastro.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 4.Itzkowitz SH, Present DH. Consensus conference: Colorectal cancer screening and surveillance in inflammatory bowel disease. Inflammatory Bowel Diseases. 2005;11:314–21. doi: 10.1097/01.mib.0000160811.76729.d5. [DOI] [PubMed] [Google Scholar]
- 5.Konda A, Duffy MC. Surveillance of patients at increased risk of colon cancer: Inflammatory bowel disease and other conditions. Gastro Clin of North America. 2008;37:191–213. doi: 10.1016/j.gtc.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 6.Risques RA, Lai LA, Brentnall TA, Li L, Feng Z, Gallaher J, Mandelson MT, Potter JD, Bronner MP, Rabinovitch PS. Ulcerative Colitis Is a Disease of Accelerated Colon Aging: Evidence From Telomere Attrition and DNA Damage. Gastro. 2008;135:410–8. doi: 10.1053/j.gastro.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loeb KR, Loeb LA. Genetic instability and the mutator phenotype. Studies in ulcerative colitis. Am J Pathol. 1999;154:1621–6. doi: 10.1016/S0002-9440(10)65415-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein C, Bernstein H, Payne CM, Dvorak K, Garewal H. Field defects in progression to gastrointestinal tract cancers. Cancer Lett. 2008;260:1–10. doi: 10.1016/j.canlet.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brentnall TA, Crispin DA, Rabinovitch PS, Haggitt RC, Rubin CE, Stevens AC, Burmer GC. Mutations in the P53 Gene - An Early Marker of Neoplastic Progression in Ulcerative-Colitis. Gastro. 1994;107:369–78. doi: 10.1016/0016-5085(94)90161-9. [DOI] [PubMed] [Google Scholar]
- 10.Salk JJ, Salipante SJ, Risques RA, Crispin DA, Li L, Bronner MP, Brentnall TA, Rabinovitch PS, Horwitz MS, Loeb LA. Clonal expansions in ulcerative colitis identify patients with neoplasia. Proc Natl Acad Sci USA. 2009;106:20871–6. doi: 10.1073/pnas.0909428106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Sullivan JN, Finley JC, Risques RA, Shen WT, Gollahon KA, Moskovitz AH, Gryaznov S, Harley CB, Rabinovitch PS. Telomere length assessment in tissue sections by quantitative FISH: image analysis algorithms. Cytometry. 2004;58A:120–31. doi: 10.1002/cyto.a.20006. [DOI] [PubMed] [Google Scholar]
- 12.Rabinovitch PS, Dziadon S, Brentnall TA, Emond MJ, Crispin DA, Haggitt RC, Bronner MP. Pancolonic chromosomal instability precedes dysplasia and cancer in ulcerative colitis. Cancer Res. 1999;59:5148–53. [PubMed] [Google Scholar]
- 13.Chen R, Rabinovitch PS, Crispin DA, Emond MJ, Koprowicz KM, Bronner MP, Brentnall TA. DNA fingerprinting abnormalities can distinguish ulcerative colitis patients with dysplasia and cancer from those who are dysplasia/cancer-free. Am J Pathol. 2003;162:665–72. doi: 10.1016/S0002-9440(10)63860-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen R, Bronner MP, Crispin DA, Rabinovitch PS, Brentnall TA. Characterization of genomic instability in ulcerative colitis neoplasia leads to discovery of putative tumor suppressor regions. Cancer Genet Cytogenet. 2005;162:99–106. doi: 10.1016/j.cancergencyto.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 15.Loo LWM, Grove DI, Williams EM, Neal CL, Cousens LA, Schubert EL, Holcomb IN, Massa HF, Glogovac J, Li CI, Malone KE, Daling JR, Delrow JJ, Trask BJ, Hsu L, Porter PL. Array comparative genomic hybridization analysis of genomic alterations in breast cancer subtypes. Cancer Res. 2004;64:8541–9. doi: 10.1158/0008-5472.CAN-04-1992. [DOI] [PubMed] [Google Scholar]
- 16.Hsu L, Self SG, Grove D, Randolph T, Wang K, Delrow JJ, Loo L, Porter P. Denoising array-based comparative genomic hybridization data using wavelets. Biostat. 2005;6:211–26. doi: 10.1093/biostatistics/kxi004. [DOI] [PubMed] [Google Scholar]
- 17.Lai W, Choudhary V, Park PJ. CGHweb: a tool for comparing DNA copy number segmentations from multiple algorithms. Bioinformatics. 2008;24:1014–5. doi: 10.1093/bioinformatics/btn067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller A, Holzmann K, Kestler HA. Visualization of genomic aberrations using Affymetrix SNP arrays. Bioinformatics. 2007;23:496–7. doi: 10.1093/bioinformatics/btl608. [DOI] [PubMed] [Google Scholar]
- 19.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome–wide expression patterns. Proc Natl Acad Sci USA. 1999;96:10943. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diskin SJ, Eck T, Greshock J, Mosse YP, Naylor T, Stoeckert CJ, Weber BL, Maris JM, Grant GR. STAC: A method for testing the significance of DNA copy number aberrations across multiple array-CGH experiments. Genome Res. 2006;16:1149–58. doi: 10.1101/gr.5076506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Risques RA, Lai LA, Himmetoglu C, Abaee A, Li L, Feng Z, Bronner MP, Al-Lahham B, Kowdley KV, Lindor KD, Rabinovitch PS, Brentnall TA. Ulcerative colitis-associated colorectal cancer arises in a field of short telomeres, senescence and inflammation. Cancer Res. 2011;71:1669–79. doi: 10.1158/0008-5472.CAN-10-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loeb LA. A mutator phenotype in cancer. Cancer Res. 2001;61:3230–9. [PubMed] [Google Scholar]
- 23.Garcia SB, Park HS, Novelli M, Wright NA. Field cancerization clonality, and epithelial stem cells: The spread of mutated clones in epithelial sheets. J Path. 1999;187:61–81. doi: 10.1002/(SICI)1096-9896(199901)187:1<61::AID-PATH247>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 24.Chen R, Rabinovitch PS, Crispin DA, Emond MJ, Bronner MP, Brentnall TA. The initiation of colon cancer in a chronic inflammatory setting. Carcinogenesis. 2005;26:1513–9. doi: 10.1093/carcin/bgi106. [DOI] [PubMed] [Google Scholar]
- 25.Garcia SB, Novelli M, Wright NA. The clonal origin and clonal evolution of epithelial tumours. Intl J of Exp Path. 2000;81:89–116. doi: 10.1046/j.1365-2613.2000.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greaves LC, Preston SL, Tadrous PJ, Taylor RW, Barron MJ, Oukrif D, Leedham SJ, Deheragoda M, Sasieni P, Novelli MR, Jankowski JAZ, Turnbull DM, Wright NA, Mcdonald SAC. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc Natl Acad Sci USA. 2006:103714–9. doi: 10.1073/pnas.0505903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bronner MP, Skacel M, Crispin DA, Hoff PD, Emond MJ, Lai LA, Tubbs RR, O’Sullivan JN, Rabinovitch PS, Brentnall TA. Array-based comparative genomic hybridization in ulcerative colitis neoplasia: single non-dysplastic biopsies distinguish progressors from non-progressors. Mod Path. 2010;23:1624–33. doi: 10.1038/modpathol.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsieh SY, Shih TC, Yeh CY, Lin CJ, Chou YY, Lee YS. Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics. 2006;6:5322–31. doi: 10.1002/pmic.200500541. [DOI] [PubMed] [Google Scholar]
- 29.Franke A, Hampe J, Rosenstiel P, Becker C, Wagner F, Hasler R, Little RD, Huse K, Ruether A, Balschun T, Wittig M, ElSharawy A, Mayr G, Albrecht M, Prescott NJ, Onnie CM, Fournier H, Keith T, Radelof U, Platzer M, Mathew CG, Stoll M, Krawczak M, Nurnberg P, Schreiber S. Systematic Association Mapping Identifies NELL1 as a Novel IBD Disease Gene. PLoS One. 2007;2:e691. doi: 10.1371/journal.pone.0000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu F, Zikusoka M, Trindade A, Dassopoulos T, Harris ML, Bayless TM, Brant SR, Chakravarti S, Kwon JH. MicroRNAs Are Differentially Expressed in Ulcerative Colitis and Alter Expression of Macrophage Inflammatory Peptide-2 alpha. Gastro. 2008;135:1624–35. doi: 10.1053/j.gastro.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 31.Tuin A, Poelstra K, Jager-Krikken A, Bok L, Raaben W, Velders MP, Dijkstra G. Role of alkaline phosphatase in colitis in man and rats. Gut. 2009;58:379–87. doi: 10.1136/gut.2007.128868. [DOI] [PubMed] [Google Scholar]
- 32.Dincer Y, Erzin Y, Himmetoglu S, Gunes KN, Bal K, Akcay T. Oxidative DNA damage and antioxidant activity in patients with inflammatory bowel disease. Digestive Diseases and Sciences. 2007;52:1636–41. doi: 10.1007/s10620-006-9386-8. [DOI] [PubMed] [Google Scholar]
- 33.Stefansson T, Ekbom A, Sparen P, Pahlman L. Increased Risk of Left Sided Colon Cancer in Patients with Diverticular-Disease. Gut. 1993;34:499–502. doi: 10.1136/gut.34.4.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stefansson T, Ekbom A, Sparen P, Pahlman L. Association between sigmoid diverticulitis and left-sided colon cancer: a nested, population-based, case control study. Scand J Gastro. 2004;39:743–7. doi: 10.1080/00365520410003272. [DOI] [PubMed] [Google Scholar]
- 35.Broome U, Bergquist A. Primary sclerosing cholangitis, inflammatory a bowel disease, and colon cancer. Sem in Liver Disease. 2006;26:31–41. doi: 10.1055/s-2006-933561. [DOI] [PubMed] [Google Scholar]
- 36.Cheng K, Raufman JP. Autocrine release of HB-EGF mediates bile acid-induced transactivation of colon cancer cell epidermal growth factor receptors (EGFR) Gastro. 2005;128:A613. doi: 10.1016/j.bcp.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 37.Raufman JP, Shant J, Guo CY, Roy S, Cheng K. Deoxycholyltaurine rescues human coron cancer cells from apoptosis by activating EGFR-dependent PI3K/Akt signaling. J Cell Physio. 2008;215:538–49. doi: 10.1002/jcp.21332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.