

Abstract
Mutations in GBA, the gene encoding glucocerebrosidase, the enzyme deficient in Gaucher disease, are common risk factors for Parkinson disease, as patients with Parkinson disease are over five times more likely to carry GBA mutations than healthy controls. Patients with GBA mutations generally have an earlier onset of Parkinson disease and more cognitive impairment than those without GBA mutations. We investigated whether GBA mutations alter the neurobiology of Parkinson disease, studying brain dopamine synthesis and resting regional cerebral blood flow in 107 subjects (38 women, 69 men). We measured dopamine synthesis with 18F-fluorodopa positron emission tomography, and resting regional cerebral blood flow with H215O positron emission tomography in the wakeful, resting state in four study groups: (i) patients with Parkinson disease and Gaucher disease (n = 7, average age = 56.6 ± 9.2 years); (ii) patients with Parkinson disease without GBA mutations (n = 11, 62.1 ± 7.1 years); (iii) patients with Gaucher disease without parkinsonism, but with a family history of Parkinson disease (n = 14, 52.6 ± 12.4 years); and (iv) healthy GBA-mutation carriers with a family history of Parkinson disease (n = 7, 50.1 ± 18 years). We compared each study group with a matched control group. Data were analysed with region of interest and voxel-based methods. Disease duration and Parkinson disease functional and staging scores were similar in the two groups with parkinsonism, as was striatal dopamine synthesis: both had greatest loss in the caudal striatum (putamen Ki loss: 44 and 42%, respectively), with less reduction in the caudate (20 and 18% loss). However, the group with both Parkinson and Gaucher diseases showed decreased resting regional cerebral blood flow in the lateral parieto-occipital association cortex and precuneus bilaterally. Furthermore, two subjects with Gaucher disease without parkinsonian manifestations showed diminished striatal dopamine. In conclusion, the pattern of dopamine loss in patients with both Parkinson and Gaucher disease was similar to sporadic Parkinson disease, indicating comparable damage in midbrain neurons. However, H215O positron emission tomography studies indicated that these subjects have decreased resting activity in a pattern characteristic of diffuse Lewy body disease. These findings provide insight into the pathophysiology of GBA-associated parkinsonism.
Keywords: brain imaging, genetic risk, positron emission tomography (PET), Parkinson disease, lysosomal storage disorders
Introduction
Once widely believed not to be hereditable (Bharucha et al., 1986), the aetiology of Parkinson disease is now known to include important genetic components, either through rare Mendelian forms or via more common susceptibility alleles (Obeso et al., 2010; Lill et al., 2012). Among the known genetic risk factors are mutations in GBA, the gene encoding glucocerebrosidase, the enzyme deficient in Gaucher disease. Gaucher disease is a recessively inherited lysosomal disorder, which, in adults, primarily manifests with hepatosplenomegaly, anaemia, thrombocytopaenia and bone involvement. The increased risk of Parkinson disease is reported in both Gaucher homozygotes and heterozygotes; based on published data from a Gaucher Disease Registry (Rosenbloom et al., 2011), among subjects with Gaucher disease, the probability of developing Parkinson disease before the age of 70 is 5–7% and 9–12% before the age of 80, compared with 1.2 and 2.6% in the general population, respectively (Elbaz et al., 2002). Moreover, patients with Parkinson disease are over five times more likely to carry GBA mutations than are healthy controls (Sidransky et al., 2009).
The identification of specific genetic risk factors for Parkinson disease and the pathology they confer can provide a window into molecular mechanisms underlying disease pathogenesis, and may inform potential new treatment approaches. Although the clinical phenotype of GBA-associated Parkinson disease is, in general, similar to Parkinson disease without GBA mutations, patients with GBA mutations tend to have a younger age at onset and more cognitive impairment (Goker-Alpan et al., 2008; Sidransky et al., 2009; Brockmann et al., 2011; Seto-Salvia et al., 2012; Alcalay et al., 2012). These phenotypic dissimilarities raise the possibility that the neurobiology of GBA-associated parkinsonism may differ from that of typical Parkinson disease.
For example, the trend towards greater cognitive impairment in GBA-associated parkinsonism could reflect greater involvement of the ‘cognitive’ dopaminergic system, which originates in the medial portion of the substantia nigra and predominantly innervates the nucleus accumbens and head of the caudate nucleus (Haber et al., 2000). These medial nigral neurons are known to be spared in typical Parkinson disease, whereas neurons located more laterally in the nigra, which are principally involved in motor processing and project to the tail of the putamen, tend to be lost early in the course of the disease (Stoessl et al., 2011). This distribution of nigral neuronal loss in the midbrain is reflected in the early loss of striatal dopaminergic terminals, which can be measured in vivo by means of 18F-fluorodopa PET. The uptake of 18F-fluorodopa in the striatal nuclei, indicated by an influx constant, Ki, reflects presynaptic dopamine synthesis (by DOPA decarboxylase) and storage. Histological studies have demonstrated that levels of striatal 18F-fluorodopa uptake correlate well with nigral cell counts in both patients with Parkinson disease and non-human primates with parkinsonism induced by the nigral toxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) (Stoessl et al., 2011). In typical Parkinson disease, decreased 18F-fluorodopa uptake is consistently localized to the tail of the putamen (Morrish et al., 1998; Stoessl et al., 2011), whereas the pattern in GBA-associated parkinsonism has not been determined beyond occasional case reports (Kono et al., 2007; Kraoua et al., 2009; Saunders-Pullman et al., 2010).
The pattern of cortical dysfunction in GBA-associated parkinsonism remains similarly undefined, and it is of interest because it could explain some of the cognitive changes not typically found in sporadic Parkinson disease (Alcalay et al., 2012). Dysfunction of cortical neurons is likely to be associated with regional changes in cortical synaptic activity and metabolism, coupled in the non-ischaemic brain, as is the case in Parkinson disease, to regional cerebral blood, which can be reproducibly measured using H215O PET (Fox et al., 1988).
To determine the degree and distribution of dopamine loss in GBA-associated Parkinson disease, as well as the pattern of resting cortical synaptic activity, we used multimodality imaging with 18F-fluorodopa and resting H215O PET to study patients with parkinsonism, comparing those with and without Gaucher disease. In addition, we studied two groups, patients with Gaucher disease and healthy carriers with GBA mutations and a strong family history of Parkinson disease, but lacking clinical manifestations of Parkinson disease. These two groups were included to screen for early brain changes that might be predictive of Parkinson disease in these high-risk individuals. As such changes are more likely in subjects with a family history of Parkinson disease, we chose to focus on at-risk individuals with a family history of Parkinson disease.
Materials and methods
Subjects
All patients and healthy controls (totalling 107 subjects, 38 females/69 males) were studied at the Clinical Centre of the National Institutes of Health Intramural Research Program. Each participant provided written informed consent and was enrolled in protocols approved by the Institutional Review Board, National Human Genome Research Institute and the Radiation Safety Committee of the National Institutes of Health. There were four study groups, two with parkinsonism: (i) those with Gaucher disease; and (ii) those without GBA mutations (sporadic Parkinson disease); and two without parkinsonism but with a parent or sibling with Parkinson disease: (iii) patients with Gaucher disease; and (iv) healthy GBA-mutation carriers. Patients with parkinsonism met UK Parkinson Disease Society Brain Bank Clinical Diagnostic Criteria for Parkinson disease (Hughes et al., 1992). All patients with Gaucher disease had the type 1 phenotype. All patients with parkinsonism, including those with GBA mutations, showed clinical improvement on dopaminergic medication. Because parkinsonism tends to have an earlier onset in GBA mutation carriers and because the patient cohorts had different sex distributions, each of the four study groups was compared with its own separate healthy control group, specially matched for age, sex and education (Table 1). All study subjects and healthy controls had extensive physical and neurological evaluations, including structural MRI studies of the brain, to exclude any other neurological or structural abnormalities (Supplementary material).
Table 1.
Demographics of patients and healthy controls in each study group
| Parkinson and Gaucher diseases | Parkinson disease without GBA mutations | Gaucher diseasea | Gaucher disease carriera | Total or average | |
|---|---|---|---|---|---|
| Patients | |||||
| Number | 7 | 11b | 14 | 7 | 39 |
| Average agec | 56.6 ± 9.2 | 62.1 ± 7.1 | 52.6 ± 12.4 | 50.1 ± 18.0 | 55.5 ± 12.3 |
| % Female | 14.3 | 27.3 | 35.7 | 57.1 | 33.3 |
| Educationd | 16.0 ± 3.1 | 18.1 ± 2.4 | 16.1 ± 2.7 | 16.7 ± 2.6 | 16.8 ± 2.7 |
| Controls | |||||
| Number | 13 | 14 | 23 | 18 | 68 |
| Average age | 56.5 ± 8.4 | 62.3 ± 7.6 | 50.3 ± 8.8 | 50.9 ± 14.9 | 53.5 ± 11.1 |
| % Female | 14.3 | 27.3 | 35.7 | 57.1 | 33.3 |
| Education | 16.4 ± 3.1 | 16.2 ± 2.7 | 16.5 ± 2.7 | 16.6 ± 3.2 | 16.5 ± 2.9 |
a No clinical signs of Parkinson disease, but first degree relative with parkinsonism.
b Eight of these patients were tested for GBA mutations; findings were similar when the non-tested patients were excluded.
c Average age and SD at the time of PET testing.
d Years of schooling, not significantly different between the groups of patients and their control groups.
Clinical phenotype and genetic testing
Each patient with parkinsonism was evaluated using uniform clinical assessments including the Unified Parkinson Disease Rating Scale (Fahn et al., 1987) and Hoehn and Yahr staging (Hoehn and Yahr, 1967). The Wechsler Adult Intelligence Scale III was used to assess cognitive performance. The University of Pennsylvania Smell Identification test was used to evaluate olfaction, and the results are summarized in Supplementary Table 1. To identify GBA mutations, genomic DNA was sequenced by selectively amplifying all exons and most intronic portions in three 1.7 to 3.0 kb fragments as previously described (Tayebi et al., 1998). The GBA genotypes identified in the study groups are listed in Supplementary Table 2.
Positron emission tomography
PET studies were performed with a GE Advance 3D scanner (septa retracted, 4.25 mm slice separation, 35 slices, axial field of view 15.3 cm). Presynaptic dopamine synthesis was measured with 18F-fluorodopa and regional cerebral blood flow with H215O during the same scanning session. All drugs that might impact the results were tapered appropriately; Parkinson disease medications were suspended at least 12 h prior to the PET study, and caffeine and nicotine intake at least 4 h prior. Sixty minutes before scanning, participants were pretreated with carbidopa (200 mg) to reduce peripheral 18F-fluorodopa metabolism and to increase tracer availability in the brain. Scanning started with two resting regional cerebral blood flow studies, each obtained following intravenous injection of 12 mCi of H215O. Then, a dose of 18F-fluorodopa averaging 14.8 ± 2.4 mCi was infused over 90 s, and 25 dynamic scans were acquired over 90 min. All scans were attenuation-corrected and reconstructed (32 planes, 6.5 mm full-width at half-maximum). Further processing was performed using SPM5 software (Wellcome Trust Centre for Neuroimaging, University College of London).
For regional cerebral blood flow analysis, after subtraction of background activity, the two individual H215O scans were normalized to an average template, scaled proportionally to a whole brain mean of 50, smoothed (10 mm3 full-width at half-maximum Gaussian kernel), and then averaged. 18F-fluorodopa data were aligned in-plane, co-registered to the regional cerebral blood flow scans and affine normalized. The kinetic rate constant, Ki, for 18F-fluorodopa uptake was calculated voxel-by-voxel by means of a linear fit based on the Patlak method, using the time activity curve in the cerebellum as the input function. The use of a cerebellar reference region may more accurately reflect striatal Ki values when using a reference region method, but may also result in smaller Ki values than when occipital reference regions are used (Moore et al., 2003). Voxel-wise group comparisons of Ki and regional cerebral blood flow were performed using a single general linear model for each measure that included all study and control groups. This allowed for a priori contrast tests comparing each of the study groups to its own age- and sex-matched control group, as well as comparing these differences (study versus its own control) across study groups. To test for significant deviations from the norm in individual patients, we also compared each participant separately with the entire healthy control group, using age and sex as nuisance covariates. Finally, to further evaluate individual contributions to the findings, we used volumes of interest outlined with the PickAtlas tool (SPM5) to extract individual mean Ki values from caudate and putamen for each study participant, and we extracted regional cerebral blood flow values from the maxima voxels in parietal cortex derived from the between-group comparisons. We also calculated an asymmetry index for the putaminal Ki values by dividing the value of the side with the higher Ki by that with the lower Ki. Analyses of demographic and non-parametric data were performed with SPSS18 (IBM). The significance level was set at P < 0.05 family-wise error corrected for multiple comparisons. Unless mentioned explicitly, all P values given were family-wise error-corrected and figures are displayed at a height threshold of T = 3.17.
Results
Clinical findings in patients with parkinsonism
Disease duration, Unified Parkinson Disease Rating Scale scores and Hoehn and Yahr stage scores were similar in the two groups of patients with parkinsonism (Table 2). Overall Wechsler Adult Intelligence Scale III IQ scores did not differ between the two groups (t = 0.69, P = 0.52), but in the Parkinson disease–Gaucher disease group, one patient scored >20 points below the estimated premorbid verbal IQ and another was found to have impaired verbal memory. More patients with both Parkinson and Gaucher diseases first presented with right-sided symptoms (Table 2).
Table 2.
Disease duration, presentation and motor and cognitive scores for the Parkinson disease groups
| Parkinson and Gaucher diseases | Parkinson disease without GBA mutations | |
|---|---|---|
| Parkinson disease duration (years) | 4.4 ± 3.1 | 5.8 ± 3.5 |
| Initial right-sided symptom | 57% | 18%* |
| Hoehn & Yahr stage | 2.4 ± 0.7 | 1.9 ± 0.7 |
| UPDRS | 27.4 ± 8.2 | 27.5 ± 10.5 |
| IQ (WAIS III) | 97.3 ± 8.4a | 108.6 ± 35.6 |
a One subject had only a verbal IQ.
*P < 0.05.
Values are presented as mean ± SD. UPDRS = Unified Parkinson Disease Rating Scale; WAIS III = Wechsler Adult Intelligence Scale III.
Dopamine synthesis in patients with parkinsonism
The decrease in striatal dopamine synthesis was similar in both Parkinson disease groups (Fig. 1, Table 3 and Supplementary Tables 3 and 4), with greatest reduction in the caudal striatum (putamen, mean Ki reduction: 44% in patients with both Parkinson and Gaucher diseases and 42% in non-GBA Parkinson disease), and less reduction in the caudate (20% and 18% loss). Findings related to laterality corresponded to the initial site of presentation in both groups (Table 3): patients with both Parkinson and Gaucher diseases showed a larger area of decreased dopamine synthesis in the left (cluster size: 2219 voxels) compared with the right (2146) hemisphere, while the opposite was true in patients with Parkinson disease without GBA mutations (left: 1932; right: 2948). Both groups showed greater asymmetry in putaminal Ki than their control groups. However, comparison of the two Parkinson disease groups, corrected by comparison with their respective control group, failed to show any difference in dopamine synthesis, even when the statistical threshold was made more liberal (P < 0.01 uncorrected).
Figure 1.

Ki values. Parametric comparison of Parkinson disease groups with their respective control groups. Colour bar indicates t values. Displayed at height threshold T = 6.26 (P < 0.001 family-wise error-corrected) to highlight areas of greatest contrast. Dopamine synthesis is decreased mainly in the tail of the putamen, with a similar distribution in both groups. GD = Gaucher disease; PD = Parkinson disease.
Table 3.
Regions of decreased dopamine synthesis (Ki values) or decreased regional cerebral blood flow in study groups compared with their respective control groups
| Neuroimaging measurement and study group | Region | Side | k | T | P-value | Maxima voxel | |
|---|---|---|---|---|---|---|---|
| Cluster-level Corrected | Voxel-level FWE-corrected | MNI coordinates (x, y, z) | |||||
| [18F]-Fluorodopa, Ki values | |||||||
| Parkinson and Gaucher diseases (Fig. 1) | Striatum | Right | 2146 | 13.1 | P < 0.0001 | P < 0.0001 | 32, −2, 2 |
| Striatum | Left | 2219 | 11.4 | P < 0.0001 | P < 0.0001 | −26, 2, 0 | |
| Parkinson disease without GBA mutations (Fig. 1) | Striatum | Right | 2948 | 14.1 | P < 0.0001 | P < 0.0001 | 32, −4, 2 |
| Striatum | Left | 1932 | 11.6 | P < 0.0001 | P < 0.0001 | −28, −8, 2 | |
| Gaucher disease, no Parkinson disease (Fig. 2) | Putamen | Left | 467 | 4.28 | P = 0.012 | P = 0.33 | −32, −2, −8 |
| H215O, relative regional cerebral blood flow | |||||||
| Parkinson and Gaucher diseases (Fig. 3) | Lateral parietal association cortex | Left | 5823 | 7.27 | P < 0.0001 | P = 0.0002 | −50, −54, 50 |
| Lateral parietal association cortex | Right | 3251 | 5.74 | P < 0.0001 | P = 0.001 | 36, −58, 64 | |
| Precuneus | Mid | 959 | 5.32 | P < 0.0001 | P = 0.006 | 0, −70, 40 | |
| Parkinson and Gaucher diseases compared with Parkinson disease without GBA mutationsa (Supplementary Fig. 2) | Precuneus | Mid | 782 | 4.66 | P = 0.011 | P = 0.065 | −2, −64, 48 |
| Inferior parietal lobule | Left | 547 | 4.61 | P = 0.038 | P = 0.076 | −50, −52, 52 | |
a This comparison was corrected by the comparison of these two patient groups with their respective control groups.
k = cluster size at height threshold T = 3.17; T = value of the Student’s t statistic; FWE = family-wise error.
Dopamine synthesis in participants without parkinsonism
In the group of patients with Gaucher disease and no parkinsonism, 18F-fluorodopa uptake was also decreased bilaterally in the striatum, reaching corrected significance (P = 0.012, cluster-corrected) in the tail of the left putamen (Table 3 and Fig. 2). This effect can be attributed to the contribution of two patients, with ages in the third and fifth decades, with 16 and 29% reduction in mean putaminal Ki, respectively, compared with their control group. While neither had increased putaminal Ki asymmetry, their mean putaminal Ki values fell two standard deviations (SD) below the mean of their control group. The younger had predominant involvement of the anteroventral putamen (Supplementary Fig. 1).
Figure 2.

Ki values. Parametric comparison of patients with Gaucher disease without parkinsonism and their controls. On the SPM ‘glass brains’ (left) and on a standard single subject template (right), striatal regions where the group of patients with Gaucher disease but no clinical manifestations of parkinsonism had lower Ki values than their control group (Table 3). PD = Parkinson disease.
Among the asymptomatic GBA mutation carriers, Ki values and striatal symmetry did not differ from their control group in either voxel based or volume of interest analyses. All control groups had similar Ki values.
Resting regional cerebral blood flow
Resting regional cerebral blood flow was significantly decreased only in the patients with Gaucher disease and parkinsonism (Table 3 and Supplementary Table 4). This group had lower regional cerebral blood flow values than its control group in both inferior parietal lobules, more pronounced on the left, and in the precuneus of both hemispheres but sparing the posterior cingulate gyrus (Fig. 3). This pattern of decreased regional cerebral blood flow was also observed when patients with both Parkinson and Gaucher diseases were compared, through their respective control groups, with patients with sporadic Parkinson disease (Supplementary Fig. 2). Among individual patients with both Parkinson and Gaucher diseases, all but two showed decreased regional cerebral blood flow in dorsal precuneus; this was not seen in any of the subjects with Parkinson disease without GBA mutations. Regional cerebral blood flow values extracted from the precuneus maximum voxel correlated significantly with Wechsler Adult Intelligence Scale III IQ scores in patients with parkinsonism (Spearman ρ = 0.52, P = 0.04), but there was no significant correlation of IQ with values extracted from the left lateral parietal maximum voxel (Spearman ρ = 0.38, P = 0.11). Among patients with sporadic Parkinson disease, regional cerebral blood flow did not differ significantly from that measured in their control group, but there was a trend (P = 0.075 cluster corrected) for decreased regional cerebral blood flow in the right inferior parietal lobule (Supplementary Fig. 3). In patients with Gaucher disease but without Parkinson disease, including the two patients with decreased putaminal dopamine synthesis, and in GBA mutation carriers, regional cerebral blood flow was normal. The four control groups did not differ in regional cerebral blood flow.
Figure 3.
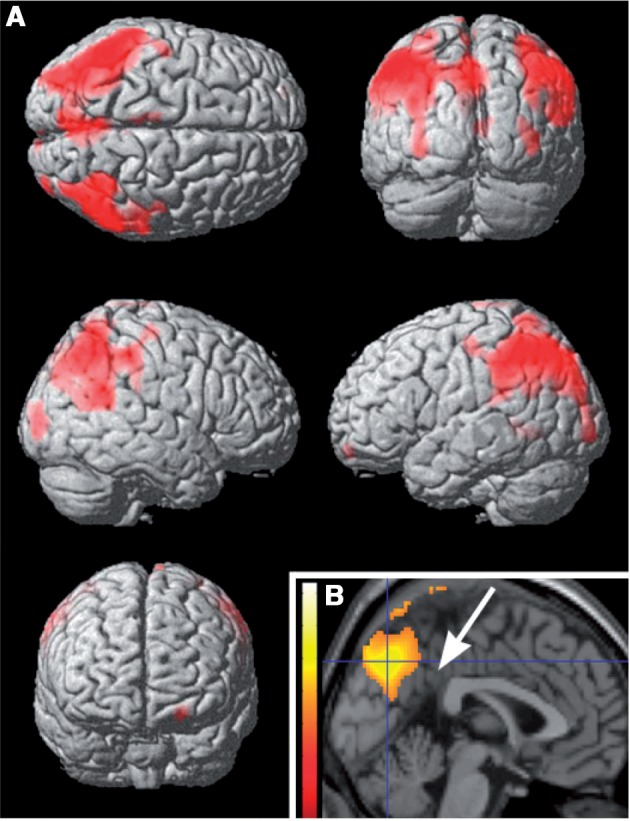
Regional cerebral blood flow in the group with both Parkinson and Gaucher diseases group compared with its control group. Regions with lower regional cerebral blood flow in the group with Parkinson and Gaucher diseases (Table 3), displayed on a rendered template in A and on a midsagittal section of a standard brain in B. Note that the precuneus is involved, but the posterior cingulate, indicated by the arrow in B, is not (‘cingulate island’ sign).
Discussion
Multimodality imaging of patients with Gaucher disease and parkinsonism yielded several findings that clarify the neurobiology of this disorder and suggest new avenues for further research. First, the pattern of dopamine synthesis in the striatum resembled the pattern seen in patients with Parkinson disease without GBA mutations: the putamen was more affected than the caudate. Reduced synthesis of dopamine in the putamen reflects the greater involvement of the neurons located more laterally in the pars compacta of the substantia nigra and is characteristic of Parkinson disease (Stoessl et al., 2011). Second, patients with Gaucher disease who lacked symptoms of Parkinson disease had, as a group, reduced putaminal dopamine synthesis. This effect was largely driven by two patients who had reduced dopamine synthesis in the putamen. The third finding of our study bears more directly on the greater prevalence of cognitive impairment in GBA-associated parkinsonism: subjects with Gaucher disease–Parkinson disease had decreased regional cerebral blood flow in the association cortex of the parietal convexity and in the precuneus, a pattern characteristic of diffuse Lewy body disease, which is accompanied by cognitive impairment (Lim et al., 2009; Silbert and Kaye, 2010; Compta et al., 2011).
The differential susceptibility of various midbrain neuronal groups to Parkinson disease is one of the most fascinating facets of the disease and is of interest because of its possible implications for the understanding of the pathophysiology and treatment of Parkinson disease (Obeso et al., 2010). The greater survival of medial midbrain neurons could relate to intrinsic properties of the neurons themselves, such as presenting receptors and cytoplasmic machinery, or to differential susceptibility to environmental and/or genetic interactions. Our study suggests that GBA mutations do not alter this differential susceptibility. Parkinson disease associated with mutations in other genes, such as SNCA or LRRK2 (Paisan-Ruiz et al., 2004), also has a pattern of reduced dopaminergic function indistinguishable from ‘sporadic’ Parkinson disease (Stoessl et al., 2011). Thus, PET imaging provides an incisive means to probe the distribution of nigral neuronal loss in vivo in GBA-associated Parkinson disease, which could be further validated through histological analysis of midbrain tissue post mortem.
Among the seven GBA mutation carriers and 14 patients with Gaucher disease but without Parkinson disease studied, all with a strong family history of Parkinson disease, only two (10%) had significantly reduced levels of dopamine synthesis. While long-term follow-up will be critical in this group, this observation may offer some reassurance to such at-risk individuals, and clearly reinforces that GBA mutations alone are not predictive of alterations in dopamine. Other genetic or environmental factors are likely to act in concert to confer the observed disease risk. Moreover, low dopamine synthesis is not necessarily predictive of developing Parkinson disease. 18F-fluorodopa PET has been used since the early 1990s to clarify the role of genetics in Parkinson disease. Applying this technique to ‘sporadic’ Parkinson disease revealed a concordance of 75% in clinically discordant monozygotic twins (Piccini et al., 1999). By measuring the rate of progression of dopamine loss in the striatum, it was calculated that the mean period between detecting decreased dopamine synthesis with PET and the onset of clinical manifestations of Parkinson disease was generally shorter than 7 years (Morrish et al., 1998). However, asymptomatic individuals carrying Parkinson disease risk mutations may behave very differently. For instance, decreased dopamine synthesis on PET has been reported in heterozygous carriers of parkin mutations (Khan et al., 2005). However, after a 5-year follow-up, only a 0.56% annual reduction in putamen 18F-fluorodopa uptake was observed, compared with the 9–12% annual loss of putamen 18F-fluorodopa uptake reported for sporadic Parkinson disease, suggesting that parkin mutation carriers with decreased dopamine synthesis are unlikely to develop clinical Parkinson disease (Pavese et al., 2009). In our patients with Gaucher disease, especially the two exhibiting abnormal dopamine synthesis, longitudinal follow-up is needed to determine the rate of 18F-fluorodopa change and whether clinical Parkinson disease develops. Subjects are being monitored for premotor features of Parkinson disease and neurocognitive changes at regular intervals. Testing whether dopamine imaging can distinguish patients with GBA mutations less likely to develop Parkinson disease from those who already have neural changes suggestive of a higher Parkinson disease risk is crucial because identifying presymptomatic stages will facilitate the evaluation of therapies aimed at preventing neurodegeneration. As with other neurodegenerative disorders such as Alzheimer’s disease, the emphasis in Parkinson disease treatment research has shifted from purely symptomatic or replacement strategies to neuroprotection of at-risk individuals (Postuma et al., 2010).
A striking finding in our study was the pattern of decreased parietal/precuneus regional cerebral blood flow, reflecting decreased synaptic activity, in the group with Parkinson and Gaucher diseases compared with those with sporadic Parkinson disease. Both Parkinson disease groups showed the pattern of reduced biparietal resting activity characteristic of Parkinson disease (Ma et al., 2007) (Fig. 3 and Supplementary Fig. 3). However, this reduction was significantly more pronounced in GBA mutation carriers and, as characteristically found in diffuse Lewy body disease, it involved the precuneus but spared the mid and posterior portions of the cingulate gyrus (‘cingulate island sign’, Fig. 3), which is affected in Alzheimer’s disease, a disorder with an otherwise similar regional cerebral blood flow pattern (Lim et al., 2009). In the group with sporadic Parkinson disease, regional cerebral blood flow was not reduced in the precuneus (Supplementary Fig. 3). Interestingly, regional cerebral blood flow in the precuneus, but not in lateral parietal cortex, was correlated with IQ, suggesting that involvement of the precuneus was critical in defining the GBA-associated pattern. This notion is further supported by inspection of individual cases: all but two patients with Gaucher disease and Parkinson disease had significantly decreased regional cerebral blood flow in the precuneus, but this was not seen in any of the patients with sporadic Parkinson disease. This observation suggests that Parkinson disease–Gaucher disease has features characteristic of diffuse Lewy body disease, a finding supported by both clinical and pathological studies of GBA-associated Parkinson disease (Goker-Alpan et al., 2008; Neumann et al., 2009). Neuropathological studies of subjects with Parkinson disease and GBA mutations demonstrate that Lewy bodies are not restricted to the nigra, as in classical Parkinson disease, but can be disseminated in the cortex, and involve hippocampal regions CA2–4 (Wong et al., 2004). Damage of neurons in these regions, which project heavily through the parahippocampus to the parietal association cortex (Lavenex et al., 2002), would reduce synaptic activity, and thereby regional cerebral blood flow, in the parietal cortex, as we found in the subjects with both Parkinson and Gaucher diseases. Dorsal precuneus, the portion affected in our group with Parkinson and Gaucher diseases, is active during visual working memory (Kochan et al., 2011); involvement of this region explain the impairment in visual working memory found in GBA-associated Parkinson disease (Alcalay et al., 2012).
Although the mechanism for the association between GBA mutations and parkinsonism is still unknown, proposed mechanisms include a gain-of-function role for mutated glucocerebrosidase enhancing α-synuclein aggregation; an enzymatic loss-of-function, where increased lysosomal glucosylceramide impacts α-synuclein processing and clearance; and a ‘bidirectional feedback loop’ fostered by lysosomal dysfunction (Goker-Alpan et al., 2010; Mazzulli et al., 2011; Westbroek et al., 2011). The aggregation of α-synuclein into cytotoxic oligomers and insoluble amyloid fibres occurs both in GBA-associated parkinsonism and other forms of Parkinson disease (Mazzulli et al., 2011). In diffuse Lewy body disease, in addition to widespread Lewy bodies, there is prominent extracellular amyloid deposition in the brain, which seems to correlate with dementia more strongly than diffuse Lewy body pathology (Compta et al., 2011). Whether the cognitive impairment and abnormal regional cerebral blood flow observed in subjects with both Parkinson and Gaucher diseases is also associated with extracellular amyloid-β plaques is an open question. It would be of interest to quantify amyloid deposition in the brain of patients with GBA mutations, either by histological studies or in vivo using amyloid imaging (Fodero-Tavoletti et al., 2007).
The main limitation of this study is the small sample size of the group of patients with parkinsonism and Gaucher disease, and thus our results must be viewed with some caution. However, Gaucher disease is a rare disorder and our findings met corrected significance, strongly suggesting their validity. Importantly, we found that striatal dopamine loss in GBA-associated Parkinson disease does not differ from sporadic Parkinson disease. Furthermore, this study reveals systems-level neurobiological abnormalities specifically in patients with Gaucher disease and parkinsonism, which may contribute to an understanding of the earlier onset and increased cognitive impairment described in this group and support its close association with diffuse Lewy body disease. The recruitment of additional subjects and sequential follow-up of GBA mutation carriers are essential to fully establish the usefulness of PET imaging as a biomarker for identifying at-risk individuals for future therapeutic trials.
Funding
Funding was provided by the Intramural Research Programs of the National Institute of Mental Health and National Human Genome Research Institute, National Institutes of Health.
Supplementary material
Supplementary material is available at Brain online.
Acknowledgements
The PET Department and the Parkinson Clinic of the National Institutes of Health Clinical Research Center contributed to making this study possible. We also acknowledge the assistance of Aideen McInerney-Leo.
References
- Alcalay RN, Caccappolo E, Mejia-Santana H, Tang MX, Rosado L, Orbe Reilly M, et al. Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology. 2012;78:1434–40. doi: 10.1212/WNL.0b013e318253d54b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharucha NE, Stokes L, Schoenberg BS, Ward C, Ince S, Nutt JG, et al. A case-control study of twin pairs discordant for Parkinson disease: a search for environmental risk factors. Neurology. 1986;36:284–8. doi: 10.1212/wnl.36.2.284. [DOI] [PubMed] [Google Scholar]
- Brockmann K, Srulijes K, Hauser AK, Schulte C, Csoti I, Gasser T, et al. GBA-associated PD presents with nonmotor characteristics. Neurology. 2011;77:276–80. doi: 10.1212/WNL.0b013e318225ab77. [DOI] [PubMed] [Google Scholar]
- Compta Y, Parkkinen L, O’Sullivan SS, Vandrovcova J, Holton JL, Collins C, et al. Lewy- and Alzheimer-type pathologies in Parkinson disease dementia: which is more important? Brain. 2011;134:1493–505. doi: 10.1093/brain/awr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz A, Bower JH, Maraganore DM, McDonnell SK, Peterson BJ, Ahlskog JE, et al. Risk tables for parkinsonism and Parkinson disease. J Clin Epidemiol. 2002;55:25–31. doi: 10.1016/s0895-4356(01)00425-5. [DOI] [PubMed] [Google Scholar]
- Fahn S, Marsden C, Calne D, Goldstein M, editors. Recent developments in Parkinson disease. Vol. 2. Florham Park, NJ: Macmillan Health Care Information; 1987. [Google Scholar]
- Fodero-Tavoletti MT, Smith DP, McLean CA, Adlard PA, Barnham KJ, Foster LE, et al. In vitro characterization of Pittsburgh compound-B binding to Lewy bodies. J Neurosci. 2007;27:10365–71. doi: 10.1523/JNEUROSCI.0630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PT, Raichle ME, Mintun MA, Dence C. Nonoxidative glucose consumption during focal physiologic neural activity. Science. 1988;241:462–4. doi: 10.1126/science.3260686. [DOI] [PubMed] [Google Scholar]
- Goker-Alpan O, Lopez G, Vithayathil J, Davis J, Hallett M, Sidransky E. The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch Neurol. 2008;65:1353–7. doi: 10.1001/archneur.65.10.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E. Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010;120:641–9. doi: 10.1007/s00401-010-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci. 2000;20:2369–82. doi: 10.1523/JNEUROSCI.20-06-02369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology. 1967;17:427–42. doi: 10.1212/wnl.17.5.427. [DOI] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–4. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NL, Scherfler C, Graham E, Bhatia KP, Quinn N, Lees AJ, et al. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology. 2005;64:134–6. doi: 10.1212/01.WNL.0000148725.48740.6D. [DOI] [PubMed] [Google Scholar]
- Kochan NA, Valenzuela M, Slavin MJ, McCraw S, Sachdev PS, Breakspear M. Impact of load-related neural processes on feature binding in visuospatial working memory. PLoS One. 2011;6:e23960. doi: 10.1371/journal.pone.0023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono S, Shirakawa K, Ouchi Y, Sakamoto M, Ida H, Sugiura T, et al. Dopaminergic neuronal dysfunction associated with parkinsonism in both a Gaucher disease patient and a carrier. J Neurol Sci. 2007;252:181–4. doi: 10.1016/j.jns.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Kraoua I, Stirnemann J, Ribeiro MJ, Rouaud T, Verin M, Annic A, et al. Parkinsonism in Gaucher disease type 1: ten new cases and a review of the literature. Mov Disord. 2009;24:1524–30. doi: 10.1002/mds.22593. [DOI] [PubMed] [Google Scholar]
- Lavenex P, Suzuki WA, Amaral DG. Perirhinal and parahippocampal cortices of the macaque monkey: projections to the neocortex. J Comp Neurol. 2002;447:394–420. doi: 10.1002/cne.10243. [DOI] [PubMed] [Google Scholar]
- Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson disease genetics: the PDGene database. PLoS Genet. 2012;8:e1002548. doi: 10.1371/journal.pgen.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SM, Katsifis A, Villemagne VL, Best R, Jones G, Saling M, et al. The 18F-FDG PET cingulate island sign and comparison to 123I-beta-CIT SPECT for diagnosis of dementia with Lewy bodies. J Nucl Med. 2009;50:1638–45. doi: 10.2967/jnumed.109.065870. [DOI] [PubMed] [Google Scholar]
- Ma Y, Tang C, Spetsieris PG, Dhawan V, Eidelberg D. Abnormal metabolic network activity in Parkinson disease: test-retest reproducibility. J Cereb Blood Flow Metab. 2007;27:597–605. doi: 10.1038/sj.jcbfm.9600358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RY, Whone AL, McGowan S, Brooks DJ. Monoamine neuron innervation of the normal human brain: an 18F-DOPA PET study. Brain Res. 2003;982:137–45. doi: 10.1016/s0006-8993(03)02721-5. [DOI] [PubMed] [Google Scholar]
- Morrish PK, Rakshi JS, Bailey DL, Sawle GV, Brooks DJ. Measuring the rate of progression and estimating the preclinical period of Parkinson disease with [18F]dopa PET. J Neurol Neurosurg Psychiatry. 1998;64:314–9. doi: 10.1136/jnnp.64.3.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson disease. Brain. 2009;132:1783–94. doi: 10.1093/brain/awp044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, et al. Missing pieces in the Parkinson disease puzzle. Nat Med. 2010;16:653–61. doi: 10.1038/nm.2165. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Pavese N, Khan NL, Scherfler C, Cohen L, Brooks DJ, Wood NW, et al. Nigrostriatal dysfunction in homozygous and heterozygous parkin gene carriers: an (18)F-Dopa PET progression study. Mov Disord. 2009;24:2260–6. doi: 10.1002/mds.22817. [DOI] [PubMed] [Google Scholar]
- Piccini P, Burn DJ, Ceravolo R, Maraganore D, Brooks DJ. The role of inheritance in sporadic Parkinson disease: evidence from a longitudinal study of dopaminergic function in twins. Ann Neurol. 1999;45:577–82. doi: 10.1002/1531-8249(199905)45:5<577::aid-ana5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Postuma RB, Gagnon JF, Montplaisir J. Clinical prediction of Parkinson disease: planning for the age of neuroprotection. J Neurol Neurosurg Psychiatry. 2010;81:1008–13. doi: 10.1136/jnnp.2009.174748. [DOI] [PubMed] [Google Scholar]
- Rosenbloom B, Balwani M, Bronstein JM, Kolodny E, Sathe S, Gwosdow AR, et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher Registry. Blood Cells Mol Dis. 2011;46:95–102. doi: 10.1016/j.bcmd.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders-Pullman R, Hagenah J, Dhawan V, Stanley K, Pastores G, Sathe S, et al. Gaucher disease ascertained through a Parkinson center: imaging and clinical characterization. Mov Disord. 2010;25:1364–72. doi: 10.1002/mds.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto-Salvia N, Pagonabarraga J, Houlden H, Pascual-Sedano B, Dols-Icardo O, Tucci A, et al. Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson disease course. Mov Disord. 2012;27:393–9. doi: 10.1002/mds.24045. [DOI] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson disease. N Engl J Med. 2009;361:1651–61. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbert LC, Kaye J. Neuroimaging and cognition in Parkinson disease dementia. Brain Pathol. 2010;20:646–53. doi: 10.1111/j.1750-3639.2009.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoessl AJ, Martin WW, McKeown MJ, Sossi V. Advances in imaging in Parkinson disease. Lancet Neurol. 2011;10:987–1001. doi: 10.1016/S1474-4422(11)70214-9. [DOI] [PubMed] [Google Scholar]
- Tayebi N, Reissner KJ, Lau EK, Stubblefield BK, Klineburgess AC, Martin BM, et al. Genotypic heterogeneity and phenotypic variation among patients with type 2 Gaucher disease. Pediatr Res. 1998;43:571–8. doi: 10.1203/00006450-199805000-00003. [DOI] [PubMed] [Google Scholar]
- Westbroek W, Gustafson AM, Sidransky E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol Med. 2011;17:485–93. doi: 10.1016/j.molmed.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. 2004;82:192–207. doi: 10.1016/j.ymgme.2004.04.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.