

Abstract
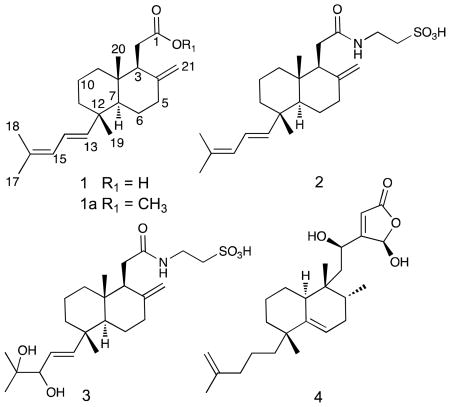
Three new bicyclic C21 terpenoids, clathric acid (1) and two N-acyl taurine derivatives, clathrimides A (2) and B (3) were isolated from the marine sponge Clathria compressa. The structures of these compounds were elucidated by interpretation of spectroscopic data. Clathric acid showed mild antibacterial activity against several Gram-positive bacteria.
Marine sponges of the genus Clathria are an abundant source of novel secondary metabolites exhibiting various biological activities and unusual chemical structures. Compounds that represent a variety of different classes have been reported including alkaloids,1 carotenoids,2 lipids,3 peptides,4 sterols,5 sugars,6 and terpenoids.7 In the course of screening pre-fractionated and semi-purified extracts of marine invertebrates in an effort to discover compounds that impact human embryonic stem cell (hESCs) growth, activity was found for the extract of the sponge Clathria compressa. A large-scale extraction yielded three unusual bicyclic C21 terpenoids clathric acid (1), and the N-acyl taurine derivatives clathrimide A (2) and B (3). We report here the isolation, structural elucidation and biological activity of these compounds. In addition we also propose that the biogenetic origin of these compounds could be from the degradation of a related γ-hydroxybutenolide sesterterpenoid.
The specimen of Clathria compressa was collected from Panama City, Florida and kept frozen until extraction. The methanolic extract was first fractionated on polymeric HP20 resin using the cyclic loading method.8 The HP20 column was eluted with 250 mL fractions of (1) 40% Me2CO/H2O (2) 75% Me2CO/H2O and finally with Me2CO. The 75% Me2CO/H2O fraction was then subjected to reversed phase HPLC on a C-18 column to obtain clathric acid (1) and clathrimide A (2). The 40% Me2CO/H2O fraction was further chromatographed on HP20SS to give clathrimide B (3).
Clathric acid (1) was obtained as a yellow amorphous powder. The molecular formula of clathric acid (1), C21H32O2, that was determined from the HRESIMS of the [M + Na]+ ion at m/z 339.2307, required six degrees of unsaturation. An initial examination of the 13C NMR data revealed one carboxylic acid (δC 180.0; IR = 1705 cm−1), and three C-C double bonds (δC 151.3, 147.0, 133.5, 127.3, 124.4, and 107.6). These data accounted for four of the six double bond equivalents, and indicated that 1 was bicyclic. An initial analysis of the NMR data (Table 1) revealed the presence of a conjugated diene with the 1H NMR spectrum revealing three olefinic proton signals at δC 6.12 (1H, dd, J = 15.5, 10.5 Hz), 5.77 (1H, d, J = 10.5 Hz), and 5.37 (1H, d, J = 15.5 Hz) that showed HSQC correlations to the olefinic carbon signals at δC 124.4 (C-14), 127.3 (C-15), and 147.0 (C-13), respectively. The observation of a UV absorption maximum at λmax = 238 nm was consistent with this assignment. HMBC correlations from the olefinic methyl signals at δC 1.75 (Me-17) and δC 1.74 (Me-18) to both C-15 (δC 124.4), a quaternary carbon at C-16 (δC 133.5) and to each other’s carbon resonance established methyl substitution of C-16 (Figure 1). The E geometry of the C-13–C-14 double bond was evident from the large 1H coupling constant (J = 15.5 Hz) between H-13 and H-14.
Table 1.
NMR Spectroscopic Data for Clathric Acid (1)a
| position | δC, mult. | δH(J in Hz) | HMBCb | ROESY |
|---|---|---|---|---|
| 1 | 180.0, C | |||
| 2a | 33.5, CH2 | 2.45, d (12.8) | 1, 3, 4, 8 | 2b, 21b, H3-20 |
| 2b | 2.33, d (12.8) | 2a, 21b | ||
| 3 | 55.1, CH | 2.35, bs | 4, 7, 8, 20 | |
| 4 | 151.3, C | |||
| 5a | 39.2, CH2 | 2.37, m | 3, 4, 6, 21 | 5b, 6a, 6b |
| 5b | 2.06, m | 5a | ||
| 6a | 26.6, CH2 | 1.60, m | 4, 8, 12 | 13 |
| 6b | 1.34, m | 5a, H3-20 | ||
| 7 | 55.3, CH | 1.32, bs | 3, 9, 19, 20 | 13 |
| 8 | 40.2, C | |||
| 9a | 40.1, CH2 | 1.69, dt (12.5, 3.0) | 9b, 10b, H3-20 | |
| 9b | 1.23, td (12.5, 3.0) | 2, 7 | 9a | |
| 10a | 20.5, CH2 | 1.65, m | 8, 12 | |
| 10b | 1.56, m | 9a, H3-19, H3-20 | ||
| 11a | 42.3, CH2 | 1.39, m | 7, 9, 19 | |
| 11b | 1.39, m | |||
| 12 | 41.4, C | |||
| 13 | 147.0, CH | 5.37, d (15.5) | 7, 11, 15, 19 | 6a, 7, 15 |
| 14 | 124.4, CH | 6.12, dd (15.5, 10.5) | 12, 15, 16 | H3-18, H3-19 |
| 15 | 127.3, CH | 5.77, d (10.5) | 13, 17, 18 | 13, H3-17 |
| 16 | 133.5, C | |||
| 17 | 26.6, CH3 | 1.75, s | 15, 16, 18 | 15 |
| 18 | 18.8, CH3 | 1.74, s | 15, 16, 17 | 14 |
| 19 | 19.2, CH3 | 1.04, s | 7, 11, 12, 13 | 10b, 14, H3-20 |
| 20 | 16.0, CH3 | 0.80, s | 3, 7, 8, 9 | 2a, 6b, 9a, 10b, H3-19 |
| 21a | 107.6, CH2 | 4.72, bs | 3, 5 | 21b |
| 21b | 4.60, bs | 3, 5, 4 | 2a, 2b, 21a |
In CD3OD, 500 MHz for 1H and 125 MHz for 13C NMR.
HMBC correlations are from proton(s) stated to the indicated carbons.
Figure 1.

Selected 2D NMR correlations for clathric acid (1).
The backbone of the decalin ring system was defined using HMBC correlations from the two methyl signals δH 1.04 and δH 0.80, which were singlets in the 1H NMR spectrum, and the proton signals of the exocyclic methylene (δH 4.72, 4.60, δC 107.6) to the methine carbon signals (H3-19 & H3-20/C-7/H3-20/C-3/CH2-21). The methyl H3-19 was also coupled to C-11, H3-20 was coupled to C-8 and C-9, and CH2-21 was coupled to C-4 and C-5. The COSY spectrum indicated coupling from H2-5 to H2-6 and H-7 and from H2-9 to H2-10 and H2-11, thereby defining the decalin ring system. An additional HMBC correlation from H3-19 to the olefinic carbon C-13 allowed connection of the conjugated diene to the decalin ring system.
Remaining to be assigned were a methylene group CH2-2 (δH 2.45, 2.33; δC 33.5) and the carboxylic acid carbon C-1 (δC 180.0). HMBC correlations from the H2-2a signal to C-3, C-4, C-8 and the carboxylic acid carbon at δ 180.0 (C-1) established the connection of the C-2 to C-3 and C-2 to C-1. To further confirm the presence of the carboxylic acid, compound 1 was methylated with CH2N2 resulting in the formation of the methyl ester 1a. The 1H NMR spectrum of 1a contained an additional methoxy signal at δH 3.64. Compound 1a showed an [M + H]+ ion at m/z 331.4 in the ESIMS corresponding to the molecular formula C22H34O2, one carbon and two hydrogens greater than clathric acid (1).
The relative configuration of 1 was determined by NOE correlations observed in a NOESY experiment (Figure 2). NOE correlations from H3-20 to H-6b, H-10b, and H3-19, together with correlations from H3-19 to H-6b, H10b and H3-20 established the trans-fused nature of the decalin ring system. An additional NOE correlation observed from H3-19 to H-14 together with correlations observed from H-13 to H-6a and H-7 established the equatorial orientation of the diene side chain at C-12. The observation of a long range W-coupling in the COSY spectrum between H3-19 and H-13 was consistent with this assignment. In a similar fashion, NOE correlations observed from H3-20 to H2-2a and from H2-2a to H2-21b established the equatorial arrangement of the carboxylic acid side chain at C-3. Thus the structure of clathric acid (1) is therefore defined as 3S*,7S*,8R*,12S*,13E.
Figure 2.

Selected NOE correlations observed for clathric acid (1).
Clathrimide A (2) was obtained as a yellow solid. The molecular formula of clathrimide A (2), C23H37NO4S, was determined by HRESIMS. The presence of an S=O stretching bands at 1202 and 1027 cm−1 in the IR spectrum and a significant [M+2] peak in the mass spectrum were consistent with presence of a sulfate group. A comparison of the NMR data (Table 2) revealed that 2 was very similar to 1, except for the presence of an additional A2X2 spin system [δH 2.92 (2H, t, J = 6.8 Hz), δc 51.5 (CH2); δH 3.55 (2H, t, J = 6.8 Hz), δc 36.7 (CH2)] consistent with a taurine group.9 This suggested that the clathrimide A (2) was the N-acyl taurine derivative of 1. An HMBC correlation observed between H2-1′ (δH 3.55) of the taurine group and the amide carbonyl carbon at δC 175.9 (C-1; IR = 1645 cm−1) confirmed the connection of the taurine to the terpenoid skeleton. The similarity of proton–proton coupling constants and 1H and 13C chemical shifts together with a ROESY spectrum of 2 showed the same relative configuration as that of clathric acid (1).
Table 2.
NMR Spectroscopic Data for Clathrimides A (2) and B (3)a
| 2 | 3 | |||
|---|---|---|---|---|
|
| ||||
| Position | δC, mult | δH(J in Hz) | δC, mult | δH(J in Hz) |
| 1 | 175.9, C | 175.9, C | ||
| 2a | 33.2, CH2 | 2.39, d (14.6) | 33.2, CH2 | 2.39, d (14.5) |
| 2b | 2.29, d (14.6) | 2.25, d (14.5) | ||
| 3 | 53.9, CH | 2.37, bd (3.6) | 53.9, CH | 2.39, bd (4.0) |
| 4 | 150.6, C | 150.5, C | ||
| 5a | 39.8, CH2 | 2.30, m | 39.8, CH2 | 2.30, m |
| 5b | 2.03, m | 2.05, m | ||
| 6a | 26.2, CH2 | 1.55, m | 25.2, CH2 | 1.65, m |
| 6b | 1.32, m | 1.35, m | ||
| 7 | 54.8, CH | 1.35, dd (11.2, 3.2) | 54.8, CH | 1.36, m |
| 8 | 38.8, C | 38.8, C | ||
| 9a | 39.8, CH2 | 1.65, bd (12.4) | 40.8, CH2 | 1.68, dt (13.1, 3.5) |
| 9b | 1.19, bd (12.4) | 1.21, td (13.0, 3.3) | ||
| 10a | 20.1, CH2 | 1.58, m | 20.1, CH2 | 1.69, bd (10.5, 3.3) |
| 10b | 1.52, m | 1.53, bd (10.5, 3.5) | ||
| 11a | 41.9, CH2 | 1.30, m | 42.6, CH2 | 1.51, dd (12.8, 3.5) |
| 11b | 1.29, m | 1.32, dd (12.8, 3.5) | ||
| 12 | 41.1, C | 40.9, C | ||
| 13 | 146.5, CH | 5.35, d (15.2) | 148.2, CH | 5.53 (d, 15.6) |
| 14 | 124.4, CH | 6.13, dd (15.2, 10.0) | 126.9, CH | 5.40 (dd, 15.6, 7.6) |
| 15 | 126.9, CH | 5.76, d (10.0) | 81.2, CH | 3.78 (d, 7.6) |
| 16 | 133.2, C | 73.9, C | ||
| 17 | 26.2, CH3 | 1.74, s | 26.2, CH3 | 1.11, s |
| 18 | 18.8, CH3 | 1.71, s | 18.7, CH3 | 1.12, s |
| 19 | 18.4, CH3 | 1.09, s | 18.7, CH3 | 0.91, s |
| 20 | 15.7, CH3 | 0.77, s | 15.6, CH3 | 0.77, s |
| 21a | 107.4, CH2 | 4.73, bs | 107.5, CH2 | 4.73, bs |
| 21b | 4.55, bs | 4.55, bs | ||
| 1′ | 36.7, CH2 | 3.55, t (6.8) | 36.7, CH2 | 3.55, t (6.8) |
| 2′ | 51.5, CH2 | 2.92, t (6.8) | 51.5, CH2 | 2.92, t (6.8) |
In CD3OD, 400 MHz for 1H and 100 MHz for 13C NMR.
Clathrimide B (3) was obtained as a yellow solid. The molecular formula of clathrimide A (2), C23H39NO6S, that was determined from the HRESIMS data, required five degrees of unsaturation. A comparison of the 1H and 13C NMR data (Table 2) revealed that 3 was similar to 2, except for changes in the proton and carbon chemical shifts of the diene side chain that suggested oxidation of the C-15–C-16 double bond to a diol. HMBC correlations from the methyl signals at δC 1.11 (3H, s, Me-17) and δH 1.12 (3H, s, Me-18) to both C-15 (δC 81.2), an oxygenated quaternary carbon at C-16 (δC 73.9) and to each other’s carbon resonance confirmed the presence of a diol. A COSY correlation observed between H-15 and H-14 together with HMBC correlations from H-15 to C-14 and C-13 further confirmed the assignment of the side chain.
The relative configuration of clathrimide B (3) was determined from NOE enhancements observed in a ROESY experiment. Due to the flexibility of the side chain, the ROESY experiment was unable to establish the configuration of C-15 relative to the decalin ring system. The configuration of clathrimide B (3) is therefore defined as 3S*,7S*,8R*,12S*,13E,15S* or 3S*,7S*,8R*,12S*,13E,15R*.
Compounds 1 – 3 were evaluated for cell growth inhibitory activities against human embryonic stem cells (BG02) using a 96-well plate real-time cell electronic sensing (RT-CES) system.10 No inhibitory activity was detected for any of the isolated compounds at 40 μM. The compounds were also examined for antimicrobial activity against Gram-positive and Gram-negative bacteria using a broth microdilution assay.11 Clathric acid (1) showed a minimum inhibition concentration (MIC) of 32 Ug/mL against Staphylococcus aureus (ATTC 6538P) and 64 μg/mL against both methicillin-resistant (ATTC 33591) and vancomycin-resistant Staphylococcus aureus (VRSA). Clathrimides A (2) and B (3) showed no activity at concentrations up to 128 μg/mL. None of the compounds were active against the Gram-negative bacteria Escherichia coli (KCTC 1923) and Klebsiella pneumonia (ATCC 10031) when tested at 128 μg/mL.
Compounds 1 – 3 are unusual C21 terpenoids with the same bicyclic arrangement seen in the sesterterpenoid dysideapalaunic acid12 and similar to the related γ-hydroxybutenolide sesterterpenoids cladocorans A and B,13,14 and dysidiolide (4).15 A number of degraded C21 terpenoids have been reported from marine sponges. Most of these are linear furanoterpenoids found in sponges belonging to the family Thorectidae and Spongiidae.16,17 It has been proposed that the biogenetic origin of these compounds originates from the hydrolysis of a related C25 tetronic acid to a 1,2-diketone followed by an oxidative cleavage to give a carboxylic acid.18 We speculate that compounds 1 – 3 could be derived from a related γ-hydroxybutenolide containing sesterterpenoid such as dysidiolide (4) via a similar hydrolysis and oxidative cleavage mechanism of tetronic acids or by a retro-aldol and oxidation sequence similar to the degradation of sugars and ascorbic acid.19 The proposed biogenetic origin is analogous to that proposed for the C21 terpenoid cavernolide from the marine sponge Fasciospongia cavernosa that was co-isolated with the related γ-hydroxybutenolide sesterterpenoid cacospongionolide B.20
Experimental Section
General Experimental Procedures
Optical rotations were measured on a Jasco P-2000 polarimeter (c g/100 mL) at 589 nm. UV spectra were obtained on a Perkin-Elmer Lambda EZ 210 UV–vis spectrophotometer. IR spectra were recorded on a Thermo Electronic Corporation Nicolet IR-100 spectrophotometer. All NMR spectra were recorded a Varian Unity-INOVA 400 or 500 spectrometer. All chemical shifts (δ) were referenced internally to the residual solvent peak (CD3OD: 1H, δ 3.30: 13C, δ 49.0; CDCl3: 1H 7.26 ppm; 13C 77.0 ppm). Short- and long-range 1H-13C correlations were determined with gradient-enhanced inverse-detected HSQC and HMBC experiments respectively. NOE correlations were detected with ROESY experiments with a 0.5 s mixing time. The high-resolution ESI mass spectra performed on an APEX II FTICR mass spectrometer equipped with a 4.7 T magnet (Bruker-Daltonics) were obtained from the University of Georgia Proteomic and Mass Spectrometry Core Facility. HPLC purifications were performed on Beckman System Gold HPLC system with a 168 UV detector and a SEDEX 85 (Sedere) evaporative light scattering detector. Thin layer chromatography (TLC) analyses were performed using Merck Kieselgel (Aufoilen) 60 F254 plates. TLC plates were visualized by spraying with 1:1 MeOH:H2SO4.
Biological Material
The sponge Clathria compressa (Schmidt, 1862) was collected by hand using SCUBA at a depth of 15–20 m at Panama City Beach, Florida. The specimen was immediately frozen and kept at −20 °C until extraction. A voucher specimen (PC01-024) has been deposited in the Department of Chemistry and Biochemistry, Florida Atlantic University, Boca Raton, Florida.
Extraction and Purification Procedures
The specimen of C. compressa (120 g wet wt.) was extracted with MeOH (3 × 350 mL) for 18 h. The third, second and then the first extracts were passed through a column of HP20 resin (2.5 × 25 cm) equilibrated with MeOH. The combined eluent was diluted with H2O (3.0 L) and passed again through the column. The column was eluted with 250 mL fractions of (1) H2O, (2) 40% Me2CO/H2O, (3) 75% Me2CO/H2O and (4) Me2CO. Fraction 3 was back-loaded onto an HP20 column to remove the H2O by passing the fraction through a column of HP20 resin (2.5 × 8.0 cm) equilibrated with H2O. The eluent was diluted with H2O (500 mL) and passed again through the column. The column was eluted with Me2CO (250 mL), and then 50% MeOH/Me2CO (250 mL), and the combined fractions concentrated to dryness. Fraction 3 was subjected to semi preparative C18 reversed phase HPLC (Gemini 5 μm; 10 × 250 mm; 4 mL/min; 20–100% CH3CN/H2O over 60 min) to give 1 (25.0 mg) and 2 (19.0 mg). Fraction 2 was concentrated to dryness and was subjected to column chromatography on HP20SS resin eluting with 50 mL fractions of (1) H2O, (2) 20% Me2CO/H2O, (3) 30% Me2CO/H2O, (4) 40% Me2CO/H2O, (5) 50% Me2CO/H2O, (6) 60% Me2CO/H2O and (7) 75% Me2CO/H2O to afforded compound 3 (15.2 mg) in fraction 4.
Clathric acid (1)
Yellow solid; [α]25D + 15.0 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 238 (4.36) nm; IR (Neat) νmax 3500, 2926, 1705, 1658, 1541, 1248 cm−1; 1H and 13C NMR (500 MHz, CD3OD), see Table 1. HRESIMS m/z 339.2307 [M + Na]+ (calcd for C21H32O2Na, 339.2300).
Methylation of Clathric Acid (1)
Clathric acid (1) (3.0 mg) was dissolved in MeOH (1 mL), and a solution of CH2N2 in EtOEt (1 mL) was added. The mixture was kept for 1 h in the dark and was then dried under nitrogen to give methyl ester 1a: 1H NMR (CDCl3) δ 6.12 (1H, dd, J = 15.5, 10.5 Hz, H-14), 5.82 (1H, d, J = 10.5 Hz, H-15), 5.40 (1H, d, J = 15.5 Hz, H-13), 4.75 (1H, s, H-21a), 4.50 (1H, s, H-21b), 3.65 (3H, s, OMe), 1.77 (3H, s, H-17), 1.76 (3H, s, H-18), 1.01 (3H, s, H-19), 0.74 (3H, s, H-20); ESIMS m/z 331.4 [M + H]+.
Clathrimide A (2)
Colorless oil; [α]25D + 11 (c 0.02, MeOH); UV (MeOH) λmax (log ε) 238 (4.05) nm; IR (Neat) νmax 2925, 2854, 1645, 1541, 1456, 1202, 1027 cm−1; 1H and 13C NMR (400 MHz, CD3OD) see Table 2. HRESIMS m/z 446.2325 [M + Na]+ (calcd for C23H37NO4SNa, 446.2336).
Clathrimide B (3)
Colorless oil; [α]25D −36 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 204 (3.77) nm; IR (Neat) νmax 3401, 2924, 2854, 1647, 1540, 1460, 1374, 1022 cm−1; 1H and 13C NMR (400 MHz, CD3OD), see Table 2. HRESIMS m/z 480.2373 [M + Na]+ (calcd for C23H39NO6SNa, 480.2390).
Antimicrobial Assay
Bacterial strains were obtained from the American Type Culture Collection (ATCC). After culturing all cells on Müller-Hinton agar at 37 °C for 24 h, the cells were suspended in Müller-Hinton broth and incubated at 37 °C for 24 h. The determinations of minimal inhibitory concentration (MIC) were done in 96-well microtiter plates using the standard microdilution broth method in sterilized 96-well flat bottomed polystyrene microtiter plates.20 Controls on each plate were media without bacteria, bacterial inoculums without antimicrobial added, bacterial inoculums to which methicillin was added and bacterial inoculums to tested compounds, in the range from 0.01 to 128 μg/mL. All the test samples were dissolved in 5% of dimethyl sulfoxide (DMSO) in H2O and were loaded in duplicate. After dilutions the final concentration of DMSO in wells was less than 0.5%. To eliminate possible influence of DMSO on bacterial growth all controls were prepared in a way that the final concentration of DMSO was the same. Plates were loaded with 90 μL of mid-logarithmic phase cells with initial 600 nm VIS absorbance of 0.001 of the tested microorganism and 10 μL aliquots of two-fold serial dilutions of the antibiotics or compounds tested. Plates were read after 20 h incubation at 37 °C with gentle shaking. The inhibition of the bacterial growth was determined by measuring VIS absorbance at 600 nm.
Supplementary Material
Acknowledgments
We thank Dr. P. Cudic and N. Bionda at the Torrey Pines Institute for performing the antibacterial screening. We also thank Dr. M. Cairelli (NIH/NLM/LHC) and T. Vansach (FAU) for assistance in collection of the sponge specimens. Mr. R. Rueda de Leon is acknowledged for isolating addition quantities of clathric acid for spectral analysis. This research was supported by the National Institutes of Health Grants (P41GM079597 and P01GM085354).
Footnotes
Supporting Information Available:
1D and 2D NMR spectroscopic data of 1 – 3 are available including 1H, 13C, COSY, HSQC, HMBC, and ROESY. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Wei X, Henriksen NM, Skalicky JJ, Harper MK, Cheatham TE, Ireland CM, Van Wagoner RM. J Org Chem. 2011;76:5515–5523. doi: 10.1021/jo200327d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanaka Y, Katayama T. Nippon Suisan Gakkaishi. 1976;42:801–805. [Google Scholar]
- 3.Xiao DJ, Deng SZ, Zeng LM. Zhongshan Daxue Xuebao, Ziran Kexueban. 2002;41:111–114. [Google Scholar]
- 4.Davis RA, Mangalindan GC, Bojo ZP, Antemano RR, Rodriguez NO, Concepcion GP, Samson SC, de Guzman D, Cruz LJ, Tasdemir D, Harper MK, Feng X, Carter GT, Ireland CM. J Org Chem. 2004;69:4170–4176. doi: 10.1021/jo040129h. [DOI] [PubMed] [Google Scholar]
- 5.Keyzers RA, Northcote PT, Berridge MV. Aust J Chem. 2003;56:279–282. [Google Scholar]
- 6.Capon RJ, MacLeod JK. J Chem Soc Chem Commun. 1987:1200–1201. [Google Scholar]
- 7.Capon RJ, Miller M, Rooney FJ. Nat Prod. 2000;63:821–824. doi: 10.1021/np990644o. [DOI] [PubMed] [Google Scholar]
- 8.Houssen WE, Jaspars M. Natural Products Isolation. In: Sarker SD, Latif Z, Gray AI, editors. Methods in Biotechnology. 2. Vol. 20. Humana Press Inc; Totowa, NJ: 2006. pp. 353–391. [Google Scholar]
- 9.Clark VC, Harinantenaina L, Zeller M, Ronto W, Rocca J, Dossey AT, Rakotondravony D, Kingston DGI, Shaw C. J Org Chem. 2012;75:473–478. doi: 10.1021/np200963r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solly K, Wang X, Xu X, Strulovici B, Zheng W. Assay Drug Dev Technol. 2004;2:363–372. doi: 10.1089/adt.2004.2.363. [DOI] [PubMed] [Google Scholar]
- 11.Otvos L, Cudic M. In: Methods in Molecular Biology. Fields GB, editor. Vol. 386. Humana Press Inc; Totowa, NJ: 2000. pp. 309–320. [Google Scholar]
- 12.(a) Nakagawa M, Ishihama M, Hamamoto Y, Endo M. Abstracts of Papers. 28th Symposium on the Chemistry of Natural Products; October 1986; Sendai, Japan: Pharmaceutical Institute, Tohoku University; p. 200. [Google Scholar]; (b) Chem Abstr. 1987;106:96126b. [Google Scholar]
- 13.Fontana A, Ciavatta ML, Cimino G. J Org Chem. 1998;63:2845–2849. [Google Scholar]
- 14.Miyaoka H, Yamanishi M, Kajiwara Y, Yamada Y. J Org Chem. 2003;68:3476–3479. doi: 10.1021/jo020743y. [DOI] [PubMed] [Google Scholar]
- 15.Gunasekera SP, McCarthy PJ, Kelly-Borges M, Lobkovsky E, Clardy J. J Am Chem Soc. 1996;118:8759–8760. [Google Scholar]
- 16.Su JH, Tseng SW, Lu MC, Liu LL, Chou Y, Sung PJ. J Nat Prod. 2011;74:2005–2009. doi: 10.1021/np2004209. [DOI] [PubMed] [Google Scholar]
- 17.Fontana A, Albarella L, Scognamiglio G, Uriz M, Cimino G. J Nat Prod. 1996;59:869–872. [Google Scholar]
- 18.Gonzalez AG, Rodraguez ML, Barrientos ASM. J Nat Prod. 1983;46:256–261. [Google Scholar]
- 19.Vranová J, Ciesarová Z. Czech J Food Sci. 2009;27:1–10. [Google Scholar]
- 20.De Rosa S, Crispino A, De Giulio A, Iodice C, Tommonaro G, Pronzato R, Sidri M. Tetrahedron. 1999;55:13805–13808. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.