

Background: A prion-like spread of α-synuclein might play a role in the pathogenesis of Parkinson disease.
Results: Extracellular α-synuclein was cleaved by plasmin. Cultured microglia and astrocytes did not take up plasmin digested extracellular α-synuclein, and were not activated.
Conclusion: Plasmin-mediated α-synuclein clearance problems might play a role in the pathogenesis of Parkinson disease.
Significance: Therapies aimed at α-synuclein clearance may lead to new therapies for Parkinson disease.
Keywords: α-synuclein, Parkinson Disease, Plasmin, Protein Aggregation, Protein Degradation
Abstract
Parkinson disease (PD) is the second most common neurodegenerative disease characterized by a progressive dopaminergic neuronal loss in association with Lewy body inclusions. Gathering evidence indicates that α-synuclein (α-syn), a major component of the Lewy body, plays an important role in the pathogenesis of PD. Although α-syn is considered to be a cytoplasmic protein, it has been detected in extracellular biological fluids, including human cerebrospinal fluid and blood plasma of healthy and diseased individuals. In addition, a prion-like spread of α-syn aggregates has been recently proposed to contribute to the propagation of Lewy bodies throughout the nervous system during progression of PD, suggesting that the metabolism of extracellular α-syn might play a key role in the pathogenesis of PD. In the present study, we found that plasmin cleaved and degraded extracellular α-syn specifically in a dose- and time- dependent manner. Aggregated forms of α-syn as well as monomeric α-syn were also cleaved by plasmin. Plasmin cleaved mainly the N-terminal region of α-syn and also inhibited the translocation of extracellular α-syn into the neighboring cells in addition to the activation of microglia and astrocytes by extracellular α-syn. Further, extracellular α-syn regulated the plasmin system through up-regulation of plasminogen activator inhibitor-1 (PAI-1) expression. These findings help to understand the molecular mechanism of PD and develop new therapeutic targets for PD.
Introduction
Parkinson disease (PD)2 is the second most common neurodegenerative disease, characterized clinically by resting tremor, rigidity, bradykinesia, and pathologically by a progressive dopaminergic neuronal loss in association with Lewy body inclusions (1).
α-Synuclein (α-Syn) is an abundant presynaptic protein that is known to be a major component of the Lewy body in PD (2). Specific mutations (A30P, E46K, and A53T) and multiplication of the wild-type gene were found in some early-onset familial PD patients (3–7). Furthermore, recent genome-wide association studies identified SNCA as a common risk factor for PD (8, 9). Animal models with transgenic overexpression of α-syn were also shown to mimic several aspects of PD, suggesting that α-syn may play a major role in the pathogenesis of PD (2).
Until recently, α-syn was considered to be a cytoplasmic protein and exert its pathogenic effects in the cytoplasm of the cells. However, monomeric and oligomeric forms of α-syn have been continuously shown to be present in human cerebrospinal fluid (CSF) and blood plasma of healthy and diseased individuals (10–12). Moreover, it was also demonstrated that α-syn can be released into the extracellular media by exocytosis (13–15) and internalized into cells by several mechanisms (16–19), implying that α-syn may function extracellularly as well as in the cytoplasmic. In support of physiological and pathological roles of extracellular α-syn, recent reports showed that extracellular α-syn regulates phagocytosis of microglia in a conformation-dependent manner (20) and stimulates the production of proinflammatory factors from astrocytes and microglia (21–26), which consequently cause dopaminergic neuronal death (27).
Recently, it was reported that human nigral neurons grafted into the putamen of patients with PD display Lewy bodies that are distinguishable from Lewy bodies seen in regions of the host brain (28, 29). Also, α-syn aggregates released from neuronal cells can be transferred to neighboring neurons, forming Lewy body-like inclusions, providing mechanistic basis for the development of Lewy body pathology in normal mesencephalic transplants in PD patients (30–32). These data suggest that a prion-like mechanism might underlie the progression of PD. Further, prevention of cell-to-cell transfer of α-syn by decreasing the levels of extracellular α-syn might delay the progression of PD.
The level of extracellular α-syn depends not only on the rate of α-syn release from neurons, but also on the rate of its removal through various clearance pathways such as proteolytic degradation, cell-mediated clearance as well as active and passive transport out of the brain. These clearance pathways could provide new therapeutic targets for PD. In the context of cell-mediated clearance, it was reported that extracellular α-syn could be cleared by neighboring cells such as neurons, astrocytes and microglia (17). In addition, in the context of proteolytic degradation, it was also reported that neurosin (33) and matrix metalloproteinases (MMPs) (34) could degrade extracellular α-syn.
Plasmin derived from its inactive zymogen form, plasminogen, by tissue type plasminogen activator (tPA) or urokinase plasminogen activator (uPA), is a serine protease that dissolves the insoluble fibrin to soluble fibrin degradation products, thus playing a central role in fibrinolysis (35). Plasmin activity is regulated by the activity of tPA and uPA as well as inhibitors of tPA and uPA including plasminogen activator inhibitor-1 (PAI-1) and neuroserpin. It is also regulated by direct inhibitors including α2-antiplasmin and α2-macroglobulin (36). Besides fibrin, plasmin also cleaves a variety of substrates including extracellular matrix components such as fibronectin, laminin, and matrix metalloproteinases (MMPs) (35). These plasmin systems are also detected in the central nervous system (CNS) (35) and have been shown to play an important role in both physiological and pathological processes in the CNS including neuronal development, synaptic plasticity and excitotoxicity (37, 38). Interestingly, plasmin is also known to degrade Aβ and block Aβ-induced neurotoxicity, thus contributing to the progression of Alzheimer disease (AD) (39–41). In the present study, we searched for the protease responsible for degrading extracellular α-syn and investigated whether the plasmin system could degrade extracellular α-syn as well as contribute α-syn-related pathogenesis of PD.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Recombinant wild type α-syn and point mutants (A30P, E46K, and A53T) were purchased from ATgen (Bundang, Korea). Plasminogen, plasmin, uPA and thrombin were purchased from Sigma-Aldrich. tPA and α2-antiplasmin were purchased from Calbiochem. Antibodies against α-syn were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and BD Bioscience (Franklin Lakes, NJ). Antibody from Santa Cruz Biotechnology was used for detecting the C-terminal region of α-syn, and antibody from BD Bioscience was used for detecting the internal region of α-syn. Antibodies against His probe and T7 tag were purchased from Santa Cruz Biotechnology and Novagen (Darmstadt, Germany), respectively.
Preparation of T7-α-Syn-His Protein
Plasmid for T7-α-syn-His protein was generated by cloning PCR product using pCDNA3.1-α-syn (42) as a template into pET21a (Novagen, Darmstadt, Germany). The construct was transformed into the Escherichia coli strain, BL21(DE3) and T7-α-syn-His protein was purified by affinity chromatography using Ni-NTA chelating agarose CL-6B (Peptron, Daejeon, Korea) according to the manufacturer's instructions.
Western Blot Analysis
α-Syn in phosphate buffered saline (PBS, pH 7.5) was incubated with indicated proteins in a total reaction volume of 20 μl at 37 °C in vitro unless otherwise stated. Digestion of α-syn was then stopped by adding SDS-sample buffer. Samples were resolved by 15% SDS-PAGE and transferred to a nitrocellulose membrane. The membranes were incubated with α-syn antibodies and HRP-conjugated secondary antibodies (Invitrogen). They were then visualized using the ECL system (Pierce).
MALDI-TOF Mass Spectrometry and Liquid Chromatography Tandem-Mass Spectrometry (LC/MS/MS)
To determine molecular mass of the α-syn fragments by plasmin, 1.735 μm α-syn was incubated with 75.3 nm plasmin for indicated times at 37 °C. The mixture of the α-syn fragments were passed through a self-packed POROS 20 R2 (Applied Biosystems, Grand Island, NY) cartridge to capture the protein. The proteins were dispensed onto a MALDI-TOF plate. The sample was allowed to air-dry at room temperature for 10 min and then MALDI-TOF mass spectrometry analysis was performed. Mass analysis of the fragments was performed on a Voyager-DE STR MALDI-TOF mass spectrometer (Applied Biosystems, Grand Island, NY) in the linear mode using a nitrogen laser (337 nm). Mass spectra were collected in the positive ion mode using an acceleration voltage of 25 kV and a delay of 800 ns. The grid voltage, low mass gate, and laser intensity were set to 96%, 1000.0 m/z, and 2,500, respectively. Each mass spectrum collected represents the sum of the data from 500 laser shots. To sequence the α-syn fragments, liquid chromatography Tandem-Mass spectrometry (LC/MS/MS) analysis was performed using integrated system consisting of an autosampler (Tempo nano LC system; MDS SCIEX, Ontario, Canada) and a hybrid quadrupole TOF MS/MS spectrometer (QStar Elite; Applied Biosystems, Gland Island, NY) equipped with a nano-electrospray ionization source and a fused silica emitter tip (New Objective, Woburn, MA). 1.735 μm α-syn was incubated with 75.3 nm plasmin for 1 h at 37 °C. The sample was then electrosprayed through a coated silica tip at an ion spray voltage of 2300 eV. For each fragment, the mean and standard deviation of the experimental molecular mass (m/z) were determined from three independent experiments. The theoretical molecular mass (m/z) was determined using an ExPASy-computed pI/MW tool program.
Transmission Electron Microscopy (TEM) and Thioflavin T Binding Assay
Oligomeric and fibrillar forms of α-syn were prepared as described previously with slight modifications (20, 43). Briefly, 1 mg/ml monomeric α-syn was incubated at 37 °C with 250 rpm of agitation for 2 weeks, and then aliquoted and stored at −80 °C until use. Oligomeric α-syn was collected after 8 h of incubation of monomeric α-syn at 37 °C with 600 rpm agitation. The status of oligomeric and fibrillar α-syn was determined by TEM analysis and/or a thioflavin T binding assay. Briefly, oligomeric and fibrillar forms of α-syn were adsorbed onto carbon-coated copper grids (200 mesh) and air dried for 1 min. After negative staining with 2% uranyl acetate for 1 min, the samples were observed with an electron microscope (H7100, Hitachi). For the thioflavin T binding assay, small aliquots of samples were taken and mixed with 5 μm thioflavin T in 500 mm glycine buffer (pH 8.5) and the fluorescence were measured using 482 nm excitation and 446 nm emission wavelengths (PerkinElmer Victor 3).
Preparation of α-Syn Overexpressing SH-SY5Y-conditioned Medium
SH-SY5Y conditioned medium was prepared as described previously (30). Briefly, differentiated SH-SY5Y cells were infected with adeno/α-syn. On day 2 of infection, the medium was replaced with serum free Dulbecco's modified Eagle's medium (DMEM) after the cells were washed twice or more times with PBS. After 24 h of incubation at 37 °C, the conditioned medium was collected and centrifuged at 10,000 × g for 30 min to remove cell debris. The conditioned medium was aliquoted into 50 μl samples and stored at −80 °C. To evaluate whether plasmin could degrade cell-derived α-syn, 40 μl of SH-SY5Y conditioned medium was incubated with indicated doses of plasmin for 12 h at 37 °C. Then, Western blot analysis for α-syn was performed.
Cell Culture
SH-SY5Y (a human dopaminergic neuron cell line), α-syn-overexpressing SH-SY5Y (18) and BV-2 (an immortalized murine microglia cell line) cells, were grown in DMEM supplemented with 10 and 5% fetal bovine serum (FBS), respectively. Rat primary microglia and astrocytes from cerebral cortices of 1-day old Sprague-Dawley rats were cultured as described previously (44, 45). Briefly, the cortices were triturated and were plated into 75 cm2 T-flasks for 2 weeks in minimum essential medium (MEM) containing 10% FBS. Then, microglia were detached from the flasks by mild shaking and filtered through a nylon mesh to remove primary astrocytes. The cells were then plated into 6-well plates for use in subsequent experiments. The purity of cultured rat primary microglia was determined by flow cytometry using anti-OX-42 antibody and its approximate percentage was 90–95% of total cultured cells. Following removal of primary microglia, primary astrocytes were prepared by trypsinization. The cells were incubated with serum-free MEM for 2 days before use to deplete microglia and meningeal cells, and the cells were then plated into 6-well plates for use in subsequent experiments. The purity of cultured rat primary astrocytes determined by GFAP staining was more than 95% of total cultured cells.
Confocal Microscopy
α-Syn-overexpressing SH-SY5Y cells were co-cultured with BV-2 cells in dual chambers for 2 h in the absence or presence of 75.3 nm plasmin. BV-2 cells cultured on poly-d-lysine-coated coverslips were then washed twice with PBS and fixed in 4% paraformaldehyde for 30 min at room temperature. The fixed cells were then washed with PBS and incubated with PBS containing 0.1% Triton X-100 for 10 min. After washing twice or more times with PBS, the cells were blocked with PBS containing 5% bovine serum albumin (BSA) for 30 min at room temperature, and then incubated overnight with α-syn antibody (Santa Cruz Biotechnology) at 4 °C. Preparations were then stained with fluorescence-conjugated secondary antibody (Jackson Immunoresearch, West Grove, PA) for 2 h, mounted with mounting solution containing Hoechst and observed under a confocal microscope (Zeiss, Germany).
Quantitative Real-time RT-PCR
The cells were plated at a density of 2–3 × 105 per well in six well plates and treated with 1 μm α-syn pretreated with 43.4 nm plasmin for indicated times. Total RNA was extracted from cells using Trizol reagent (Invitrogen), and cDNA was prepared using avian myeloblastosis virus reverse transcriptase (Promega) according to the manufacturer's instructions. cDNA samples were analyzed by the Rotor-Gene SYBR Green PCR Master mix kit on Rotor-Gene cyclers (Qiagen, Valencia, CA) with specific primers: human genes (plasminogen, NM_000301.2, tissue plasminogen activator: NM_000930.3, urokinase plasminogen activator: NM_002658.3, plasminogen activator inhibitor-1: M16006.1, GAPDH: NM_002046.3) and rat genes (plasminogen: NM_053491.2, tissue plasminogen activator: NM_013151.2, urokinase plasminogen activator: NM_013085.3, plasminogen activator inhibitor-1: M24067.1, TNF-α: NM_012675.3, IL-1β: NM_031512.2, and GAPDH: NM017008.3). All values were calculated using the delta Ct method and expressed as a change relative to expression of GAPDH mRNA.
IL-1β and TNF-α ELISA
The cells were plated at a density of 2–3 × 105 per well in six well plates and treated with 1 μm α-syn for 12 h. IL-1β and TNF-α in cell-free culture media were determined using a rat IL-1β ELISA kit (Antigenix America Inc.) and rat TNF ELISA Set (BD Bioscience) according to manufacturer's instructions.
Statistical Analysis
All values are expressed as means ± S.E. Statistical significance was evaluated using an unpaired t test (Graphpad Software, San Diego, CA).
RESULTS
Plasmin specifically cleaves recombinant α-syn- To evaluate whether plasmin could cleave α-syn, recombinant α-syn was incubated with a serial dose of plasmin for indicated times and then Western blots were performed. As shown in Fig. 1, A and B, plasmin cleaved recombinant α-syn in a dose- and time- dependent manner. In this experiment, we used anti-α-syn antibody which detects the internal region of α-syn. To validate these cleaved patterns observed, we used a different anti-α-syn antibody, which detects the C-terminal region of α-syn, and also performed Coomassie Blue staining without Western blot. We also observed similar cleaved bands in both experiments as shown in Fig. 1, A and B (supplemental Fig. S1). In addition, we observed that plasminogen with tPA or uPA also cleaved recombinant α-syn, but tPA and uPA alone did not cleave recombinant α-syn. Also, plasmin with α2-antiplasmin, an inhibitor of plasmin, did not cleave recombinant α-syn (Fig. 1C), suggesting that these phenomena are solely dependent on plasmin activity, and not on other contaminated proteases. Thrombin (10 units/ml), another serine protease, also did not cleave recombinant α-syn, and even 100 units/ml thrombin had little effect (Fig. 1D), suggesting that plasmin cleaved α-syn specifically.
FIGURE 1.

Plasmin cleaves recombinant α-syn specifically. A, after incubation of 1.735 μm α-syn with plasmin at the indicated molar ratios for 12 h at 37 °C. B, after incubation of 1.735 μm α-syn with plasmin at the 1:0.04 molar ratio for the indicated times at 37 °C, C, after incubation of 1.735 μm α-syn with indicated proteins (62.5 ng/ml plasminogen, 150 units/ml tPA, 100 units/ml uPA, or 25 μg/ml α2-antiplasmin) for 12 h at 37 °C, D, after incubation of 1.735 μm α-syn with indicated dose of thrombin or PI (10 units/ml or 100 units/ml thrombin, 1:1000 PI) for 12 h at 37 °C, the samples were loaded into SDS-PAGE gels, then Western blots for α-syn were performed. PI indicates complete protease inhibitors mixture (Roche Molecular Diagnostics, Mannheim, Germany).
PD-associated Mutants of α-Syn Are Also Degraded by Plasmin
The α-syn point mutants (A30P, E46K, and A53T) are found in patients with a rare early-onset familial form of PD (3–5). These mutant forms of α-syn appear to have slightly different properties than the wild-type with respect to in vitro aggregation patterns, membrane binding properties and cellular cytotoxicity (46–48). When we evaluated whether point mutants of α-syn were also cleaved by plasmin, we observed that recombinant point mutants of α-syn were also cleaved by plasmin in a similar pattern and degree as wild-type α-syn (Fig. 2).
FIGURE 2.
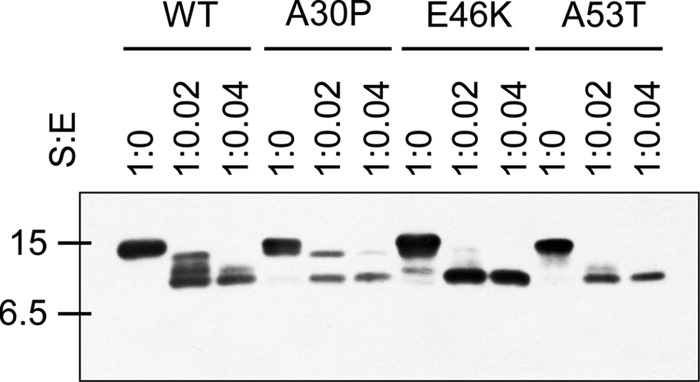
PD-associated mutants of α-syn are cleaved by plasmin. After incubation of 1.735 μm wild-type, A30P, E46K, or A53T α-syn with plasmin at the indicated molar ratios for 12 h at 37 °C, the samples were loaded into an SDS-PAGE gel, then Western blot for α-syn was performed.
Plasmin Cleavage Sites within α-Syn
To identify the region of plasmin cleavage within α-syn, first, we generated recombinant α-syn fused with N-terminal T7 and C-terminal His tags (Fig. 3A). As shown in Fig. 3A, when we incubated T7-α-syn-His protein with plasmin, we detected fragmented α-syn by Western blot using anti-α-syn and anti-His antibodies, but not in Western blots using anti-T7 antibody, suggesting that α-syn fragments observed in Western blot contained the C-terminal region of α-syn. Accordingly, plasmin cleaved mainly the N-terminal region of α-syn into small fragments. Next, to further characterize the sequence of plasmin cleavage within α-syn, we performed MALDI-TOF Mass Spectrometric analysis. Fig. 3B shows that the MALDI-TOF mass spectra of plasmin-cleaved α-syn from 0–12 h. At the time 0 point, we observed a major band of 14,455.9 ± 7.2 Da, which is compatible with the calculated molecular mass of intact α-syn (14,460.2 Da). At 30 min time point, we observed 6 major peaks which were 13,317.97 ± 6.7, 11,224.04 ± 5.6, 10,062.45 ± 5, 8,557.12 ± 4.3, 6,419.29 ± 3.2, and 4,831.66 ± 2.4 Da without intact α-syn, and we still observed one major band of 4,830.36 ± 2.4 Da at the 12 h time point. To further identify the exact sequence of plasmin cleavage sites within α-syn, we performed LC/MS/MS analysis. It showed that the 6 major peaks corresponded to 11–140, 33–140, 44–140, 59–140, 81–140, and 98–140 of α-syn (Fig. 3C). We also observed that all plasmin cleavage sites of α-syn were after lysine residues, which is in agreement of general plasmin cleavage sites (49). In addition, the plasmin cleavage sites of α-syn were mainly within the KTKEGV repeat regions (Fig. 3D). Additional LC/MS/MS data analyses are available in supplemental Table S1.
FIGURE 3.

Plasmin cleavage sites within α-syn. A, diagram for recombinant T7-α-syn-His protein. After incubation of 1.735 μm recombinant T7-α-syn-His protein with plasmin at the indicated molar ratios for 12 h at 37 °C, Western blots for α-syn, T7-tag, and His tag were performed. B, after incubation of 1.735 μm α-syn with 75.3 nm plasmin for indicated times at 37 °C, MALDI-TOF Mass spectrometry was performed as described under “Experimental Procedures.” C, characteristics of the indicated fragments including sequencing region, observed molecular mass (Da) analyzed by LC/MS/MS, and theoretical molecular mass (Da). D, sequence of human α-syn. Arrows indicate the cleavage sites for plasmin, and underlining indicates imperfect KTKEGV repeats.
Oligomeric and Fibrillar α-Syn Are Degraded by Plasmin
Monomeric α-syn is prone to aggregation, and aggregated oligomeric and fibrillar α-syn has different characteristics from monomeric α-syn (50). In addition, the conversion of α-syn from soluble monomers to aggregated, insoluble forms is a key event in the pathogenesis of PD (51). It was also reported that oligomeric α-syn as well as monomeric α-syn is detected in the CSF of patients with PD (10) and fibrillar α-syn as well as monomeric α-syn is secreted from neurons (14).
Thus, to evaluate whether plasmin also cleaves oligomeric and/or fibrillar forms of α-syn, we performed two independent experiments. In electron microscopic (EM) analysis, typical oligomeric and fibrillar forms of α-syn were detected in the control samples, respectively. However, when we incubated oligomeric and fibrillar α-syn with plasmin, the oligomeric and fibrillar forms of α-syn were significantly decreased (Fig. 4, A and C). In addition, we also observed that oligomeric and fibrillar forms of α-syn were cleaved by plasmin in a dose-dependent manner by Western blot analysis (Fig. 4, B and D). The thioflavin T binding assay for fibrillar α-syn also showed similar results as seen EM and Western blot analysis (Fig. 4E), suggesting that plasmin could also cleave the extracellular oligomeric and fibrillar forms of α-syn.
FIGURE 4.

Oligomeric and fibrillar α-syn are degraded by plasmin. After incubation of 1.735 μm oligomeric (A) and fibrillar α-syn (C) with plasmin at the indicated molar ratios for 3 and 12 h, respectively, at 37 °C, the samples, which were prepared as described under “Experimental Procedures,” were observed by electron microscopy. Scale bar indicates 1 μm (A) and 200 nm (C). After incubation of 1.735 μm oligomeric (B) and fibrillar α-syn (D) in the presence or absence of plasmin for 12 h at 37 °C, the samples were loaded into SDS-PAGE gels, then Western blots for α-syn were performed. E, after incubation of 1.735 μm fibrillar α-syn with plasmin at the indicated molar ratios for 12 h at 37 °C, the samples were analyzed with thioflavin T binding assay.
Cell-derived α-Syn Is Degraded by Plasmin
To evaluate whether cell-derived α-syn is also cleaved by plasmin, we first used culture supernatant from α-syn-overexpressing SH-SY5Y cells. As shown in Fig. 5A, when we performed Western blots for α-syn, we could detect cell-derived α-syn in the culture supernatant, which is in agreement with previous findings (15). Next, we incubated with culture supernatant from α-syn-overexpressing SH-SY5Y cells with plasmin. Cell-derived α-syn was also cleaved by plasmin in a dose- and time-dependent manner. In addition, using a co-culture system, we previously observed that α-syn from one cell is propagated into the neighboring cell (18). When plasmin was added into the co-culture system, propagation of α-syn from α-syn-overexpressing SH-SY5Y cells into BV-2 cells (a murine microglial cell line) was significantly inhibited (Fig. 5B). Plasmin treatment in a co-culture system did not show any cytotoxicity in our experimental condition (data not shown), suggesting that cell-derived extracellular α-syn was also cleaved and degraded by plasmin and its propagation into neighboring cells was also inhibited by plasmin.
FIGURE 5.

Cell-derived α-syn is also cleaved and degraded by plasmin. A, conditioned media containing cell-derived α-syn obtained as described under “Experimental Procedures” were incubated with the indicated doses of plasmin for 12 h at 37 °C or with 6.25 μg/ml plasmin for the indicated times at 37 °C, and then Western blot for α-syn was performed. Recombinant α-syn in the absence or presence of plasmin (1:0.04 substrate/enzyme molar ratio) for 12 h at 37 °C was loaded as the control. SCM indicates SH-SY5Y-conditioned media. B, after co-culture of BV-2 cells with α-syn-overexpressing SH-SY5Y cells for 2 h in the presence and absence of 75.3 nm plasmin, BV-2 cells were stained with α-syn antibody (red). Then, the cells were observed by confocal microscopy. BV-2 cells cultured in the absence of α-syn-overexpressing SH-SY5Y cells are indicated by “control.” Scale bar indicates 10 μm.
α-Syn Degraded by Plasmin Loses Its Effects on Cytokine Expression of Primary Microglia and Astrocytes
We and others previously observed that α-syn could activate microglia and astrocytes (21–26). To evaluate the effect of plasmin on α-syn-induced microglia/astrocytes activation, we pre-incubated α-syn with plasmin, and then added these α-syn solutions to rat primary microglia and astrocytes. We observed that α-syn without pretreatment of plasmin activated microglia and astrocytes, so the expression/secretion of TNF-α and IL-1β was increased. However, α-syn pretreated with plasmin lost its effect on TNF-α and IL-1β expression/secretion of microglia/astrocytes significantly (Fig. 6, A and B), suggesting that plasmin degraded α-syn, and degraded α-syn did not affect neighboring cells.
FIGURE 6.

α-Syn cleaved by plasmin loses its effect on the activation of primary microglia and astrocytes. Rat primary microglia (A) and astrocytes (B) were incubated with 1 μm α-syn with or without preincubation of 43.4 nm plasmin (1:0.04 substrate/enzyme molar ratio) for 1 or 2 h (real-time RT-PCR) or 12 h (ELISA), and then real-time RT-PCR and ELISA for TNF-α (a) and IL-1β (b) were performed. **, p < 0.01 against control.
Exogenously Added α-Syn Increases the mRNA Expression of PAI-1, but Not the Expression of Plasminogen, tPA, and uPA in SH-SY5Y Cells, Rat Primary Astrocytes and Microglia
To evaluate whether extracellular α-syn could affect the expression of the plasmin system in the brain, we incubated SH-SY5Y cells (human dopaminergic neuronal cell line), rat primary astrocytes and microglia with α-syn, then analyzed the mRNA expression level of the components of the plasmin system including plasminogen, tPA, uPA, and PAI-1 by real-time RT-PCR. As shown in Fig. 7, we observed that plasminogen, tPA, uPA and PAI-1 were expressed in the neuronal cell line, primary astrocytes, and microglia. However, the expression of plasminogen was not detected in rat primary microglia under our experimental conditions. Moreover, exogenously added α-syn did not induce the expression of plasminogen, tPA, and uPA in the three distinct cells. Interestingly, α-syn induced the expression of PAI-1 in the three distinct cells equally, suggesting that extracellular α-syn may decrease the activity of the plasmin system by increasing the expression of PAI-1 in the brain.
FIGURE 7.

Exogenously added α-syn induces the mRNA expression of PAI-1, but not plasminogen, tPA and uPA in SH-SY5Y cells, rat primary astrocytes, and microglia. After SH-SY5Y cells, rat primary astrocytes and microglia were treated with 1 μm α-syn for 3 h, and then real-time RT-PCR for plasminogen, tPA, uPA, and PAI-1 were performed. *, p < 0.05, **, p < 0.01 against control.
DISCUSSION
Many neurodegenerative diseases such as AD, PD, Huntington disease (HD), and prion disease share some common characteristics. They show typical protein aggregate deposition, and the processes from monomeric forms of each protein such as Aβ, tau, α-syn, huntingtin, and prion to fibrillar forms have been considered to play a significant role in the pathogenesis of each neurodegenerative disease (52–54). Additionally, recent studies suggest that such aggregation prone proteins can spread into neighboring cells, and this spreading might also play a role in the pathogenesis of AD, PD, and HD like that of prion disease (55). Accordingly, a number of studies searching for therapeutics of these neurodegenerative diseases focus on how protein aggregate formation can be inhibited, how the aggregates can be eliminated, and how their spreading can be efficiently halted.
α-Syn, which is a major component of Lewy bodies found in PD and is considered to be a cytosolic protein, has been continuously reported to be found extracellularly (10–12), and the molecular mechanisms of its release and uptake in cells have also been elucidated (13–19). In addition, its potential extracellular effects, including activation of neighboring astrocytes and microglia, has been also increasingly reported (21–26). Interestingly, reports on its propagation to neighboring cells and the association between its phenomena and the pathogenesis of PD have received much attention in order to further understand the pathogenesis of PD (30–32). Therefore, the regulation of extracellular α-syn levels may play an important role for the treatment of PD.
To our knowledge, four proteases including calpain, cathepsin D, MMPs, and neurosin, have been reported to be able to cleave and degrade α-syn (34, 56–58). Among them, neurosin and MMPs can cleave and degrade extracellular α-syn (33, 34). In the present study, we observed that plasmin also cleaved extracellular α-syn. Based on our observation that other serine proteases such as tPA, uPA, and thrombin, which could be found in the CNS, did not cleave α-syn, the effect of plasmin on α-syn cleavage appears to be specific. Plasmin, which is synthesized in the liver and plays a role in the coagulation system, has now been found in the CNS and reportedly expressed in neurons and astrocytes (41). Interestingly, plasmin also cleaves several forms of Aβ, which play a key role in the pathogenesis of AD (40, 59). In the present study, we observed that plasmin also cleaved oligomeric and fibrillar α-syn as well as monomeric α-syn. Considering that the processes for conversion of monomeric α-syn to oligomers, and further to the fibrillar form can play a significant role in the pathogenesis of PD, it is a very important finding for development of a therapeutic strategy of PD, since plasmin can cleave the monomeric, oligomeric, and fibrillar forms of α-syn.
MMP-3 has been reported to cleave T54, E57, A78, Q79, A91, and G93 within the α-syn sequence (34) while neurosin cleaves K80, K97, E114, and D121 sequence (60). On the contrary, plasmin cleaves α-syn from its N-terminal region and subsequently amino acids below the lysine residue within α-syn sequence (K10, K32, K43, K58, K80, and K97) specifically (Fig. 3). It was reported that MMP cleavage of α-syn further induces the aggregation of α-syn, which establishes a detrimental effect of MMPs on the pathogenesis of PD (34, 61). In our previous report, the N-terminal region of α-syn plays an important role on microglial/macrophage activation (26). In agreement of our previous findings, α-syn cleaved by plasmin lost its effect on activation of neighboring astrocytes and microglia (Fig. 6). In addition, the KTKEGV imperfect repeat within the α-syn sequence was reported to play an important role on internalization of α-syn into cells (16). Based on previous findings and our data showing that plasmin cleaved primarily at the KTKEGV repeat region of α-syn sequence, its internalization into neighboring cells was also significantly inhibited by plasmin cleavage (Fig. 5), although we could not exclude the additional possibility that plasmin may also cleave a certain receptor for internalization of α-syn into cells. This suggests that plasmin may have a beneficial effect on the pathogenesis of PD in terms of extracellular α-syn propagation, although the pathological roles of plasmin in the brain are still controversial (35).
In the present study, we also observed that exposure of α-syn to SH-SY5Y cells, a dopaminergic neuronal cell line, as well as rat primary astrocytes and microglia did not induce the expression of plasminogen, tPA, and uPA. However, it induced the expression of plasmingoen activator inhibitor-1 (PAI-1), a natural inhibitor of tPA and uPA (62), suggesting that extracellular α-syn might reduce the activity of the plasmin system, which is in agreement with a recent report showing that α-syn reduced tPA activity with only marginal changes in tPA mRNA in primary astrocytes and microglia (63).
Currently, there is no report on the association between the plasmin system in the CNS and PD. However, in AD, tPA activity has been shown to be decreased in AD models, and its activity is proposed to be controlled by substantial increases in PAI-1 (64). In addition, PAI-1 protein levels increase in the CSF of AD patients (65), and PAI-1 mRNA is increased in APP transgenic mice (66). Brain plasmin activity was reported to also be reduced in AD brains (67). In prion disease, tPA was reported to accelerate the cleavage of prion protein by plasmin, implying that the plasmin system may be involved in the pathogenesis of prion disease (68). Thus, it suggests that the plasmin system in the brain may be also involved in the pathogenesis of PD as well as AD and prion diseases. Further studies will be needed to elucidate the role of the plasmin system in the pathogenesis of PD.
In conclusion, plasmin cleaved and degraded extracellular α-syn specifically in a dose- and time- dependent manner. Oligomeric and fibrillar forms of α-syn as well as monomeric α-syn were also cleaved by plasmin. Plasmin cleaved mainly the N-terminal region of α-Syn. Thus, plasmin-cleaved α-syn fragments did not activate neighboring astrocytes and/or microglia. Additionally, they were not transferred into neighboring cells, suggesting that it could prevent further propagation of α-syn. Exogenously added α-syn induced PAI-1 expression, not plasminogen, tPA, and uPA expression in the brain. These observations may help to elucidate the pathogenesis of PD and develop the therapeutic strategies of PD.
Supplementary Material
This research was supported in part by Basic Science Research Program (2010-0003623), Mid-career Researcher Program (2011-0016603), and MRC program (R13-2003-019) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology.

This article contains supplemental Fig. S1 and Table S1.
- PD
- Parkinson disease
- α-syn
- α-synuclein
- PAI
- plasminogen activator inhibitor
- MMP
- matrix metalloproteinase
- PA
- plasminogen activator.
REFERENCES
- 1. Shulman J. M., De Jager P. L., Feany M. B. (2011) Parkinson's disease: genetics and pathogenesis. Annu. Rev. Pathol. 6, 193–222 [DOI] [PubMed] [Google Scholar]
- 2. Surguchov A. (2008) Molecular and cellular biology of synucleins. Int. Rev. Cell. Mol. Biol. 270, 225–317 [DOI] [PubMed] [Google Scholar]
- 3. Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L., Riess O. (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108 [DOI] [PubMed] [Google Scholar]
- 4. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Di Iorio G., Golbe L. I., Nussbaum R. L. (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 5. Zarranz J. J., Alegre J., Gómez-Esteban J. C., Lezcano E., Ros R., Ampuero I., Vidal L., Hoenicka J., Rodriguez O., Atarés B., Llorens V., Gomez Tortosa E., del Ser T., Muñoz D. G., de Yebenes J. G. (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 [DOI] [PubMed] [Google Scholar]
- 6. Chartier-Harlin M. C., Kachergus J., Roumier C., Mouroux V., Douay X., Lincoln S., Levecque C., Larvor L., Andrieux J., Hulihan M., Waucquier N., Defebvre L., Amouyel P., Farrer M., Destée A. (2004) α-Synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364, 1167–1169 [DOI] [PubMed] [Google Scholar]
- 7. Singleton A. B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., Lincoln S., Crawley A., Hanson M., Maraganore D., Adler C., Cookson M. R., Muenter M., Baptista M., Miller D., Blancato J., Hardy J., Gwinn-Hardy K. (2003) α-Synuclein locus triplication causes Parkinson's disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- 8. Satake W., Nakabayashi Y., Mizuta I., Hirota Y., Ito C., Kubo M., Kawaguchi T., Tsunoda T., Watanabe M., Takeda A., Tomiyama H., Nakashima K., Hasegawa K., Obata F., Yoshikawa T., Kawakami H., Sakoda S., Yamamoto M., Hattori N., Murata M., Nakamura Y., Toda T. (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 41, 1303–1307 [DOI] [PubMed] [Google Scholar]
- 9. Simón-Sánchez J., Schulte C., Bras J. M., Sharma M., Gibbs J. R., Berg D., Paisan-Ruiz C., Lichtner P., Scholz S. W., Hernandez D. G., Krüger R., Federoff M., Klein C., Goate A., Perlmutter J., Bonin M., Nalls M. A., Illig T., Gieger C., Houlden H., Steffens M., Okun M. S., Racette B. A., Cookson M. R., Foote K. D., Fernandez H. H., Traynor B. J., Schreiber S., Arepalli S., Zonozi R., Gwinn K., van der Brug M., Lopez G., Chanock S. J., Schatzkin A., Park Y., Hollenbeck A., Gao J., Huang X., Wood N. W., Lorenz D., Deuschl G., Chen H., Riess O., Hardy J. A., Singleton A. B., Gasser T. (2009) Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 41, 1308–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. El-Agnaf O. M., Salem S. A., Paleologou K. E., Curran M. D., Gibson M. J., Court J. A., Schlossmacher M. G., Allsop D. (2006) Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson's disease. FASEB J. 20, 419–425 [DOI] [PubMed] [Google Scholar]
- 11. Miller D. W., Hague S. M., Clarimon J., Baptista M., Gwinn-Hardy K., Cookson M. R., Singleton A. B. (2004) α-Synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62, 1835–1838 [DOI] [PubMed] [Google Scholar]
- 12. Mollenhauer B., Locascio J. J., Schulz-Schaeffer W., Sixel-Döring F., Trenkwalder C., Schlossmacher M. G. (2011) α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol. 10, 230–240 [DOI] [PubMed] [Google Scholar]
- 13. Emmanouilidou E., Melachroinou K., Roumeliotis T., Garbis S. D., Ntzouni M., Margaritis L. H., Stefanis L., Vekrellis K. (2010) Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jang A., Lee H. J., Suk J. E., Jung J. W., Kim K. P., Lee S. J. (2010) Non-classical exocytosis of α-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 113, 1263–1274 [DOI] [PubMed] [Google Scholar]
- 15. Lee H. J., Patel S., Lee S. J. (2005) Intravesicular localization and exocytosis of α-synuclein and its aggregates. J. Neurosci. 25, 6016–6024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ahn K. J., Paik S. R., Chung K. C., Kim J. (2006) Amino acid sequence motifs and mechanistic features of the membrane translocation of α-synuclein. J. Neurochem. 97, 265–279 [DOI] [PubMed] [Google Scholar]
- 17. Lee H. J., Suk J. E., Bae E. J., Lee J. H., Paik S. R., Lee S. J. (2008) Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int. J. Biochem. Cell. Biol. 40, 1835–1849 [DOI] [PubMed] [Google Scholar]
- 18. Park J. Y., Kim K. S., Lee S. B., Ryu J. S., Chung K. C., Choo Y. K., Jou I., Kim J., Park S. M. (2009) On the mechanism of internalization of α-synuclein into microglia: roles of ganglioside GM1 and lipid raft. J. Neurochem. 110, 400–411 [DOI] [PubMed] [Google Scholar]
- 19. Volpicelli-Daley L. A., Luk K. C., Patel T. P., Tanik S. A., Riddle D. M., Stieber A., Meaney D. F., Trojanowski J. Q., Lee V. M. (2011) Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 72, 57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park J. Y., Paik S. R., Jou I., Park S. M. (2008) Microglial phagocytosis is enhanced by monomeric α-synuclein, not aggregated α-synuclein: implications for Parkinson's disease. Glia. 56, 1215–1223 [DOI] [PubMed] [Google Scholar]
- 21. Klegeris A., Giasson B. I., Zhang H., Maguire J., Pelech S., McGeer P. L. (2006) α-Synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB. J. 20, 2000–2008 [DOI] [PubMed] [Google Scholar]
- 22. Lee H. J., Suk J. E., Patrick C., Bae E. J., Cho J. H., Rho S., Hwang D., Masliah E., Lee S. J. (2010) Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285, 9262–9272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roodveldt C., Christodoulou J., Dobson C. M. (2008) Immunological features of α-synuclein in Parkinson's disease. J. Cell. Mol. Med. 12, 1820–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee E. J., Woo M. S., Moon P. G., Baek M. C., Choi I. Y., Kim W. K., Junn E., Kim H. S. (2010) α-Synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J. Immunol. 185, 615–623 [DOI] [PubMed] [Google Scholar]
- 25. Alvarez-Erviti L., Couch Y., Richardson J., Cooper J. M., Wood M. J. (2011) α-Synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci. Res. 69, 337–342 [DOI] [PubMed] [Google Scholar]
- 26. Lee S. B., Park S. M., Ahn K. J., Chung K. C., Paik S. R., Kim J. (2009) Identification of the amino acid sequence motif of α-synuclein responsible for macrophage activation. Biochem. Biophys. Res. Commun. 381, 39–43 [DOI] [PubMed] [Google Scholar]
- 27. Zhang W., Wang T., Pei Z., Miller D. S., Wu X., Block M. L., Wilson B., Zhou Y., Hong J. S., Zhang J. (2005) Aggregated α-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J. 19, 533–542 [DOI] [PubMed] [Google Scholar]
- 28. Kordower J. H., Chu Y., Hauser R. A., Freeman T. B., Olanow C. W. (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat. Med. 14, 504–506 [DOI] [PubMed] [Google Scholar]
- 29. Li J. Y., Englund E., Holton J. L., Soulet D., Hagell P., Lees A. J., Lashley T., Quinn N. P., Rehncrona S., Björklund A., Widner H., Revesz T., Lindvall O., Brundin P. (2008) Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503 [DOI] [PubMed] [Google Scholar]
- 30. Desplats P., Lee H. J., Bae E. J., Patrick C., Rockenstein E., Crews L., Spencer B., Masliah E., Lee S. J. (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hansen C., Angot E., Bergström A. L., Steiner J. A., Pieri L., Paul G., Outeiro T. F., Melki R., Kallunki P., Fog K., Li J. Y., Brundin P. (2011) α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luk K. C., Song C., O'Brien P., Stieber A., Branch J. R., Brunden K. R., Trojanowski J. Q., Lee V. M. (2009) Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20051–20056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tatebe H., Watanabe Y., Kasai T., Mizuno T., Nakagawa M., Tanaka M., Tokuda T. (2010) Extracellular neurosin degrades α-synuclein in cultured cells. Neurosci. Res. 67, 341–346 [DOI] [PubMed] [Google Scholar]
- 34. Sung J. Y., Park S. M., Lee C. H., Um J. W., Lee H. J., Kim J., Oh Y. J., Lee S. T., Paik S. R., Chung K. C. (2005) Proteolytic cleavage of extracellular secreted {α}-synuclein via matrix metalloproteinases. J. Biol. Chem. 280, 25216–25224 [DOI] [PubMed] [Google Scholar]
- 35. Sheehan J. J., Tsirka S. E. (2005) Fibrin-modifying serine proteases thrombin, tPA, and plasmin in ischemic stroke: a review. Glia 50, 340–350 [DOI] [PubMed] [Google Scholar]
- 36. Schaller J., Gerber S. S. (2011) The plasmin-antiplasmin system: structural and functional aspects. Cell. Mol. Life Sci. 68, 785–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dotti C. G., Galvan C., Ledesma M. D. (2004) Plasmin deficiency in Alzheimer's disease brains: causal or casual? Neurodegener Dis. 1, 205–212 [DOI] [PubMed] [Google Scholar]
- 38. Syrovets T., Simmet T. (2004) Novel aspects and new roles for the serine protease plasmin. Cell. Mol. Life Sci. 61, 873–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Nostrand W. E., Porter M. (1999) Plasmin cleavage of the amyloid β-protein: alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry 38, 11570–11576 [DOI] [PubMed] [Google Scholar]
- 40. Tucker H. M., Kihiko M., Caldwell J. N., Wright S., Kawarabayashi T., Price D., Walker D., Scheff S., McGillis J. P., Rydel R. E., Estus S. (2000) The plasmin system is induced by and degrades amyloid-β aggregates. J. Neurosci. 20, 3937–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barker R., Love S., Kehoe P. G. (2010) Plasminogen and plasmin in Alzheimer's disease. Brain Res. 1355, 7–15 [DOI] [PubMed] [Google Scholar]
- 42. Kim K. S., Park J. Y., Jou I., Park S. M. (2010) Regulation of Weibel-Palade body exocytosis by α-synuclein in endothelial cells. J. Biol. Chem. 285, 21416–21425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bhak G., Lee J. H., Hahn J. S., Paik S. R. (2009) Granular assembly of α-synuclein leading to the accelerated amyloid fibril formation with shear stress. PLoS One 4, e4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim K. S., Park J. Y., Jou I., Park S. M. (2009) Functional implication of BAFF synthesis and release in gangliosides-stimulated microglia. J. Leukoc. Biol. 86, 349–359 [DOI] [PubMed] [Google Scholar]
- 45. Lee J. H., Park S. M., Kim O. S., Lee C. S., Woo J. H., Park S. J., Joe E. H., Jou I. (2009) Differential SUMOylation of LXRα and LXRβ mediates transrepression of STAT1 inflammatory signaling in IFN-γ-stimulated brain astrocytes. Mol. Cell. 35, 806–817 [DOI] [PubMed] [Google Scholar]
- 46. Conway K. A., Harper J. D., Lansbury P. T. (1998) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320 [DOI] [PubMed] [Google Scholar]
- 47. Li J., Uversky V. N., Fink A. L. (2001) Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry 40, 11604–11613 [DOI] [PubMed] [Google Scholar]
- 48. Ostrerova-Golts N., Petrucelli L., Hardy J., Lee J. M., Farer M., Wolozin B. (2000) The A53T α-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci. 20, 6048–6054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weinstein M. J., Doolittle R. F. (1972) Differential specificities of the thrombin, plasmin and trypsin with regard to synthetic and natural substrates and inhibitors. Biochim Biophys. Acta 258, 577–590 [DOI] [PubMed] [Google Scholar]
- 50. Norris E. H., Giasson B. I., Lee V. M. (2004) α-Synuclein: normal function and role in neurodegenerative diseases. Curr. Top Dev. Biol. 60, 17–54 [DOI] [PubMed] [Google Scholar]
- 51. Cookson M. R. (2005) The biochemistry of Parkinson's disease. Annu. Rev. Biochem. 74, 29–52 [DOI] [PubMed] [Google Scholar]
- 52. Frost B., Diamond M. I. (2010) Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 11, 155–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goedert M., Clavaguera F., Tolnay M. (2010) The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 33, 317–325 [DOI] [PubMed] [Google Scholar]
- 54. Miller G. (2009) Neurodegeneration. Could they all be prion diseases? Science 326, 1337–1339 [DOI] [PubMed] [Google Scholar]
- 55. Lee S. J., Desplats P., Sigurdson C., Tsigelny I., Masliah E. (2010) Cell-to-cell transmission of non-prion protein aggregates. Nat. Rev. Neurol. 6, 702–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Iwata A., Maruyama M., Akagi T., Hashikawa T., Kanazawa I., Tsuji S., Nukina N. (2003) α-Synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum. Mol. Genet 12, 2625–2635 [DOI] [PubMed] [Google Scholar]
- 57. Dufty B. M., Warner L. R., Hou S. T., Jiang S. X., Gomez-Isla T., Leenhouts K. M., Oxford J. T., Feany M. B., Masliah E., Rohn T. T. (2007) Calpain-cleavage of α-synuclein: connecting proteolytic processing to disease-linked aggregation. Am. J. Pathol. 170, 1725–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sevlever D., Jiang P., Yen S. H. (2008) Cathepsin D is the main lysosomal enzyme involved in the degradation of α-synuclein and generation of its C-terminally truncated species. Biochemistry 47, 9678–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Miners J. S., Barua N., Kehoe P. G., Gill S., Love S. (2011) Aβ-degrading enzymes: potential for treatment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 70, 944–959 [DOI] [PubMed] [Google Scholar]
- 60. Kasai T., Tokuda T., Yamaguchi N., Watanabe Y., Kametani F., Nakagawa M., Mizuno T. (2008) Cleavage of normal and pathological forms of α-synuclein by neurosin in vitro. Neurosci. Lett. 436, 52–56 [DOI] [PubMed] [Google Scholar]
- 61. Levin J., Giese A., Boetzel K., Israel L., Högen T., Nübling G., Kretzschmar H., Lorenzl S. (2009) Increased α-synuclein aggregation following limited cleavage by certain matrix metalloproteinases. Exp. Neurol. 215, 201–208 [DOI] [PubMed] [Google Scholar]
- 62. Liu R. M., van Groen T., Katre A., Cao D., Kadisha I., Ballinger C., Wang L., Carroll S. L., Li L. (2011) Knockout of plasminogen activator inhibitor 1 gene reduces amyloid β peptide burden in a mouse model of Alzheimer's disease. Neurobiol. Aging. 32, 1079–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Joo S. H., Kwon K. J., Kim J. W., Kim J. W., Hasan M. R., Lee H. J., Han S. H., Shin C. Y. (2010) Regulation of matrix metalloproteinase-9 and tissue plasminogen activator activity by α-synuclein in rat primary glial cells. Neurosci. Lett. 469, 352–356 [DOI] [PubMed] [Google Scholar]
- 64. Melchor J. P., Pawlak R., Strickland S. (2003) The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-β (Aβ) degradation and inhibits Aβ-induced neurodegeneration. J. Neurosci. 23, 8867–8871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sutton R., Keohane M. E., VanderBerg S. R., Gonias S. L. (1994) Plasminogen activator inhibitor-1 in the cerebrospinal fluid as an index of neurological disease. Blood Coagul. Fibrinolysis. 5, 167–171 [DOI] [PubMed] [Google Scholar]
- 66. Cacquevel M., Launay S., Castel H., Benchenane K., Chéenne S., Buée L., Moons L., Delacourte A., Carmeliet P., Vivien D. (2007) Ageing and amyloid-β peptide deposition contribute to an impaired brain tissue plasminogen activator activity by different mechanisms. Neurobiol. Dis. 27, 164–173 [DOI] [PubMed] [Google Scholar]
- 67. Ledesma M. D., Da Silva J. S., Crassaerts K., Delacourte A., De Strooper B., Dotti C. G. (2000) Brain plasmin enhances APP α-cleavage and Aβ degradation and is reduced in Alzheimer's disease brains. EMBO Rep. 1, 530–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kornblatt J. A., Marchal S., Rezaei H., Kornblatt M. J., Balny C., Lange R., Debey M. P., Hui Bon Hoa G., Marden M. C., Grosclaude J. (2003) The fate of the prion protein in the prion/plasminogen complex. Biochem. Biophys. Res. Commun. 305, 518–522 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.