

Abstract
A series of new HIV-1 protease inhibitors (PIs) were designed using a general strategy that combines computational structure-based design with substrate-envelope constraints. The PIs incorporate various alcohol-derived P2 carbamates with acyclic and cyclic heteroatomic functionalities into the (R)-hydroxyethylamine isostere. Most of the new PIs show potent binding affinities against wild-type HIV-1 protease and three multidrug resistant (MDR) variants, in particular inhibitors containing 2,2-dichloroacetamide, pyrrolidinone, imidazolidinone, and oxazolidinone moieties at P2 are the most potent with Ki values in the picomolar range. Several new PIs exhibit nanomolar antiviral potencies against patient-derived wild-type viruses from HIV-1 clades A, B, and C and two MDR variants. Crystal structure analyses of four potent inhibitors revealed that carbonyl groups of the new P2 moieties promote extensive hydrogen bond interactions with the invariant Asp-29 residue of the protease. These structure-activity relationship findings can be utilized to design new PIs with enhanced enzyme inhibitory and antiviral potencies.
Introduction
The human immunodeficiency virus type 1 (HIV-1) protease plays an important role in the viral replication by processing the viral Gag and Gag-Pol polyproteins into functional and structural proteins essential for viral assembly and maturation. Inhibition of HIV-1 protease leads to the generation of immature virus particles. Hence, HIV-1 protease has emerged as a promising target for therapeutic intervention in HIV/AIDS patients. The addition of protease inhibitors (PIs) to the highly active antiretroviral therapy (HAART) with reverse-transcriptase inhibitors resulted in an unprecedented success of HIV/AIDS chemotherapy.1–4 However, rapid emergence of drug-resistant HIV-1 variants, transmission of these resistant viral strains and the adverse effects of currently used HIV-1 PIs remain critical factors limiting the clinical effectiveness of antiretroviral therapies.5–7 Therefore, to alleviate these problems, there is a need for the development of new PIs with improved activity against drug resistant variants and excellent pharmacokinetic and safety profiles.
Crystal structure analyses of HIV-1 protease in complex with its peptide substrates suggest that substrate specificity depends on matching a defined shape within the binding site. This shape has been termed as the substrate envelope.8 Among the FDA approved PIs, amprenavir9 (1, APV, Figure 1) containing tetrahydrofuran (THF) as the P2 ligand fits reasonably well within the substrate envelope. The structurally similar non-peptidic PI, darunavir10–13 (2, DRV, Figure 1) with bis-tetrahydrofuran (bis-THF) as the P2 ligand exerts high in vitro and in vivo antiviral potency against wild-type HIV-1 as well as multidrug resistant (MDR) strains. It is reasoned that the additional interactions of bis-THF moiety in inhibitor 2 with the backbone atoms of Asp-29 and Asp-30 residues in protease result in its potent activity against MDR HIV-1 variants.13, 14 However, inhibitor 2, though significantly more potent than 1, contains a structurally complex and stereochemically defined P2 ligand, requiring multiple steps for synthesis. In addition to inhibitor 2, a number of highly potent PIs incorporate the bis-THF moiety or similar bicyclic ether and flexible poly-ether based P2 ligands that mimic bis-THF.15–20
Figure 1.

Chemical structures of amprenavir 1, darunavir 2, lead compound 3, and designed inhibitors 4 and 5.
We have been pursuing a general strategy combining structure-based design with substrate-envelope constraints to develop novel HIV-1 PIs against clinically relevant MDR protease variants with reduced structural complexity and hence cost effectiveness. We have recently reported a series of novel PIs that incorporate stereochemically defined N-phenyloxazolidinone-5-carboxamides as P2 ligands into the (R)-(hydroxyethylamino)sulfonamide isostere, the core scaffold of inhibitors 1 and 2, and possess impressive enzyme affinities and antiviral activity against both wild-type and a panel of MDR HIV-1 variants.21, 22 Moreover, crystal structure analysis of the inhibitor 3 in complex with wild-type HIV-1 protease confirmed that the carbonyl group of the oxazolidinone ring mimics the hydrogen-bond interactions of THF/bis-THF moieties of inhibitors 1 and 2, respectively.
Since the carbonyl oxygen of ester, amide, and lactone functionalities can also act as hydrogen bond acceptors, it is expected that the above functionalities could mimic the critical hydrogen binding interactions of bis-THF with the protease enzyme, and the presence of other heteroatoms in the ligand would make additional interactions with the side chain residues of the protease in the S2 binding pocket. Thus, we envisioned that P2 ligands containing ester, amide, urea and lactone with flexible carbon chain may accommodate amino acid side chain variations and interact strongly with enzyme backbone. Based on this precept, we selected as P2 ligands alcohols 6a-j containing structurally related five membered heterocyclic structures pyrrolidone, dihydrofuranone, imidazolidone, oxazolidone and some acyclic ketone, ester, amide subunits for the design of new PIs. To investigate the potential of these functionalized P2 ligands in combination with (R)-(hydroxyethylamino)sulfonamide isostere, we have designed and synthesized a series of new PIs (Figure 1).
In the present study, we report the structure-based computational design, synthesis and biological evaluation of a series of HIV-1 PIs incorporating a variety of P2 carbamates derived from acyclic and cyclic alcohols. Structure-activity relationship (SAR) studies with variations at P2, P22 and P12 positions resulted in the generation of several highly potent HIV-1 PIs. Compounds with potent enzymatic activity have been further evaluated against a panel of MDR HIV-1 protease variants and for antiviral activity in cellular assays against patient derived wild-type HIV-1 viruses from clades A, B, and C and two MDR HIV-1 variants. Crystal structures of the four most potent new inhibitors 13c, 13g, 13f and 14j in complex with wild-type HIV-1 protease are also discussed.
Computational Design
The aim of inverse design is to consider a large combinatorial library of potential inhibitors and identify a set of molecules with high affinity against a protein target that are capable of binding within the substrate envelope. The first step in the inverse design procedure is to select and prepare the protein target structure. Previous work on HIV-1 protease has shown that improved results can be achieved by using a crystal structure containing a ligand that is similar to the molecules in the combinatorial library.23 The ligand is removed from the complex to yield a structure that is amenable for inverse design. In this work, based on the (R)-hydroxyethylamine isostere of inhibitors 1 and 2, the 2-bound HIV-1 protease structure (PDB accession code 1T3R) was used.
Once selected, the protein structure was subjected to a standard preparation protocol. Coordinates were taken from the PDB and the ligand was removed from the bound state. Three tightly bound water molecules (HOH1201, HOH1202, and HOH1203) were retained, with all other water molecules removed. The protein and water hydrogen-atom positions were then built using the HBUILD facility of the CHARMM27 program package24 with the CHARMM22 energy function.25 Histidine residues were checked for orientations and protonation state, which resulted in the two His69 residues being flipped and assigned as epsilon protonated. The residues lysine, arginine, aspartate, glutamate, cysteine and tyrosine were also analyzed to check their protonation state. There was no evidence of any unusual protonation states and thus all lysine and arginine residues were assigned as positively charged, all aspartate and glutamate residues were assigned as negatively charged, and all cysteine and tyrosine residues were assigned as neutral. After minimization of the hydrogen-atom positions, the resultant structures were then rotated into a new coordinate frame and the protein atoms were assigned PARSE charges26 for use with Delphi.27, 28 The next stage involved creating van der Waals and electrostatic grids for grid-based energy calculations. The primary scoring function contains three components: a van der Waals term, a screened electrostatic interaction term, and desolvation penalties for the ligand and the protein. Grids for van der Waals energies are computed by placing every type of parameterized CHARMM atom type at each grid-point and computing its van der Waals interaction energy with the protein. For any charged atoms at the grid points, the electrostatic binding free energy was calculated using the linearized Poisson–Boltzmann equation29 using a locally modified version of the DelPhi computer program.30
The next step in the inverse design procedure is to generate the volume in which the designed inhibitors must lie. When attempting to design molecules that are not susceptible to resistance mutations, this volume is the substrate envelope. The substrate envelope is generated from the consensus volume of a set of naturally occurring substrates. In this case, the substrate envelope was created from five aligned HIV-1 protease–substrate peptide complexes and then transferred to a 2-bound HIV-1 protease structure.23 After selecting and preparing the protein structure and the design shape, the next stage is to place the core scaffold in an ensemble of low-energy conformations within the site. The core scaffold in this case is the (R)-hydroxyethylamine isostere shown in Figure 1. A systematic set of conformations was created by rotating each rotatable torsion angle in increments of 30°. Each conformation was then subjected to systematic placement in the active site with a translational enumeration of 0.25 Å and a rotational enumeration such that the maximum arc length of atoms from the centroid swept out a distance of 1.0 Å between orientations. Scaffold placements were accepted if all atom centers were contained within the substrate envelope, but discarded if their calculated van der Waals binding energy was greater than zero or any two nonbonded atoms’ 0.75-scaled radii overlapped.
Each designed inhibitor is comprised of the (R)-hydroxyethylamine isostere with three side group substituents, termed R1, R2 and R3. These substituents are derived from primary amines, sulfonyl chlorides, and alcohols respectively (Scheme 2). The substituents used in the inverse design procedure were taken from the ZINC database of commercially available compounds.31 Every acceptable scaffold pose was used for a separate design with the set of side group substituents at R1, R2 and R3 using the guaranteed combinatorial search algorithms dead-end elimination (DEE)32 and A*.33 The DEE/A* algorithm places all side group substituents in all conformations and combinations on each scaffold pose to yield an energy-ranked list of 1000 unique molecules. The ranked list was then reduced to a computationally feasible size by removing compounds with an energy of 25.0 kcal/mol or higher than the lowest energy result. The resulting top-ranked compounds from the combinatorial search were reevaluated using more accurate physics-based energy functions to identify candidate molecules for synthesis and testing. The most sophisticated energy function included a geometry optimization using CHARMM, performed on the ligand in a rigid protein structure for 1,000,000 steps using the adopted basis Newton-Raphson method. The binding free energies were then estimated using a molecular mechanics and Poisson–Boltzmann/Surface Area (MM-PB/SA) method.30 This includes the sum of van der Waals interactions with the protein, an electrostatic binding free energy calculated by solving the linearized Poisson–Boltzmann equation in both the bound and unbound states and a surface area burial term. A surface tension of 5 cal/Å2 was used for the surface area burial term.30 The compounds proposed for synthesis were among the highest scoring compounds using the most sophisticated energy function.
Scheme 2.

Synthesis of Designed Protease Inhibitors 13a-j–16a-ja
aReagents and conditions: (a) EtOH, 80 °C, 3 h; (b) aq. Na2CO3, CH2Cl2, 0 °C to rt, 6 h; (c) Et3N, CH2Cl2, 0 °C to rt, 5 h; (d) TFA, CH2Cl2, rt, 1 h; (e) Et3N, THF, 0 °C to rt, 4–8 h.
Chemistry
As outlined in Scheme 1, activated mixed carbonates 7a-j of cyclic and acyclic alcohols used in the synthesis of designed inhibitors were prepared following the literature procedure.19 It is to be noted that attempts to use one standard activation condition was not very efficient and resulted in poor yields. Therefore, depending on the functional groups present in the P2 alcohol fragment, two different activating agents have been used. Accordingly, alcohols 6a-c, 6f-j were reacted with p-nitrophenylchloroformate and N-methylmorpholine in THF at 25 °C to provide corresponding carbonates 7a-c, 7f-j in 58–79% yields. Alcohols 6d-e were converted to corresponding succinimidyl carbonates 7d-e by treatment with N,N′-succinimidylcarbonate and triethylamine in acetonitrile in 35–41% isolated yields.
Scheme 1.

Synthesis of Activated Carbonate Intermediates 7a–ja
aReagents and conditions: (a) N-methylmorpholine, p-nitrophenylchloroformate, 0 °C to rt, 1 h; (b) N,N′-disuccimidyl carbonate, Et3N, CH3CN, 0 °C to rt, 8 h.
The synthesis of (R)-(hydroxyethylamino)sulfonamide isosteres 12 was carried out following the reported methods21, 23 as outlined in Scheme 2. Regioselective ring opening of commercially available epoxide 8, (1S,2S)-(1-oxiranyl-2-phenylethyl)carbamic acid tert-butyl ester, with two primary amines, (S)-2-methylbutyl amine and n-pentyl amine, in ethanol provided β-amino alcohols 10a-b, respectively. Depending upon the chemical properties of selected sulfonyl chlorides, two different methods were used for the synthesis of (R)-(hydroxyethylamino)sulfonamide intermediates 12a-d. Reactions of the amino alcohols 10a-b with p-(methoxy)phenylsulfonyl chloride 11a were carried out using aqueous sodium carbonate (Na2CO3) and 6-benzothiazolesulfonyl chloride 11b using diisopropylethylamine in anhydrous CH2Cl2, and furnished the corresponding sulfonamides 12a-d. Subsequent removal of the Boc protection using trifluoroacetic acid in CH2Cl2 furnished the corresponding free amino alcohols. Selective alkoxycarbonylation of the aliphatic amines in amino alcohols 12a-d using either activated p-nitrophenyl carbonate or succinimidyl carbonate 7a-j of selected alcohols under basic conditions provided the desired inhibitors 13a-j–16a-j in 49–81% yields (Scheme 2).
Results and Discussion
We used computational structure-based design combined with substrate-envelope constraints to design a series of novel HIV-1 PIs. The designed PIs incorporate various alcohol-derived P2 carbamates with acyclic and cyclic heteroatomic functionalities into the (R)-hydroxyethylamine isostere to enhance the ligand-receptor interactions in the S2 binding pocket of the protease. The (S)-2-methylbutyl and n-pentyl groups were used as P1′ ligands in combination with 4-methoxyphenyl and 6-benzothiazole groups as P2′ ligands. Inhibitory activities of new PIs were determined against wild-type HIV-1 protease (Q7K) using a fluorescence resonance energy transfer (FRET) method34 in a semi-high throughput format. Chemical structures of all new inhibitors along with their inhibitory activities (Ki values) are presented in Table 1; each Ki value denotes the mean of at least three independent determinations.
Table 1.
Inhibitory Activities of Compounds against Wild-type HIV-1 Protease

| |||||
|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 |
|
|
| 12b |
|
3,4-S-CH=N- |
|
0.243 | |
| 13a |
|
4-OCH3 |
|
0.873 | |
| 13b |
|
4-OCH3 |
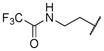
|
11.95 | |
| 13c |
|
4-OCH3 |
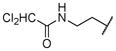
|
0.003 | |
| 13d |
|
4-OCH3 |
|
0.426 | |
| 13e |
|
4-OCH3 |
|
2.01 | |
| 13f |
|
4-OCH3 |

|
0.028 | |
| 13g |
|
4-OCH3 |

|
0.048 | |
| 13h |
|
4-OCH3 |
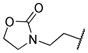
|
0.003 | |
| 13i |
|
4-OCH3 |
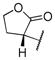
|
35.11 | |
| 13j |
|
4-OCH3 |
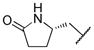
|
0.055 | |
| 14a |
|
3,4-S-CH=N- |
|
0.590 | |
| 14b |
|
3,4-S-CH=N- |
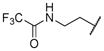
|
9.13 | |
| 14c |
|
3,4-S-CH=N- |
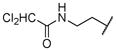
|
0.060 | |
| 14d |
|
3,4-S-CH=N- |
|
0.303 | |
| 14e |
|
3,4-S-CH=N- |
|
0.230 | |
| 14f |
|
3,4-S-CH=N- |
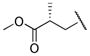
|
1.392 | |
| 14g |
|
3,4-S-CH=N- |
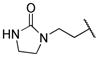
|
0.005 | |
| 14h |
|
3,4-S-CH=N- |
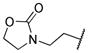
|
0.005 | |
| 14i |
|
3,4-S-CH=N- |

|
121.5 | |
| APV | 0.007 | ||||
| 14j |
|
3,4-S-CH=N- |
|
0.003 | |
| 15a |
|
4-OCH3 |
|
17.24 | |
| 15b |
|
4-OCH3 |

|
31.44 | |
| 15d |
|
4-OCH3 |
|
6.98 | |
| 15e |
|
4-OCH3 |
|
2.955 | |
| 15f |
|
4-OCH3 |
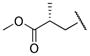
|
0.406 | |
| 15g |
|
4-OCH3 |
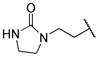
|
0.398 | |
| 15h |
|
4-OCH3 |
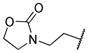
|
0.008 | |
| 15i |
|
4-OCH3 |
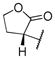
|
65.89 | |
| 15j |
|
4-OCH3 |
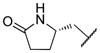
|
0.624 | |
| 16a |
|
3,4-S-CH=N- |
|
11.39 | |
| 16b |
|
3,4-S-CH=N- |

|
19.67 | |
| 16c |
|
3,4-S-CH=N- |
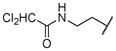
|
0.001 | |
| 16d |
|
3,4-S-CH=N- |
|
3.375 | |
| 16e |
|
3,4-S-CH=N- |
|
1.29 | |
| 16f |
|
3,4-S-CH=N- |
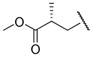
|
3.345 | |
| 16g |
|
3,4-S-CH=N- |
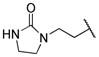
|
0.217 | |
| 16h |
|
3,4-S-CH=N- |
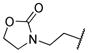
|
0.017 | |
| 16i |
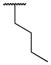
|
3,4-S-CH=N- |

|
42.11 | |
| 16j |
|
3,4-S-CH=N- |
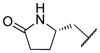
|
0.027 | |
| DRV | 0.0002 | ||||
As predicted, introduction of cyclic and acyclic P2 carbamates with multiple hydrogen bond donor and acceptor parts provided a series of highly potent HIV-1 PIs. Compounds 13-16c, 13-16g and 13-16h, and 13-16j, containing 2,2-dichloroacetamide, imidazolidinone, oxazolidinone, and pyrrolidinone moities, respectively, and an extended carbon chain at P2, were highly potent against wild-type protease with Ki vales in the low nanomolar (nM) to low picomolar (pM) range. In particular, compounds 13c and 16c showed impressive protease inhibitory activities with Ki values of 0.003 nM and 0.001 nM, respectively, which are comparable to that of structurally related drugs 1 and 2. Inhibitors 13-15h with an oxazolidinone substituent at P2 also displayed highly potent inhibitory activities against wild-type HIV-1 protease (Ki = 0.003, 0.005, and 0.008 nM, respectively). Similarly, inhibitors 14g and 14J with imidazolidinone, and pyrrolidinone moities, respectively, also showed highly potent inhibitory activities. Conversely, introduction of trifluoromethylacetamide, (R)-5-hexan-2-one and dihydrofuranone groups at the P2 carbamate in 13-16b, 13-16d and 13-16i resulted in weak inhibitory activity. Among the inhibitors 13-16a and 13-16f containing carbamates of (R)-methyl 3-hydroxybutanoate and its positional isomer, (R)-methyl 3-hydroxy-2-methylpropanoate, respectively, only compounds 13-16f showed better enzyme inhibitory potency. Same trend was observed for compounds 13-16e which incorporate bis-carbamate P2 substituent. These SAR data suggest that flexible heterocyclic moieties are preferred at P2.
A subset of compounds with high binding affinity against wild-type protease was further tested against three MDR protease variants. These MDR variants were selected by examining the Stanford HIV-1 Drug Resistance Database (http://hivdb.stanford.edu), which contains sequences of viral isolates from HIV-1-infected patients. The selected MDR variants (M1: L10I, G48V, I54V, L63P, V82A; M2: L10I, L63P, A71V, G73S, I84V, L90M; M3: I50V, A71V) represent the pattern of resistance mutations observed under the selective pressure of three or more currently prescribed PIs.35 The Ki values of selected inhibitors are presented in Table 2 with drugs 1, 2 used as control. Most of the PIs in the current series retained high affinity against the three mutant variants (M1-M3), consistent with their wild-type protease inhibitory activities. Importantly, compounds 13c, 13f, 13j and 14j exhibited excellent potencies in the low nM ranges against all three MDR protease variants. In particular, compound 14j with Ki value of 0.003 nM against wild-type HIV-1 protease exhibited consistently higher potencies against all three MDR variants (MDR Ki = 0.240–0.377 nM).
Table 2.
Inhibitory Activities of Compounds against MDR Variants of HIV-1 Proteasea
| Compd |
Ki (nM)
|
|||
|---|---|---|---|---|
| Wt | M1 | M2 | M3 | |
| 13a | 0.873 | 4.725 | 17.32 | 4.503 |
| 13c | 0.003 | 0.400 | 0.563 | 0.044 |
| 13f | 0.028 | 0.301 | 0.372 | 0.297 |
| 13g | 0.048 | 1.974 | 3.256 | 1.056 |
| 13h | 0.003 | 0.750 | 1.056 | 0.333 |
| 13j | 0.055 | 0.284 | 0.540 | 0.770 |
| 14a | 0.590 | 11.72 | 12.04 | 24.19 |
| 14c | 0.060 | 1.481 | 1.269 | 0.265 |
| 14d | 0.303 | 4.228 | 13.37 | 3.238 |
| 14e | 0.230 | 3.986 | 3.198 | 2.547 |
| 14g | 0.005 | 0.599 | 1.367 | 0.328 |
| 14i | 121.5 | 375.2 | 453.1 | 305.7 |
| 14j | 0.003 | 0.299 | 0.240 | 0.377 |
| 15f | 0.406 | 2.801 | 1.665 | 1.795 |
| 16a | 11.39 | 180.0.0 | 45.92 | 119.6 |
| 16c | 0.001 | 1.345 | 1.074 | 0.057 |
| 16j | 0.027 | 7.020 | 1.126 | 1.009 |
| APV | 0.007 | 0.041 | 0.195 | 0.030 |
| DRV | 0.0002 | 0.004 | 0.027 | 0.001 |
Wt: Q7K; M1: L10I, G48V, I54V, L63P, V82A; M2: L10I, L63P, A71V, G73S, I84V, L90M; M3: I50V, A71V.
Selected PIs were further examined for antiviral potency in cellular assays against patient derived wild-type viruses from HIV-1 clades A, B, C and two MDR HIV-1 variants; drugs 1 and 2 were used as controls (Table 3). Most of the tested compounds exhibited significant antiviral potencies with the EC50 values in the low nM range against both wild-type and drug-resistant HIV-1 strains. Among these, three close analogues 13f, 13j and 14g exhibited modest antiviral potency. Compounds 16 with the most potent protease binding affinity showed lower activity in cellular assays against both wild-type and MDR HIV-1 variants. Interestingly, compound 14j with a cyclic pyrrolidinone group at the P2 position and 6-benzothiazole sulfonamide at the P2’ position was the most potent against all the viruses tested (EC50 = 15.5–35.6 nM).
Table 3.
Antiviral Activity of Protease Inhibitors against Wild-Type and MDR HIV-1 Strainsa

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | EC50 (nM)
|
|||||
| WT | WT-A | WT-B | WT-C | MDR | MDR1 | ||||
| 13c |
|
4-OCH3 |

|
39.1 | 51.3 | 30.7 | 84.6 | 158.7 | 123.0 |
| 13f |
|
4-OCH3 |

|
30.9 | 24.9 | 20.6 | 36.3 | 56.0 | 41.8 |
| 13g |
|
4-OCH3 |

|
84.4 | 95.4 | 57.3 | 134.8 | 132.1 | 113.3 |
| 13h |
|
4-OCH3 |
|
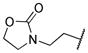
40.5 | 43.3 | 27.8 | 57.3 | 74.4 | 56.1 |
| 13j |
|
4-OCH3 |
|
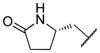
43.9 | 64.5 | 38.5 | 44.1 | 72.2 | 37.1 |
| 14c |
|
3,4-S-CH=N- |
|
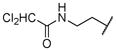
43.0 | 64.1 | 28.6 | 88.0 | 189.3 | 143.9 |
| 14e |
|
3,4-S-CH=N- |
|
197.6 | 231.1 | 183.2 | 331.3 | 917.9 | 755.9 |
| 14g |
|
3,4-S-CH=N- |
|
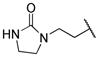
34.8 | 31.4 | 21.6 | 49.3 | 88.1 | 50.4 |
| 14h |
|
3,4-S-CH=N- |

|
33.3 | 21.2 | 12.0 | 36.2 | 73.6 | 32.2 |
| 14j |
|
3,4-S-CH=N- |
|
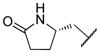
15.5 | 25.1 | 16.4 | 13.2 | 35.6 | 21.1 |
| 15h |
|
4-OCH3 |

|
77.1 | 126.4 | 52.4 | 158.2 | 265.4 | 299.7 |
| 16c |
|
3,4-S-CH=N- |
|
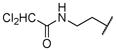
38.3 | 79.2 | 30.3 | 67.0 | 285.1 | 253.9 |
| 16g |
|
3,4-S-CH=N- |

|
83.8 | 99.7 | 53.9 | 129.8 | 274.5 | 236.8 |
| 16h |
|
3,4-S-CH=N- |
|
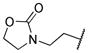
73.0 | 96.3 | 41.1 | 120.5 | 420.0 | 259.4 |
| 16j |
|
3,4-S-CH=N- |
|
58.9 | 82.7 | 44.8 | 96.5 | 174.2 | 129.6 |
| APV | 3.0 | 2.6 | 1.8 | 3.7 | 17.5 | 15.6 | |||
| DRV | 0.25 | 0.3 | 0.15 | 0.18 | 0.6 | 0.23 | |||
Antiviral assays were carried out by Monogram Biosciences. WT, wild-type HIV-1 control; WT-A, WT-B, WT-C, patient-derived strains of wild-type HIV-1 from clades A, B, and C, respectively; MDR, drug-resistant HIV-1 control MDRC4; MDR1, drug-resistant HIV-1 variant with protease mutations M46I, I54V, V82A, and L90M.
In general, inhibitors with a flexible heterocyclic moiety at P2 were considerably more potent than their inflexible and acyclic counterparts in both enzyme inhibitory and antiviral assays. These results revealed the compatibility of most of the selected P2 substituents with established P12 and P22 substituents. Thus, further optimization of these potent compounds may lead to the development of novel HIV-1 protease inhibitors active against drug-resistant HIV-1.
Crystal Structures of HIV-1 Protease Complexes
Crystal structures of four potent inhibitors 13c, 13f, 13g and 14j have been determined in complex with wild-type HIV-1 protease. The R3 group in the 13c-protease complex was disordered and the R1 and R2 groups have similar interactions as in 13f and 13g, hence this structure is not discussed in detail. The backbone atoms of the protease in the three structures, 13f, 13g and 14j, superpose well within 2.12 Å to 2.59 Å. The scaffold of the inhibitors along with the R1 and R2 groups also superpose well onto each other. The conformation of the protease, including the orientations of the side chains, is similar in the regions where the three inhibitors superpose well onto each other. Both the benzothiazole and the 4-methoxyphenyl R2 substituents in the three structures form hydrogen bonding interactions with the backbone amide NH of the Asp-30 residue as predicted computationally and previously observed (Figure 2).
Figure 2.

Hydrogen bond interactions between the wild-type protease and inhibitors in complexes of (a) 13f, (b) 13g and (c) 14j. The two monomers of the protease and the inhibitor are shown in cyan, magenta, and orange, respectively; nitrogen, oxygen, and sulfur atoms are in blue, red, and yellow, respectively.
The R3 groups of 13f, 13g and 14j make different contacts in each case, providing interesting structural basis for their distinct enzymatic activities. Compound 13f contains a methyl ester at R3 which forms a bifurcated hydrogen bond with the backbone amide NHs of Asp-29 and Asp-30 through its carbonyl oxygen (See Figure 2a). The methyl group of the ester packs within the hydrophobic P2 pocket containing residues Val-32, Ile-47 and Ile84. The side chains of Val32 and Ile84 in 13f adopt a different conformation compared to 13g and 14j structures, probably to accommodate the methyl group while the backbone of the protease is similar in all the structures. The ester oxygen atom of 13f does not make a hydrogen bond and thus it would be interesting to test the corresponding keto compound. Compound 13g contains a cyclic urea at R3 in which the carbonyl group forms two hydrogen bonds, one with the backbone amide NH of Asp-29 and one with a structural water molecule held in place by the backbone carbonyl of Gly27 as shown in Figure 2b. The nitrogen NH of the urea appears to be less important. Compound 14j contains a pyrrolidone in which the carbonyl group also forms hydrogen bonds with the backbone amide NH of Asp-29 and with the structural water molecule held by Gly-27. This structural water could be considered in future design efforts. In addition, the pyrrolidinone NH forms bridging hydrogen bond interactions through a water molecule to the backbone amide NH of Asp-30. Overall, the three picomolar inhibitors, 13f, 13g and 14j, appear to lock in the active site by forming hydrogen bonds to both sides of the floor of the active site at Asp29 and/or Asp30, which was also observed for other picomolar inhibitors from previous studies.36
HIV-1 protease is highly plastic and adapts to the conformation of the inhibitor to which it binds. The conformation of the protease in all the three structures is similar except minor differences in the side chains where the R3 groups of the inhibitors are different and have different conformations. The number of hydrogen bonds is similar in all the three inhibitor complexes. The binding affinities of the three inhibitors can not be explained by the number of hydrogen bonds alone, emphasizing that the binding affinity is a product of both enthalpic interactions (both hydrophilic and hydrophobic) and the entropy of binding.37, 38
Conclusions
In summary, we have described a series of highly potent HIV-1 PIs based on the (hydroxyethylamino)sulfonamide isostere that incorporate various P2 carbamates of acyclic and cyclic alcohols. A number of analogues possessing the (R)-2-methylpropanoate, (S)-5-(methyl)pyrrolidin-2-one, ethyloxazolidin-2-one and ethylimidazolidin-2-one at the P2 exhibited excellent binding affinities against wild-type HIV-1 protease and a panel of three MDR variants. In general, PIs incorporating the flexible cyclic P2 moieties with multiple polar atoms showed highly potent inhibitory activities against wild-type protease and MDR variants. Moreover, several compounds exhibited antiviral activities with EC50 values in the nM range. Notably, inhibitor 14j with the (S)-5-(methyl)pyrrolidin-2-one P2 moiety consistently exhibited potent activity in both enzymatic and cellular assays. The X-ray crystal structure analysis of 14j bound to HIV-1 protease revealed enhanced backbone interactions with the protease enzyme. The carbonyl oxygen and NH of the pyrrolidone P2 ligand are involved in direct and water-mediated hydrogen bonding interactions with the backbone NHs of Asp-29 and Asp-30 in the S2 subsite. These extensive interactions of 14j may be responsible for its impressive activity against MDR protease variants and for its antiviral potency in cellular assays. The results from the current study support the compatibility of many selected P2 ligands and the structure-activity relationship data can be utilized to design new protease inhibitors with better enzyme inhibitory and antiviral potencies.
Experimental Section
General
Thin-layer chromatography (TLC) was performed on silica gel (60 F-254) coated plates (Merck KGA) and spots were visualized with UV light. Flash column chromatography was performed using silica gels (230–400 mesh, Merck KGA). Proton and carbon nuclear magnetic resonance (1H NMR and 13C NMR) spectra were normally carried out in CDCl3 solutions on a JEOL JNM-ECS spectrometer, operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR. Trimethylsilane (TMS) was used as an internal standard. Chemical shifts are given in ppm relative to the trimethylsilane (TMS) solvent signal and coupling constant (J) values are reported in Hertz (Hz). Low-resolution mass spectra (ESI) were obtained using Waters Micromass Model ZQ 4000 using methanol solvent. Tetrahydrofuran (THF) was distilled from sodium/benzophenone. Anhydrous dichloromethane, benzene, toluene and N,N-dimethylformamide (DMF), and all other solvents and reagents were purchased from commercial vendors and were used as received. Analytical reversed-phase high performance liquid chromatography (HPLC) was performed on a Waters Separation Module 2695 system equipped with an auto-sampler and a Waters 996 photodiode array detector. Purity of the final compounds was determined using chromatographic systems: column, YMC-Pack Pro C18 (particle size = 5 μm, pore size = 12 nm, dimensions = 150 mm × 4.6 mm); mobile phase A, water; mobile phase B, methanol. Using a flow rate of 1.0 mL/min, gradient elution was performed from 20% B to 100% B over 10 min. In every case, 10 μL of a 1 mM solution was injected. Purity data and the corresponding retention times of all final compounds have been presented in the Supporting Information.
(R)-methyl 3-((((2S,3R)-3-hydroxy-4-(4-methoxy-N-((S)-2-methylbutyl)phenylsulfonamido)-1-phenylbutan-2-yl)carbamoyl)oxy)butanoate (13a)
To a solution of the tert-butyl ((2S,3R)-3-((hydroxy-4-(4-methoxy-N-((S)-2-methylbutyl)phenylsulfonamido)-1-phenylbutan-2-yl)carbamate 12a (0.02 g, 0.384 mmol) in CH2Cl2 (7 mL) was added trifluoroacetic acid (2 mL) and the resulting mixture was stirred at room temperature for 1.5 h. After completion of reaction, the reaction mixture was concentrated under reduced pressure, and the residue was dissolved in toluene (10 mL) and again evaporated under reduced pressure to dryness. The resulting de-protected amine product was dissolved in dry CH2Cl2 (6 mL) and the solution was cooled 0 °C. Diisopropylethyl amine (0.2 mL, 1.21 mmol) was added and after 10 min the activated carbonate 7a solution in dry CH2Cl2 was slowly added. The resulting reaction mixture was stirred at 0 °C for 4 h, allowed to warm to room temperature, and stirred until reaction was complete. Water (5 mL) and EtOAc (20 mL) were added and layers were separated; the organic layer was washed with saturated aqueous NaCl solution (10 mL), dried (Na2SO4), filtered, and concentrated. The residue was purified by flash chromatography on silica gel, eluting with a mixture of 1:3 EtOAc/hexane, to afford the target compound 13a (0.17 g, 81%) as a colorless semi-solid. 1H NMR (400 MHz, CDCl3) δ 7.68-7.62 (m, 2H), 7.25-7.13 (m, 5H), 6.94-6.88 (m, 2H), 5.87 (d, J = 8.5 Hz, 1H), 5.19-5.10 (m, 1H), 4.13-4.03 (m, 2H), 3.84-3.74 (m, 4H), 3.61 (s, 3H), 3.05 (dd, J = 14.9, 4.3 Hz, 1H), 2.99 (d, J = 8.5 Hz, 1H), 2.97-2.86 (m, 2H), 2.75 (dd, J = 13.4, 7.3 Hz, 1H), 2.58-2.51 (m, 1H), 2.51-2.44 (m, 1H), 2.43-2.37 (m, 1H), 2.36-2.30 (m, 1H), 2.29-2.19 (m, 1H), 1.15 (d, J = 4.8 Hz, 3H), 1.01-0.96 (m, 1H), 0.82-0.73 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 170.67, 162.97, 155.71, 137.49, 129.75, 129.53 (2C), 129.45 (2C), 128.47 (2C), 126.46, 114.30 (2C), 72.32, 67.97, 57.33, 55. 58, 54.84, 53.62, 51.63, 40.72, 35.47, 33.39, 26.35, 20.07, 16.84, 11.00; MS (ESI) m/z 587.16 (M + Na)+.
2-(2,2,2-trifluoroacetamido)ethyl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-((S)-2-methylbutyl)phenylsulfonamido)-1-phenylbutan-2-yl)carbamate (13b)
The title compound was obtained from 12a and 7b as described for 13a in 71% yield after flash-chromatography (1:4 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 8.5 Hz, 1H), 7.66-7.59 (m, 2H), 7.23-7.11 (m, 5H), 6.92-6.87 (m, 2H), 4.29 (t, J = 7.8 Hz, 1H), 4.11-3.97 (m, 1H), 3.86-3.83 (m, 1H), 3.81-3.74 (m, 4H), 3.57-3.41 (m, 1H), 3.13 (dd, J = 15.2, 9.2 Hz, 1H), 3.05-2.79 (m, 4H), 2.73-2.67 (m, 1H), 2.44-2.31 (m, 1H), 1.75 (bs, 1H), 1.57-1.48 (m, 1H), 1.44-1.35 (m, 1H), 1.20-1.14 (m, 1H); 1.04-0.92 (m, 1H), 0.85-0.72 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 162.95, 155.42, 151.56, 137.28, 129.43, 129.36 (2C), 129.24 (2C), 128.42 (2C), 128.39, 126.53, 114.28 (2C), 72.26, 62.25, 55.53, 54.73, 53.21, 42.30, 35.86, 33.33, 29.62, 26.39, 16.84, 10.94; MS (ESI) m/z 626.31 (M + Na)+.
2-(2,2-dichloroacetamido)ethyl (2S,3R)-3-hydroxy-4-(4-methoxy-N-((S)-2-methylbutyl)phenylsulfonamido)-1-phenylbutan-2-ylcarbamate (13c)
The title compound was obtained from 12a and 7c as described for 13a in 57% yield after flash-chromatography (1:4 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 9.2 Hz, 2H), 7.24-7.09 (m, 5H), 6.99 (bs, 1H), 6.91 (d, J = 8.5 Hz, 2H), 5.89 (s, 1H), 5.04 (d, J = 9.2 Hz, 1H), 4.09-3.97 (m, 2H), 3.86-3.76 (m, 5H), 3.74-3.56 (m, 2H), 3.47-3.33 (m, 2H), 3.14-2.67 (m, 5H), 1.67-1.46 (m, 2H), 1.05-0.95 (m, 1H), 0.83-0.70 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 164.30, 163.05, 156.36, 137.46, 129.63 (2C), 129.42 (2C), 128.84 (2C), 128.48, 126.58, 114.33 (2C), 72.24, 66.22, 63.01, 57.13, 55.60, 55.07, 53.35, 40.28, 35.34, 33.34, 26.41, 16.86, 11.00; MS (ESI) m/z 640.19 (M−1+ Na)+, 642.19 (M+1+ Na)+.
(R)-5-oxohexan-2-yl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-((S)-2-methylbutyl)phenylsulfonamido)-1-phenylbutan-2-yl)carbamate (13d)
The title compound was obtained from 12a and 7d as described for 13a in 69% yield after flash-chromatography (1:3 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 7.66-7.60 (m, 2H), 7.25-7.10 (m, 5H), 6.95-6.86 (m, 2H), 4.69 (d, J = 7.9 Hz, 1H), 4.63-4.54 (m, 1H), 3.88-3.67 (m, 6H), 3.10-2.89 (m, 4H), 2.89-2.67 (m, 2H), 2.35 (t, J = 6.7 Hz, 1H), 2.02 (s, 3H), 1.79-1.50 (m, 4H), 1.47-1.36 (m, 1H), 1.09-0.93 (m, 4H), 0.84-0.72 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 207.90, 163.00, 156.21, 137.63, 129.78, 129.44 (4C), 128.47 (2C), 126.47, 114.31 (2C), 79.35, 72.36, 70.93, 57.23, 55.60, 54.88, 53.50, 39.47, 35,27, 33.39, 29.88, 26.40, 20.18, 16.88, 11.01; MS (ESI) m/z 585.40 (M + Na)+.
2-((tert-butoxycarbonyl)amino)ethyl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-((S)-2-methylbutyl)phenylsulfonamido)-1-phenylbutan-2-yl)carbamate (13e)
The title compound was obtained from 12a and 7e as described for 13a in 51% yield after flash-chromatography (1:3 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 8.5 Hz, 2H), 7.30-7.12 (m, 5H), 6.93 (d, J = 7.9 Hz, 2H), 5.15-4.92 (m, 1H), 4.80 (bs, 1H), 4.01-3.88 (m, 2H), 3.85-3.73 (m, 5H), 3.65-3.61 (m, 1H), 3.43-3.29 (m, 1H), 3.28-3.15 (m, 3H), 3.08-2.91 (m, 4H), 1.58-1.44 (m, 1H), 1.43-1.28 (m, 10H), 1.03-0.93 (m, 1H), 0.85-0.69 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 162.96, 156.45, 155.81, 137.63, 131.01, 129.72 (2C), 129.40 (2C), 128.41 (2C), 126.46, 114.26 (2C), 79.51, 72.27, 62.36, 57.09, 55.54, 53.34, 43.03, 41.96, 35.27, 33.30, 28.30 (3C), 26.40, 16.83, 10.96; MS (ESI) m/z 630.52 (M + Na)+.
(R)-methyl 3-((((2S,3R)-3-hydroxy-4-(4-methoxy-N-((S)-2-methylbutyl)phenylsulfonamido)-1-phenylbutan-2-yl)carbamoyl)oxy)-2-methylpropanoate (13f)
The title compound was obtained from 12a and 7f as described for 13a in 63% yield after flash-chromatography (1:3 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 7.64-7.60 (m, 2H), 7.25-7.11 (m, 5H), 6.92-6.88 (m, 2H), 4.82-4.79 (m, 1H), 4.08-3.97 (m, 2H), 3.80 (s, 3H), 3.77-3.60 (m, 3H), 3.57 (s, 3H), 3.07-2.79 (m, 5H), 2.75-2.58 (m, 2H), 1.58-1.35 (m, 2H), 1.04 (d, J = 6.7 Hz, 3H), 1.02-0.93 (m, 1H), 0.83-0.72 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 174.23, 162.96, 156.05, 137.43, 129.70, 129.46 (2C), 129.43 (2C), 128.46 (2C), 126.56, 114.26 (2C), 72.27, 65.96, 57.26, 55.56, 54.92, 53.57, 51.79, 39.23, 35.36, 33.36, 26.33, 16.82, 13.51, 10.95; MS (ESI) m/z 587.35 (M + Na)+.
2-(2-oxoimidazolidin-1-yl)ethyl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-((S)-2-methylbutyl)phenylsulfonamido)-1-phenylbutan-2-yl)carbamate (13g)
The title compound was obtained from 12a and 7g as described for 13a in 51% yield after flash-chromatography (1:4 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.5 Hz, 2H), 7.23-7.07 (m, 5H), 6.89 (d, J = 8.5 Hz, 2H), 5.30 (m, 1H), 5.00 (bs, 1H), 4.17-3.97 (m, 2H), 3.96-3.88 (m, 1H), 3.87-3.79 (m, 2H), 3.78 (s, 3H), 3.35-3.16 (m, 6H), 3.06-2.97 (m, 2H), 2.96-2.84 (m, 2H), 2.83-2.69 (m, 2H), 1.63-1.48 (m, 1H), 1.41-1.26 (m, 1H), 1.05-0.85 (m, 1H), 0.81-0.70 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 162.80, 162.75, 155.98, 137.86, 129.97, 129.38 (2C), 129.27 (2C), 128.28 (2C), 126.24, 114.15 (2C), 72.04, 62.10, 56.76, 55.50, 55.00, 52.96, 45.25, 42.80, 38.09, 34.98, 33.18, 26.46, 16.79, 10.96; MS (ESI) m/z 599.32 (M + Na)+.
2-(2-oxooxazolidin-3-yl)ethyl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-((S)-2-methylbutyl)phenylsulfonamido)-1-phenylbutan-2-yl)carbamate (13h)
The title compound was obtained from 12a and 7h as described for 13a in 68% yield after flash-chromatography (1:3 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 7.69-7.67 (m, 2H), 7.25-7.14 (m, 5H), 6.95-6.63 (m, 2H), 5.05 (d, J = 7.9 Hz, 1H), 4.34-3.99 (m, 4H), 3.93-3.66 (m, 6H), 3.48-3.27 (m, 4H), 3.14-2.92 (m, 4H), 2.91-2.73 (m, 2H), 2.68-2.50 (m, 1H), 1.51-1.34 (m, 1H), 1.13-0.91 (m, 1H), 0.94-0.74 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 162.90, 158.51, 155.83, 137.64, 129.86, 129.38 (2C), 129.32 (2C), 128.36 (2C), 126.35, 114.23 (2C), 72.06, 61.82, 57.02, 55.55, 55.00, 53.16, 44.81, 43.60, 35.21, 33.28, 26.38, 20.92, 16.78, 10.96; MS (ESI) m/z 599.86 (M + Na)+.
(R)-2-oxotetrahydrofuran-3-yl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-((S)-2-methylbutyl)phenylsulfonamido)-1-phenylbutan-2-yl)carbamate (13i)
The title compound was obtained from 12a and 7i as described for 13a in 64% yield after flash-chromatography (1:3 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 7.76-7.69 (m, 2H), 7.31-7.15 (m, 5H), 7.02-6.96 (m, 2H), 4.60 (t, J = 5.5 Hz, 1H), 4.33-4.21 (m, 1H), 3.87 (s, 3H), 3.84-3.76 (m, 1H), 3.75-3.56 (m, 2H), 3.38-3.14 (m, 3H), 3.13-3.01 (m, 1H), 2.99-2.80 (m, 2H), 2.10-1.96 (m, 1H), 1.95-1.65 (m, 3H), 1.63-1.51 (m, 1H), 1.50-1.36 (m, 1H), 1.13-0.97 (m, 1H), 0.91-0.78 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 173.73, 163.05, 155.59, 136.73, 129.88, 129.59 (2C), 129.22, 129.09, 128.65 (2C), 127.00, 114.36 (2C), 76.35, 70.88, 57.21, 55.64, 52.79, 33.45, 32.90, 32.61, 32.13, 31.95, 26.64, 16.84, 10.08; MS (ESI) m/z 571.34 (M + Na)+.
((S)-5-oxopyrrolidin-2-yl)methyl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-((S)-2-methylbutyl)phenylsulfonamido)-1-phenylbutan-2-yl)carbamate (13j)
The title compound was obtained from 12a and 7j as described for 13a in 59% yield after flash-chromatography (1:3 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 8.5 Hz, 2H), 7.32-7.11 (m, 6H), 6.92 (d, J = 8.5 Hz, 2H), 5.60 (d, J = 8.5 Hz, 1H), 4.15-3.99 (m, 2H), 3.69-3.60 (m, 7H), 3.18-2.68 (m, 4H), 2.38-2.18 (m, 2H), 2.15-2.04 (m, 1H), 1.71-1.28 (m, 4H), 1.11-0.86 (m, 2H), 0.89-0.71 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 178.63, 162.76, 155.82, 137.84, 129.96, 129.34 (2C), 129.28 (2C), 128.24 (2C), 126.25, 114.12 (2C), 72.01, 67.20, 56.65, 55.45, 55.15, 53.20, 52.95, 34.95, 33.14, 29.64, 26.44, 22.67, 16.78, 10.92; MS (ESI) m/z 584.01 (M + Na)+.
(R)-methyl 3-((((2S,3R)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamoyl)oxy)butanoate (14a)
The title compound was obtained from 12b and 7a as described for 13a in 46% yield after flash-chromatography (1:2 EtOAc/hexane) as colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 1H), 8.40 (d, J = 1.8 Hz, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.83 (dd, J = 8.5, 1.8 Hz, 1H), 7.25–7.15 (m, 5H), 6.02 (d, J = 7.9 Hz, 1H), 5.18-5.09 (m, 1H), 4.14-4.05 (m, 2H), 3.82-3.76 (m, 1H), 3.60 (s, 3H), 3.10 (dd, J = 14.9, 3.9 Hz, 1H), 3.08-3.03 (m, 1H), 3.02-2.92 (m, 1H), 2.94-2.83 (m, 2H), 2.76 (dd, J = 14.1, 7.0 Hz, 1H), 2.56-2.47 (m, 1H), 2.42-2.35 (m, 1H), 2.29-2.19 (m, 1H), 1.44-1.32 (m, 1H), 1.13 (d, J = 6.1 Hz, 3H), 1.01-0.96 (m, 1H), 0.83-0.73 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 172.05, 170.72, 157.89, 155.46, 137.66, 135.87, 134.26, 129.31 (2C), 128.54 (2C), 126.58, 124.84, 124.25, 122.24, 72.29, 67.57, 57.10, 54.17, 53.06, 51.78, 40.29, 35.03, 33.35, 26.47, 19.70, 16.79, 11.03; MS (ESI) m/z 614.26 (M + Na)+.
2-(2,2,2-trifluoroacetamido)ethyl ((2S,3R)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (14b)
The title compound was obtained from 12b and 7b as described for 13a in 66% yield after flash-chromatography (1:2 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 8.40 (d, J = 1.8 Hz, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.85-7.78 (m, 2H), 7.23-7.11 (m, 6H), 4.30 (t, J = 8.0 Hz, 2H), 4.07-3.99 (m, 1H), 3.88-3.78 (m, 3H), 3.56 (d, J = 3.7 Hz, 1H), 3.15 (dd, J = 15.3, 9.2 Hz, 1H), 3.06-2.94 (m, 2H), 2.89-2.78 (m, 2H), 1.57-1.49 (m, 1H), 1.44-1.36 (m, 1H), 1.06-0.94 (m, 2H), 0.87-0.70 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 157.94, 155.50, 155.44, 151.66, 137.35, 134.32, 132.74, 129.43 (2C), 129.23, 128.50 (2C), 126.65, 124.88, 124.31, 122.34, 71.90, 62.27, 57.03, 54.83, 53.11, 42.30, 35.95, 33.27, 26.37, 16.86, 10.98; MS (ESI) m/z 653.37 (M + Na)+.
2-(2,2-dichloroacetamido)ethyl ((2S,3R)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (14c)
The title compound was obtained from 12b and 7c as described for 13a in 61% yield after flash-chromatography (1:2 EtOAc/hexane) as foam. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 8.37 (d, J = 2.1 Hz, 1H), 8.17 (dd, J = 8.5, 2.4 Hz, 1H), 7.81 (dd, J = 8.5, 1.8 Hz, 1H), 7.31-7.08 (m, 5H), 6.98 (bs, 1H), 5.81 (s, 1H), 5.12 (bs, 1H), 5.07 (d, J = 7.9 Hz, 1H), 4.09-3.97 (m, 2H), 3.87-3.81 (m, 1H), 3.75-3.58 (m, 2H), 3.47-3.33 (m, 2H), 3.14-2.67 (m, 5H), 1.66-1.45 (m, 2H), 1.06-0.93 (m, 1H), 0.86-0.72 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 163.43, 157.20, 156.87, 154.57, 135.52, 135.02, 133.42, 128.50, 128.28, 127.92, 127.60, 126.48, 123.81, 123.44, 121.25, 72.24, 65.37, 63.43, 56.02, 54.80, 54.71, 47.29, 34.97, 25.91, 16.95, 14.16, 11.32; MS (ESI) m/z 666.99 (M−1+ Na)+, 668.93 (M+1+ Na)+.
(R)-5-oxohexan-2-yl ((2S,3R)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (14d)
The title compound was obtained from 12b and 7d as described for 13a in 55% yield after flash-chromatography (1:2 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 8.39 (d, J = 1.2 Hz, 1H), 8.17 (d, J = 9.2 Hz, 1H), 7.82 (dd, J = 8.5, 1.8 Hz, 1H), 7.23-7.06 (m, 6H), 4.72 (d, J = 7.9 Hz, 1H), 4.63-4.55 (m, 1H), 3.87-3.67 (m, 3H), 3.15-2.99 (m, 3H), 2.97-2.91 (m, 1H), 2.89-2.79 (m, 1H), 2.33 (t, J = 7.3 Hz, 1H), 2.00 (s, 3H), 1.79-1.51 (m, 4H), 1.45-1.34 (m, 1H), 1.07-0.92 (m, 4H), 0.85-0.72 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 207.93, 157.96, 156.23, 155.47, 137.56, 135.76, 134.28, 129.40 (2C), 128.49 (2C), 126.53, 124.80, 124.30, 122.22, 72.22, 71.02, 56.98, 55.01, 53.24, 39.44, 35.20, 33.33, 29.83, 29.77, 26.39, 20.14, 16.83, 11.00; MS (ESI) m/z 612.06 (M + Na)+.
2-((tert-butoxycarbonyl)amino)ethyl ((2S,3R)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (14e)
The title compound was obtained from 12b and 7e as described for 13a in 49% yield after flash-chromatography (1:2 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 8.38 (m, 1H), 8.14 (d, J = 8.5 Hz, 1H), 7.82 (dd, J = 8.5, 1.8 Hz, 1H), 7.24-7.11 (m, 5H), 4.86 (d, J = 6.7 Hz, 1H), 4.65 (bs, 1H), 3.98-3.88 (m, 2H), 3.86-3.74 (m, 2H), 3.70 (bs, 1H), 3.30-2.92 (m, 6H), 2.89-2.75 (m, 2H), 1.62-1.47 (m, 2H), 1.36 (s, 9H), 1.06-0.93 (m, 1H), 0.85-0.73 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 157.95, 156.27, 155.78, 155.56, 137.47, 135.67, 134.34, 129.47 (2C), 128.58 (2C), 126.68, 124.83, 124.38, 122.26, 79.51, 72.20, 64.36, 57.14, 55.09, 53.41, 39.95, 35.27, 33.40, 28.35 (3C), 26.41, 16.87, 11.02; MS (ESI) m/z 657.37 (M + Na)+.
(R)-methyl 3-((((2S,3R)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamoyl)oxy)-2-methylpropanoate (14f)
The title compound was obtained from 12b and 7f as described for 13a in 68% yield after flash-chromatography (1:2 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 1H), 8.37 (d, J = 1.2 Hz, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.82 (dd, J = 8.5, 1.8 Hz, 1H), 7.24-7.11 (m, 5H), 4.86 (d, J = 7.9 Hz, 1H), 4.08-3.97 (m, 2H), 3.82-3.68 (m, 3H), 3.56 (s, 3H), 3.14-2.92 (m, 4H), 2.89-2.72 (m, 2H), 2.69-2.57 (m, 1H), 1.62-1.47 (m, 1H), 1.46-1.36 (m, 1H), 1.06-0.96 (m, 4H), 0.82-0.70 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 174.21, 157.98, 156.11, 155.45, 137.36, 135.62, 134.25, 129.41 (2C), 128.49 (2C), 126.54, 124.80, 124.25, 122.20, 72.23, 66.01, 57.11, 55.05, 53.41, 51.83, 39.21, 35.35, 33.33, 26.34, 16.78, 13.48, 10.95; MS (ESI) m/z 614.30 (M + Na)+.
2-(2-oxoimidazolidin-1-yl)ethyl ((2S,3R)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (14g)
The title compound was obtained from 12b and 7g as described for 13a in 57% yield after flash-chromatography (1:2 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 1H), 8.41-8.38 (m, 1H), 8.16 (d, J = 9.2 Hz, 1H), 7.84 (dd, J = 8.5, 1.8 Hz, 1H), 7.24-7.09 (m, 6H), 5.28 (m, 1H), 4.81 (bs, 1H), 4.20-4.00 (m, 2H), 3.97-3.55 (m, 3H), 3.48-3.21 (m, 5H), 3.19-3.06 (m, 2H), 3.01-2.87 (m, 3H), 2.79-2.71 (m, 1H), 1.64-1.44 (m, 1H), 1.37-3.25 (m, 1H), 1.07-0.82 (m, 1H), 0.85-0.70 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 162.77, 157.93, 156.05, 155.36, 137.78, 136.03, 134.20, 129.28 (2C), 128.39 (2C), 126.39, 124.89, 124.16, 122.27, 71.76, 61.85, 56.54, 55.15, 52.71, 45.10, 42.86, 38.11, 34,89, 33.18, 26.50, 16.83, 11.03; MS (ESI) m/z 626.34 (M + Na)+.
2-(2-oxooxazolidin-3-yl)ethyl ((2S,3R)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (14h)
The title compound was obtained from 12b and 7h as described for 13a in 55% yield after flash-chromatography (1:2 EtOAc/hexane) as a colorless semi-solid. 1H NMR (400 MHz, CDCl3) δ 9.12 (s, 1H), 8.46 (d, J = 1.4 Hz, 1H), 8.22 (d, J = 8.7 Hz, 1H), 7.89 (dd, J = 8.7, 1.8 Hz, 1H), 7.34-7.14 (m, 6H), 5.07 (d, J = 8.6 Hz, 1H), 4.30-4.07 (m, 3H), 4.08-3.94 (m, 1H), 3.94-3.77 (m, 1H), 3.56-3.36 (m, 3H), 3.24-3.13 (m, 2H), 3.10-3.05 (m, 1H), 3.02-2.97 (m, 2H), 2.94-2.83 (m, 2H), 1.69-1.54 (m, 1H), 1.51-1.32 (m, 1H), 1.10-0.89 (m, 1H), 0.85-0.78 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 158.57, 157.95, 155.90, 155.42, 137.52, 135.88, 134.24, 129.35 (2C), 128.83, 128.46, 126.48, 124.86, 124.21, 122.24, 71.78, 61.79, 56.86, 55.10, 54.72, 52.95, 44.60, 43.65, 35.15, 33.29, 26.39, 16.79, 11.01; MS (ESI) m/z 626.90 (M + Na)+.
(R)-2-oxotetrahydrofuran-3-yl ((2S,3R)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (14i)
The title compound was obtained from 12b and 7i as described for 13a in 62% yield after flash-chromatography (1:2 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 9.15 (s, 1H), 8.45 (d, J = 3.7 Hz, 1H), 8.18 (dd, J = 8.5, 5.5 Hz, 1H), 7.87-7.81 (m, 1H), 7.26-7.12 (m, 6H), 4.58 (d, J = 4.9 Hz, 1H), 4.50-4.36 (m, 1H), 4.36-4.23 (m, 1H), 3.85-3.56 (m, 3H), 3.37-3.07 (m, 3H), 3.05-2.91 (m, 2H), 1.61 (bs, 1H), 1.50-1.33 (m, 2H), 1.27-1.19 (m, 1H), 1.12-0.96 (m, 2H), 0.87-0.73 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 173.72, 158.10, 155.55, 155.46, 136.59, 135.82, 134.31, 129.06 (2C), 128.65 (2C), 127.03, 124.88, 124.30, 122.43, 76.34, 70.77, 57.04, 52.66, 33.37, 32.50, 32.17, 29.66, 28.68, 26.58, 16.79, 11.05; MS (ESI) m/z 598.32 (M + Na)+.
((S)-5-oxopyrrolidin-2-yl)methyl ((2S,3R)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (14j)
The title compound was obtained from 12b and 7j as described for 13a in 60% yield after flash-chromatography (1:2 EtOAc/hexane) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.12 (s, 1H), 8.39 (s, 1H), 8.14 (d, J = 8.5 Hz, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.24-7.09 (m, 6H), 5.62 (d, J = 9.2 Hz, 1H), 4.16-3.94 (m, 2H), 3.93-3.51 (m, 3H), 3.24-3.03 (m, 2H), 3.01-2.83 (m, 3H), 2.81-2.69 (m, 1H), 2.33-1.98 (m, 4H), 1.69-1.42 (m, 2H), 1.39-1.26 (m, 1H), 1.06-0.89 (m, 1H), 0.80-0.69 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 177.85, 156.99, 155.01, 154.27, 136.75, 134.97, 133.13, 128.92 (2C), 128.09 (2C), 125.37, 123.83, 123.04, 121.16, 71.81, 66.25, 55.37, 54.22, 52.23, 51.67, 33.86, 32.09, 28.68, 25.47, 21.82, 16.06, 10.16; MS (ESI) m/z 611.29 (M + Na)+.
(R)-methyl 3-((((2S,3R)-3-hydroxy-4-(4-methoxy-N-pentylphenylsulfonamido)-1-phenylbutan-2-yl)carbamoyl)oxy)butanoate (15a)
The title compound was obtained from 12c and 7a as described for 13a in 63% yield after flash-chromatography (1:3 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 7.69-7.62 (m, 2H), 7.21-7.12 (m, 5H), 6.92-6.86 (m, 2H), 6.01 (d, J = 8.5 Hz, 1H), 5.20-5.12 (m, 1H), 4.16-3.97 (m, 2H), 3.86-3.76 (m, 4H), 3.60 (s, 3H), 3.20 (dd, J = 14.6, 4.9 Hz, 1H), 3.05-2.87 (m, 3H), 2.54-2.42 (m, 3H), 1.50-1.31 (m, 2H), 1.25-1.10 (m, 8H), 0.78 (t, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.95, 167.71, 162.92, 137.87, 130.46, 129.31 (2C), 129.25 (2C), 128.49 (2C), 126.48, 114.28 (2C), 72.27, 67.57, 55.57, 54.41, 51.77, 50.41, 40.35, 35.04, 30.99, 29.62, 28.81, 21.26, 19.76, 13.93; MS (ESI) m/z 587.16 (M + Na)+.
2-(2,2,2-trifluoroacetamido)ethyl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-pentylphenylsulfonamido)-1-phenylbutan-2-yl)carbamate (15b)
The title compound was obtained from 12c and 7b as described for 13a in 56% yield after flash-chromatography (1:3 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.5 Hz, 1H), 7.68-7.59 (m, 2H), 7.24-7.12 (m, 5H), 6.92-6.86 (m, 2H), 4.29 (t, J = 7.9 Hz, 1H), 4.08-3.96 (m, 1H), 3.91-3.75 (m, 5H), 3.57-3.42 (m, 1H), 3.14-3.07 (m, 1H), 3.05-2.96 (m, 2H), 2.94-2.77 (m, 3H), 2.44-2.33 (m, 1H), 1.68 (bs, 1H), 1.48-1.32 (m, 2H), 1.22-1.09 (m, 5H), 0.77 (t, J = 7.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.92, 155.42, 151.65, 137.39, 130.31, 129.43 (2C), 129.36 (2C), 129.24, 128.42 (2C), 126.53, 114.28 (2C), 72.00, 62.52, 55.58, 54.75, 52.13, 50.34, 42.29, 35.80, 28.69, 28.13, 22.13, 13.89; MS (ESI) m/z 626.32 (M + Na)+.
(R)-5-oxohexan-2-yl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-pentylphenylsulfonamido)-1-phenylbutan-2-yl)carbamate (15d)
The title compound was obtained from 12c and 7d as described for 13a in 71% yield after flash-chromatography (1:3 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 7.66-7.61 (m, 2H), 7.25-7.10 (m, 5H), 6.94-6.86 (m, 2H), 4.89-4.81 (m, 1H), 4.75 (d, J = 7.7 Hz, 1H), 4.64-4.56 (m, 1H), 3.92-3.67 (m, 5H), 3.18-2.99 (m, 3H), 2.98-2.92 (m, 1H), 2.89-2.79 (m, 1H), 2.36 (t, J = 7.3 Hz, 1H), 2.03 (s, 3H), 1.75-1.58 (m, 2H), 1.48-1.35 (m, 2H), 1.53-1.25 (m, 2H), 1.26-1.04 (m, 4H), 1.02 (d, J = 6.1 Hz, 3H), 0.78 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 207.96, 168.71, 162.97, 151.14, 137.74, 130.30, 129.44, 129.31 (2C), 128.44 (2C), 126.74, 114.30 (2C), 79.35, 72.27, 71.06, 55.50, 52.23, 50.40, 39.46, 38,56, 35.20, 29.86, 28.74, 25.42, 22.17, 19.81, 13.90; MS (ESI) m/z 585.40 (M + Na)+.
2-((tert-butoxycarbonyl)amino)ethyl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-pentylphenylsulfonamido)-1-phenylbutan-2-yl)carbamate (15e)
The title compound was obtained from 12c and 7e as described for 13a in 62% yield after flash-chromatography (1:3 EtOAc/hexane) as a colorless viscous liquid. 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.5 Hz, 2H), 7.26-7.13 (m, 5H), 6.90 (d, J = 9.2 Hz, 2H), 5.03-4.76 (m, 1H), 4.68 (bs, 1H), 3.99-3.87 (m, 2H), 3.85-3.73 (m, 5H), 3.66-3.59 (m, 1H), 3.44-2.92 (m, 7H), 2.89-2.78 (m, 1H), 1.46-1.31 (m, 10H), 1.25-1.06 (m, 5H), 0.78 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.98, 156.36, 155.78, 137.67, 130.23, 129.46 (2C), 129.32 (2C), 128.49 (2C), 126.54, 114.32 (2C), 79.48, 72.20, 64.23, 55.59, 55.07, 52.27, 50.43, 39.93, 35.27, 28.74, 28.34 (3C), 28.17, 22.17, 13.90; MS (ESI) m/z 630.53 (M + Na)+.
(R)-methyl 3-((((2S,3R)-3-hydroxy-4-(4-methoxy-N-pentylphenylsulfonamido)-1-phenylbutan-2-yl)carbamoyl)oxy)-2-methylpropanoate (15f)
The title compound was obtained from 12c and 7f as described for 13a in 61% yield after flash-chromatography (1:3 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 7.66-7.60 (m, 2H), 7.25-7.11 (m, 5H), 6.92-6.86 (m, 2H), 4.87 (d, J = 7.9 Hz, 1H), 4.09-3.97 (m, 2H), 3.83-3.71 (m, 5H), 3.69-3.61 (m, 1H), 3.57 (s, 3H), 3.11-2.90 (m, 4H), 2.87-2.78 (m, 1H), 2.68-2.56 (m, 1H), 1.45-1.34 (m, 2H), 1.24-1.07 (m, 5H), 1.04 (d, J = 7.3 Hz, 3H), 0.77 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 174.25, 162.93, 156.18, 137.55, 130.21, 129.44 (2C), 129.30 (2C), 128.45 (2C), 126.43, 114.27 (2C), 72.22, 65.99, 55.56, 55.01, 52.36, 51.80, 50.45, 39.24, 35.31, 28.71, 28.18, 22.14, 13.87, 13.52; MS (ESI) m/z 587.36 (M + Na)+.
2-(2-oxoimidazolidin-1-yl)ethyl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-pentylphenylsulfonamido)-1-phenylbutan-2-yl)carbamate (15g)
The title compound was obtained from 12c and 7g as described for 13a in 70% yield after flash-chromatography (1:3 EtOAc/hexane) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 9.2 Hz, 2H), 7.24-7.10 (m, 5H), 6.89 (d, J = 9.2 Hz, 2H), 5.16 (d, J = 8.5 Hz, 1H), 4.75 (bs, 1H), 4.14-4.02 (m, 2H), 3.95-3.89 (m, 1H), 3.86-3.79 (m, 2H), 3.78 (s, 3H), 3.39-3.13 (m, 5H), 3.05-2.99 (m, 1H), 2.95-2.71 (m, 3H), 2.68-2.59 (m, 1H), 1.54-1.26 (m, 2H), 1.23-0.99 (m, 6H), 0.73 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.94, 162.74, 156.23, 137.92, 130.61, 129.41 (2C), 129.37 (2C), 128.49 (2C), 126.45, 114.31 (2C), 71.94, 62.12, 55.64, 55.09, 51.84, 50.23, 45.27, 43.02, 38.20, 34.95, 28.84, 28.12, 22.24, 13.98; MS (ESI) m/z 599.38 (M + Na)+.
2-(2-oxooxazolidin-3-yl)ethyl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-pentylphenylsulfonamido)-1-phenylbutan-2-yl)carbamate (15h)
The title compound was obtained from 12c and 7h as described for 13a in 67% yield after flash-chromatography (1:3 EtOAc/hexane) as a colorless semi-solid. 1H NMR (400 MHz, CDCl3) δ 7.71-7.67 (m, 2H), 7.27-7.17 (m, 5H), 6.96-6.62 (m, 2H), 5.10 (d, J = 8.2 Hz, 1H), 4.25-4.12 (m, 3H), 3.92-3.78 (m, 5H), 3.74-3.65 (m, 1H), 3.52-3.31 (m, 4H), 3.21-2.95 (m, 5H), 2.92-2.79 (m, 1H), 1.59-1.42 (m, 2H), 1.28-1.12 (m, 5H), 0.82 (t, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.92, 158.59, 155.98, 137.77, 130.40, 129.36, 129.31 (3C), 128.41 (2C), 126.40, 114.29 (2C), 71.94, 61.87, 55.57, 55.14, 51.93, 50.28, 44.85, 43.68, 38.84, 35.17, 28.75, 28.11, 22.16, 13.90; MS (ESI) m/z 599.86 (M + Na)+.
(R)-2-oxotetrahydrofuran-3-yl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-pentylphenylsulfonamido)-1-phenylbutan-2-yl)carbamate (15i)
The title compound was obtained from 12c and 7i as described for 13a in 50% yield after flash-chromatography (1:3 EtOAc/hexane) as a colorless semi-solid. 1H NMR (400 MHz, CDCl3) δ 7.76-7.69 (m, 2H), 7.30-7.15 (m, 5H), 7.01-6.96 (m, 2H), 4.62 (t, J = 5.5 Hz, 1H), 4.53-4.41 (m, 1H), 4.34-4.21 (m, 1H), 3.87 (s, 3H), 3.78-3.64 (m, 2H), 3.36-3.18 (m, 3H), 3.17-3.00 (m, 3H), 2.14-1.76 (m, 3H), 1.54-1.40 (m, 2H), 1.31-1.10 (m, 5H), 0.85 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.80, 163.01, 155.67, 136.72, 130.40, 129.43 (2C), 129.09 (2C), 128.65 (2C), 127.00, 114.34 (2C), 76.38, 70.99, 57.19, 56.88, 56.64, 51.69, 50.34, 32.36, 32.15, 28.77, 28.17, 22.21, 13.95; MS (ESI) m/z 571.34 (M + Na)+.
((S)-5-oxopyrrolidin-2-yl)methyl ((2S,3R)-3-hydroxy-4-(4-methoxy-N-pentylphenylsulfonamido)-1-phenylbutan-2-yl)carbamate (15j)
The title compound was obtained from 12c and 7j as described for 13a in 56% yield after flash-chromatography (1:3 EtOAc/hexane) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 8.5 Hz, 2H), 7.23-7.09 (m, 5H), 6.93-6.86 (m, 3H), 5.47 (d, J = 7.9 Hz, 1H), 4.10-3.93 (m, 2H), 3.91-3.81 (m, 2H), 3.81-3.70 (m, 4H), 3.65 (dd, J = 11.0, 7.3 Hz 1H), 3.16-2.99 (m, 3H), 2.94 (dd, J = 14.0, 4.3 Hz 1H), 2.82-2.71 (m, 1H), 2.31-2.15 (m, 2H), 2.14-2.02 (m, 1H), 1.66-1.53 (m, 1H), 1.51-1.29 (m, 2H), 1.26-1.03 (m, 5H), 0.77 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 178.75, 162.92, 156.03, 138.96, 129.91, 129.56 (2C), 129.49 (2C), 128.36 (2C), 126.43, 114.30 (2C), 71.27, 66.36, 54.61, 54.25, 52.35, 50.85, 49.11, 36.03, 28.74, 27.81, 27.02, 21.86, 21.21, 13.06; MS (ESI) m/z 583.88 (M + Na)+.
(R)-methyl 3-((((2S,3R)-3-hydroxy-4-(N-pentylbenzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamoyl)oxy)butanoate (16a)
The title compound was obtained from 12d and 7a as described for 13a in 51% yield after flash-chromatography (1:2 EtOAc/hexane) as a colorless semi-solid. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 8.43 (d, J = 1.8, Hz, 1H), 8.18 (d, J = 8.5 Hz, 1H), 7.87 (dd, J = 8.5, 1.8 Hz, 1H), 7.26-7.11 (m, 5H), 6.03 (d, J = 8.5, Hz, 1H), 5.21-5.11 (m, 1H), 4.18-4.09 (m, 2H), 3.87-3.79 (m, 1H), 3.60 (s, 3H), 3.29 (dd, J = 14.6, 4.8 Hz, 1H), 3.06-2.86 (m, 3H), 2.62-2.50 (m, 1H), 2.45-2.21 (m, 2H), 1.50-1.36 (m, 2H), 1.24-1.03 (m, 8H), 0.76 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.27, 170.75, 157.86, 155.49, 137.77, 136.30, 134.32, 129.30 (2C), 128.60 (2C), 126.62, 124.78, 124.33, 122.16, 72.18, 67.62, 54.68, 51.81, 50.39, 40.34, 35.13, 29.66, 28.79, 28.21, 22.18, 19.75, 13.90; MS (ESI) m/z 614.26 (M + Na)+.
2-(2,2,2-trifluoroacetamido)ethyl ((2S,3R)-3-hydroxy-4-(N-pentylbenzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (16b)
The title compound was obtained from 12d and 7b as described for 13a in 63% yield after flash-chromatography (1:2 EtOAc/hexane) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 8.40 (d, J = 1.8 Hz, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.85-7.80 (m, 2H), 7.23-7.13 (m, 5H), 4.30 (t, J = 7.9 Hz, 2H), 4.08-4.00 (m, 1H), 3.88-3.80 (m, 3H), 3.51 (d, J = 3.1 Hz, 1H), 3.21-2.98 (m, 5H), 1.50-1.37 (m, 2H), 1.22-0.99 (m, 6H), 0.73 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 157.92, 155.49, 155.44, 152.86, 137.28, 136.17, 134.31, 129.44 (2C), 128.51 (3C), 126.66, 124.78, 124.35, 122.22, 72.02, 62.28, 54.95, 52.11, 50.36, 42.31, 35.89, 28.71, 28.19, 22.15, 13.88; MS (ESI) m/z 653.37 (M + Na)+.
2-(2,2-dichloroacetamido)ethyl ((2S,3R)-3-hydroxy-4-(N-pentylbenzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (16c)
The title compound was obtained from 12d and 7c as described for 13a in 58% yield after flash-chromatography (1:2 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 8.44-8.33 (m, 1H), 8.20-8.15 (m, 1H), 7.83 (dd, J = 9.2, 1.8 Hz, 1H), 7.27-7.10 (m, 5H), 6.97 (bs, 1H), 5.91-5.78 (m, 1H), 5.08 (d, J = 9.2 Hz, 1H), 4.09-3.97 (m, 2H), 3.92-3.81 (m, 2H), 3.68-3.53 (m, 1H), 3.49-3.23 (m, 3H), 3.16-2.81 (m, 3H), 1.53-1.25 (m, 2H), 1.23-0.99 (m, 6H), 0.73 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 163.47, 157.12, 155.61, 154.64, 136.62, 135.22, 133.48, 128.51 (2C), 127.68 (2C), 125.80, 123.81, 123.51, 121.23, 71.21, 65.36, 62.22, 54.38, 51.23, 49.45, 39.37, 34.43, 27.83, 27.27, 21.26, 12.98; MS (ESI) m/z 666.92 (M−1+ Na)+, 668.93 (M+1+ Na)+.
(R)-5-oxohexan-2-yl ((2S,3R)-3-hydroxy-4-(N-pentylbenzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (16d)
The title compound was obtained from 12d and 7d as described for 13a in 65% yield after flash-chromatography (1:2 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 8.40 (d, J = 1.8 Hz, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.83 (dd, J = 8.5, 1.8 Hz, 1H), 7.23-7.06 (m, 5H), 4.81 (d, J = 8.5 Hz, 1H), 4.64-4.56 (m, 1H), 3.92-3.67 (m, 3H), 3.21-3.04 (m, 1H), 3.01-2.92 (m, 1H), 2.89-2.79 (m, 2H), 2.35 (t, J = 7.3 Hz, 1H), 2.01 (s, 3H), 1.76-1.59 (m, 2H), 1.48-1.36 (m, 2H), 1.33-1.25 (m, 2H), 1.23-1.03 (m, 5H), 1.02 (d, J = 6.1 Hz, 3H), 0.79 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 207.99, 157.92, 156.35, 155.45, 137.68, 136.24, 134.28, 129.37 (2C), 128.46 (2C), 126.50, 124.68, 124.31, 122.07, 72.17, 71.01, 55.08, 52.07, 50.24, 39.44, 35.13, 29.81, 29.78, 28.69, 28.14, 22.11, 20.13, 13.84; MS (ESI) m/z 612.43 (M + Na)+.
2-((tert-butoxycarbonyl)amino)ethyl ((2S,3R)-3-hydroxy-4-(N-pentylbenzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (16e)
The title compound was obtained from 12d and 7e as described for 13a in 54% yield after flash-chromatography (1:2 EtOAc/hexane) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 8.39 (m, 1H), 8.14 (d, J = 8.5 Hz, 1H), 7.82 (dd, J = 8.5, 1.8 Hz, 1H), 7.22-7.09 (m, 5H), 4.85 (d, J = 6.7 Hz, 1H), 4.65 (bs, 1H), 3.97-3.88 (m, 2H), 3.85-3.76 (m, 2H), 3.70-2.62 (m, 1H), 3.29-3.02 (m, 6H), 3.01-2.92 (m, 1H), 2.88-2.78 (m, 1H), 1.49-1.32 (m, 10H), 1.23-1.05 (m, 5H), 0.75 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 157.94, 156.38, 155.79, 155.50, 137.59, 136.14, 134.33, 129.43 (2C), 128.56 (2C), 126.64, 124.72, 123.37, 122.12, 79.32, 72.13, 64.34, 55.17, 52.23, 50.38, 39.93, 35.22, 28.85, 28.24 (3C), 28.04, 22.16, 13.87; MS (ESI) m/z 657.36 (M+Na)+.
(R)-methyl 3-((((2S,3R)-3-hydroxy-4-(N-pentylbenzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamoyl)oxy)-2-methylpropanoate (16f)
The title compound was obtained from 12d and 7f as described for 13a in 48% yield after flash-chromatography (1:2 EtOAc/hexane) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 8.39 (d, J = 1.8 Hz, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.82 (dd, J = 8.5, 1.8 Hz, 1H), 7.26-7.12 (m, 6H), 4.92-4.81 (m, 1H), 4.09-3.98 (m, 2H), 3.93-3.51 (m, 3H), 3.57(s, 3H), 3.19-3.02 (m, 3H), 2.99-2.93 (m, 1H), 2.89-2.81 (m, 1H), 2.68-2.59 (m, 1H), 1.47-1.36 (m, 1H), 1.22-1.08 (m, 5H), 1.04 (d, J = 7.3 Hz, 3H), 0.77 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 174.26, 157.94, 156.25, 155.47, 137.49, 136.10, 134.29, 129.42 (2C), 128.51 (2C), 126.56, 124.71, 124.31, 122.11, 72.23, 66.06, 55.15, 52.31, 51.81, 50.40, 39.26, 35.32, 28.68, 28.22, 22.12, 13.84, 13.51; MS (ESI) m/z 614.36 (M + Na)+.
2-(2-oxoimidazolidin-1-yl)ethyl ((2S,3R)-3-hydroxy-4-(N-pentylbenzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (16g)
The title compound was obtained from 12d and 7g as described for 13a in 57% yield after flash-chromatography (1:2 EtOAc/hexane) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 1H), 8.42 (d, J = 1.2 Hz, 1H), 8.14 (d, J = 8.5 Hz, 1H), 7.84 (dd, J = 8.5, 1.8 Hz, 1H), 7.21-7.08 (m, 6H), 5.54 (d, J = 8.5 Hz, 1H), 5.11 (s, 1H), 4.17-4.06 (m, 1H), 3.97-3.80 (m, 3H), 3.50-3.41 (m, 1H), 3.38-3.18 (m, 4H), 3.18-3.01 (m, 3H), 3.00-2.87 (m, 1H), 2.79-2.65 (m, 1H), 1.54-1.33 (m, 2H), 1.23-0.99 (m, 6H), 0.73 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.92, 158.00, 156.06, 155.22, 137.97, 136.43, 134.13, 129.18 (2C), 128.24 (2C), 126.20, 124.67, 124.05, 122.03, 71.78, 61.67, 55.10, 51.39, 49.72, 42.74, 38.05, 34.73, 28.62, 27.77, 25.32, 22.01, 13.76; MS (ESI) m/z 626.34 (M + Na)+.
2-(2-oxooxazolidin-3-yl)ethyl ((2S,3R)-3-hydroxy-4-(N-pentylbenzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (16h)
The title compound was obtained from 12d and 7h as described for 13a in 66% yield after flash-chromatography (1:2 EtOAc/hexane) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.18 (s, 1H), 8.47 (d, J = 1.8 Hz, 1H), 8.23-8.20 (m, 1H), 7.92-7.89 (m, 1H), 7.29-7.18 (m, 6H), 5.01 (d, J = 7.3 Hz, 1H), 4.23-4.17 (m, 2H), 4.05-3.81 (m, 3H), 3.69 (bs, 1H), 3.52-3.34 (m, 4H), 3.29-3.10 (m, 3H), 3.07-2.97 (m, 1H), 2.92-2.78 (m, 1H), 1.57-1.41 (m, 2H), 1.29-1.10 (m, 5H), 0.81 (t, J = 4.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 158.72, 158.04, 156.08, 155.46, 137.81, 136.43, 134.33, 129.37 (2C), 128.49 (2C), 126.50, 124.82, 124.29, 122.19, 71.83, 61.93, 55.29, 51.80, 50.16, 44.70, 43.72, 35.14, 29.26, 28.77, 28.18, 22.19, 13.92; MS (ESI) m/z 626.85 (M + Na)+.
(R)-2-oxotetrahydrofuran-3-yl ((2S,3R)-3-hydroxy-4-(N-pentylbenzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (16i)
The title compound was obtained from 12d and 7i as described for 13a in 53% yield after flash-chromatography (1:2 EtOAc/hexane) as an amorphous white solid. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 8.42 (d, J = 1.8 Hz, 1H), 8.14 (d, J = 8.5 Hz, 1H), 7.85 (dd, J = 8.5, 1.8 Hz, 1H), 7.23-7.09 (m, 5H), 4.55 (t, J = 5.6 Hz, 1H), 4.42-4.34 (m, 1H), 4.30-4.21 (m, 1H), 3.77 (dd, J = 3.7, 9.2 Hz, 1H), 3.70-3.57 (m, 2H), 3.37-3.30 (m, 1H), 3.29-3.16 (m, 2H), 3.15-3.06 (m, 3H), 2.02-1.90 (m, 1H), 1.89-1.75 (m, 1H), 1.50-1.35 (m, 2H), 1.23-1.04 (m, 5H), 0.76 (t, J = 7.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.78, 158.11, 155.60, 155.35, 136.58, 136.32, 134.27, 129.03 (2C), 128.61 (2C), 126.99, 124.73, 124.25, 122.23, 76.35, 70.88, 57.04, 56.82, 51.61, 50.22, 32.35, 28.69, 28.08, 22.13, 14.15, 13.84; MS (ESI) m/z 598.13 (M + Na)+.
((S)-5-oxopyrrolidin-2-yl)methyl ((2S,3R)-3-hydroxy-4-(N-pentylbenzo[d]thiazole-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (16j)
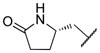
The title compound was obtained from 12d and 7j as described for 13a in 68% yield after flash-chromatography (1:2 EtOAc/hexane) as foam. 1H NMR (400 MHz, CDCl3) δ 9.12 (s, 1H), 8.40 (s, 1H), 8.14 (d, J = 8.5 Hz, 1H), 7.83 (d, J = 8.5 Hz, 1H), 7.25-7.08 (m, 6H), 4.36-4.00 (m, 2H), 3.96-3.68 (m, 3H), 3.66-3.53 (m, 1H), 3.31-3.05 (m, 4H), 2.99-2.87 (m, 1H), 2.83-2.66 (m, 1H), 2.30-1.99 (m, 3H), 1.67-1.30 (m, 3H), 1.25-1.04 (m, 5H), 0.72 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 178.89, 157.96, 155.92, 155.34, 137.86, 136.45, 134.22, 129.27 (2C), 128.37 (2C), 126.41, 124.73, 124.16, 122.07, 71.81, 67.28, 55.21, 53.29, 51.58, 49.84, 34.82, 29.71, 28.68, 27.88, 22.72, 22.09, 13.81; MS (ESI) m/z 611.29 (M + Na)+.
HIV-1 Protease Inhibition Assays
The protease inhibitory activities were determined by a fluorescence resonance energy transfer (FRET) method22, 23, 34 in a semi-high throughput format. Protease substrate (Arg-Glu(EDANS)-Ser-Gln-Asn-Tyr-Pro-Ile-Val-Gln Lys- (DABCYL)-Arg) was purchased from Molecular Probes. The expression, isolation, and purification of wild-type and mutant HIV-1 protease used for binding experiments were carried out as previously described.39 Wild-type HIV-1 protease (Q7K) and its MDR variants (M1: L10I, G48V, I54V, L63P, V82A; M2: L10I, L63P, A71V, G73S, I84V, L90M; M3: I50V, A71V) were desalted through PD-10 columns (Amersham Biosciences). Sodium acetate (20 mM, pH 5) was used as the elution buffer. Apparent protease concentrations were around ~50 nM as estimated by UV spectrophotometry at 280 nm. Fluorescence measurements were carried out on EnVisoin plate reader (PerkinElmer). Excitation and emission filters were 340 and 492 nm, respectively. All inhibitors were dissolved in DMSO and diluted to 12 appropriate concentrations in 96-well plate (8 inhibitors/plate). Protease (2 μL) and inhibitor or DMSO (1 μL) were added into a half area 96-well assay plate containing 37 μM HIV-1 substrate buffer [0.1 M sodium acetate, 1 M sodium chloride, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM dithiothreitol (DTT), 2% DMSO, and 1 mg/mL bovine serum albumin (BSA) with an adjusted pH of 4.7]. All pipetting was performed by a liquid handling system (Tecan Genesis and TeMo). Protease substrate (40 μL) was quickly injected into the assay plate with one row at a time by Envision injector to initiate the cleavage reaction. Each reaction was recorded for 5 min. Inhibitor binding dissociation constant (Ki) values were obtained by nonlinear regression fitting (GraFit 5, Erithacus software) of the plot of initial velocity as a function of inhibitor concentrations based on the Morrison equation.40 The initial velocities were derived from the linear range of reaction curves.
Antiviral Assays
Drug susceptibility assays were carried out by Monogram Biosciences against three patient-derived strains of wild-type HIV-1 from clades A, B, and C and against two multidrug-resistant HIV-1 variants. Assays were carried out according to protocols detailed by Petropoulos et al.41 and on the website http://www.monogramvirology.com. Genbank accession numbers for the PR/RT regions of the six HIV-1 strains used in antiviral assays are as follows: WT-control, HQ179654; WT-A, HQ179655; WT-B, HQ179656; WT-C, HQ179657; MDR, HQ179658; MDR1, HQ179659.
Protein Crystallography
The expression, isolation and purification of wild-type HIV-1 protease used for crystallization experiments were carried out as previously described.39 Co-crystals of the inhibitors with the wild-type protease were grown at room temperature by hanging drop vapor diffusion method. Protease concentration of 1.6 mg/ml was used with 3-fold molar excess of inhibitors to set the crystallization drops. The reservoir solution consisted of 126 mM phosphate buffer at pH 6.2, 63 mM sodium citrate and 24–29% ammonium sulfate.
The crystals used for data collection were mounted in Mitegen Micromounts and flash frozen over a nitrogen stream. Intensity data for 13c wild-type protease complex were collected at −80 °C on an in-house Rigaku X-ray generator equipped with an R-axis IV image plate. 180 frames were collected per crystal with an angular separation of 1° and no overlap between frames. Intensity data for protease complexes of 13g, 13f and 14j were collected under cryogenic conditions at BioCARS 14-BMC beamline at Argonne National Laboratory (Advanced Photon Source, Chicago, IL). Diffraction images for all the four complexes were indexed and scaled using the program HKL2000.42
The crystal structures were solved and refined with the programs within the CCP4 interface.43 Structure solutions for all the wild-type protease-inhibitor complexes were obtained with the molecular replacement package AMoRe44 using 1F7A45 as the starting model. Model building was performed using the interactive graphics program Coot.46 Conjugate gradient refinement using Refmac547 was performed by incorporating Schomaker and Trueblood tensor formulation of TLS (translation, libration, screw-rotation) parameters.48–50 The working R (Rfactor) and its cross validation (Rfree) were monitored throughout the refinement. The data collection and refinement statistics are shown in Table 4.
Table 4.
X-ray Crystallographic Data Collection and Refinement Statistics for Complexes of 13c, 13f, 13g and 14j with the Wild-type HIV-1 Protease
| 13c | 13f | 13g | 14j | |
|---|---|---|---|---|
| Resolution (Å) | 1.78 | 1.40 | 1.40 | 1.55 |
| Space group | P212121 | P212121 | P212121 | P212121 |
| a (Å) | 50.73 | 50.83 | 50.71 | 50.73 |
| b (Å) | 57.76 | 57.97 | 57.93 | 57.85 |
| c (Å) | 61.74 | 62.00 | 61.82 | 61.69 |
| Z | 4 | 4 | 4 | 4 |
| Rmerge (%) | 4.3 | 5.9 | 5.4 | 5.6 |
| Completeness (%) | 98.7 | 99.6 | 99.1 | 97.9 |
| Total no. of reflections | 121403 | 252434 | 244283 | 169795 |
| No. of unique reflections | 17839 | 36681 | 36239 | 26519 |
| Rfree (%) | 21.7 | 18.9 | 20.4 | 19.6 |
| Rfactor (%) | 16.0 | 17.3 | 17.9 | 17.0 |
| RMSD in: | ||||
| Bond lengths (Å) | 0.009 | 0.009 | 0.009 | 0.009 |
| RMS Angle (°) | 1.310 | 1.296 | 1.368 | 1.393 |
| Temperature (°C) | −80 | −80 | −80 | −80 |
| PDB ID | 4DJO | 4DJP | 4DJQ | 4DJR |
Supplementary Material
Acknowledgments
This research was supported in part by grants from the National Institutes of General Medical Sciences of the NIH (GM066524, GM082209, AI41404, AI43198). We thank Kaneka USA for their generous gifts of chiral epoxides, Ellen A. Nalivaika for providing the wild-type and MDR HIV-1 proteases, the AIDS Research and Reference Reagent Program (NIAD, NIH) for reference protease inhibitors, Monogram Biosciences for antiviral testing, and members of the Rana, Tidor and Schiffer laboratories for helpful discussions, particularly Dr. Huricha Baigude for his help in HPLC data collection. In addition, we would like to thank Kuan Hung Lin for his help in the refinement of the wild-type protease complex of 13c and Shivender Shandilya for collecting the data on complexes of 13f, 13g and 14j with the wild-type protease at BioCARS beamline of the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source for x-ray data collection was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. DE-AC02-06CH11357. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under grant number RR007707.
Footnotes
Supporting Information Available: Detailed experimental procedures for the synthesis of intermediate activated carbonates (7a-j), (R)-hydroxyethylamines (10a-b) and (R)-(hydroxyethylamino) sulfonamides (12a-d), characterization data for intermediates, results of HPLC analysis for all final compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Bartlett JA, DeMasi R, Quinn J, Moxham C, Rousseau F. Overview of the effectiveness of triple combination therapy in antiretroviral-naive HIV-1 infected adults. AIDS. 2001;15:1369–1377. doi: 10.1097/00002030-200107270-00006. [DOI] [PubMed] [Google Scholar]
- 2.Gulick RM, Mellors JW, Havlir D, Eron JJ, Meibohm A, Condra JH, Valentine FT, McMahon D, Gonzalez C, Jonas L, Emini EA, Chodakewitz JA, Isaacs R, Richman DD. 3-Year suppression of HIV viremia with indinavir, zidovudine, and lamivudine. Ann Intern Med. 2000;133:35–39. doi: 10.7326/0003-4819-133-1-200007040-00007. [DOI] [PubMed] [Google Scholar]
- 3.Palella FJ, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD, The HIVOSI. Declining morbidity mortality among patients with advanced human immunodeficiency virus infection. N Engl J Med. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 4.Hogg RS, Heath KV, Yip B, Craib KJP, O’Shaughnessy MV, Schechter MT, Montaner JSG. Improved survival among HIV-infected individuals following initiation of antiretroviral therapy. JAMA. 1998;279:450–454. doi: 10.1001/jama.279.6.450. [DOI] [PubMed] [Google Scholar]
- 5.Waters L, Nelson M. Why do patients fail HIV therapy? Int J Clin Pract. 2007;61:983–990. doi: 10.1111/j.1742-1241.2007.01383.x. [DOI] [PubMed] [Google Scholar]
- 6.Condra JH, Schleif WA, Blahy OM, Gabryelski LJ, Graham DJ, Quintero JC, Rhodes A, Robbins HL, Roth E, Shivaprakash M, Titus D, Yang T, Tepplert H, Squires KE, Deutsch PJ, Emini EA. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature. 1995;374:569–571. doi: 10.1038/374569a0. [DOI] [PubMed] [Google Scholar]
- 7.Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- 8.Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure. 2002;10:369–381. doi: 10.1016/s0969-2126(02)00720-7. [DOI] [PubMed] [Google Scholar]
- 9.Kim EE, Baker CT, Dwyer MD, Murcko MA, Rao BG, Tung RD, Navia MA. Crystal structure of HIV-1 protease in complex with VX-478, a potent and orally bioavailable inhibitor of the enzyme. J Am Chem Soc. 1995;117:1181–1182. [Google Scholar]
- 10.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang Y-F, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Surleraux DLNG, Tahri A, Verschueren WG, Pille GME, de Kock HA, Jonckers TH, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune M-P, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PBTP. Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J Med Chem. 2005;48:1813–1822. doi: 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]
- 12.De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Bethune MP. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob Agents Chemother. 2005;49:2314–2321. doi: 10.1128/AAC.49.6.2314-2321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck PBTP, de Bethune MP, Schiffer CA. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol. 2004;78:12012–12021. doi: 10.1128/JVI.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kovalevsky AY, Tie Y, Liu F, Boross PI, Wang Y-F, Leshchenko S, Ghosh AK, Harrison RW, Weber IT. Effectiveness of nonpeptide clinical inhibitor TMC-114 on HIV-1 protease with highly drug resistant mutations D30N, I50V, and L90M. J Med Chem. 2006;49:1379–1387. doi: 10.1021/jm050943c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Surleraux DLNG, de Kock HA, Verschueren WG, Pille GME, Maes LR, Peeters A, Vendeville S, De Meyer S, Azijn H, Pauwels R, de Bethune MP, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PBTP. Design of HIV-1 protease inhibitors active on multidrug-resistant virus. J Med Chem. 2005;48:1965–1973. doi: 10.1021/jm049454n. [DOI] [PubMed] [Google Scholar]
- 16.Miller JF, Andrews CW, Brieger M, Furfine ES, Hale MR, Hanlon MH, Hazen RJ, Kaldor I, McLean EW, Reynolds D, Sammond DM, Spaltenstein A, Tung R, Turner EM, Xu RX, Sherrill RG. Ultra-potent P1 modified arylsulfonamide HIV protease inhibitors: The discovery of GW0385. Bioorg Med Chem Lett. 2006;16:1788–1794. doi: 10.1016/j.bmcl.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 17.He G-X, Yang Z-Y, Williams M, Callebaut C, Cihlar T, Murray BP, Yang C, Mitchell ML, Liu H, Wang J, Arimilli M, Eisenberg E, Stray KM, Tsai LK, Hatada M, Chen X, Chen JM, Wang Y, Lee MS, Strickley RG, Iwata Q, Zheng X, Kim CU, Swaminathan S, Desai MC, Lee WA, Xu L. Discovery of GS-8374, a potent human immunodeficiency virus type 1 protease inhibitor with a superior resistance profile. MedChemComm. 2011;2:1093–1098. [Google Scholar]
- 18.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. Structure-based design of novel HIV-1 protease inhibitors to combat drug resistance. J Med Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh AK, Gemma S, Baldridge A, Wang Y-F, Kovalevsky A, Yu, Koh Y, Weber IT, Mitsuya H. Flexible cyclic ethers/polyethers as novel P2-ligands for HIV-1 protease inhibitors: Design, synthesis, biological evaluation, and protein–ligand X-ray studies. J Med Chem. 2008;51:6021–6033. doi: 10.1021/jm8004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh AK, Chapsal BD, Baldridge A, Steffey MP, Walters DE, Koh Y, Amano M, Mitsuya H. Design and synthesis of potent HIV-1 protease inhibitors incorporating hexahydrofuropyranol-derived high affinity P2 ligands: structure–activity studies and biological evaluation. J Med Chem. 2011;54:622–634. doi: 10.1021/jm1012787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ali A, Reddy GSKK, Cao H, Anjum SG, Nalam MNL, Schiffer CA, Rana TM. Discovery of HIV-1 protease inhibitors with picomolar affinities incorporating N-aryl-oxazolidinone-5-carboxamides as novel P2 ligands. J Med Chem. 2006;49:7342–7356. doi: 10.1021/jm060666p. [DOI] [PubMed] [Google Scholar]
- 22.Ali A, Reddy GSKK, Nalam MNL, Anjum SG, Cao H, Schiffer CA, Rana TM. Structure-based design, synthesis, and structure–activity relationship studies of HIV-1 protease inhibitors incorporating phenyloxazolidinones. J Med Chem. 2010;53:7699–7708. doi: 10.1021/jm1008743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altman MD, Ali A, Reddy GSKK, Nalam MNL, Anjum SG, Cao H, Chellappan S, Kairys V, Fernandes MX, Gilson MK, Schiffer CA, Rana TM, Tidor B. HIV-1 Protease inhibitors from inverse design in the substrate envelope exhibit subnanomolar binding to drug-resistant variants. J Am Chem Soc. 2008;130:6099–6113. doi: 10.1021/ja076558p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 25.Momany FA, Rone R. Validation of the general purpose QUANTA ®3.2/CHARMm® force field. J Comput Chem. 1992;13:888–900. [Google Scholar]
- 26.Sitkoff D, Sharp KA, Honig B. Accurate calculation of hydration free energies using macroscopic solvent models. J Phys Chem. 1994;98:1978–1988. [Google Scholar]
- 27.Gilson MK, Honig B. Calculation of the total electrostatic energy of a macromolecular system: Solvation energies, binding energies, and conformational analysis. Proteins. 1988;4:7–18. doi: 10.1002/prot.340040104. [DOI] [PubMed] [Google Scholar]
- 28.Nicholls A, Honig B. A rapid finite difference algorithm, utilizing successive over-relaxation to solve the Poisson–Boltzmann equation. J Comput Chem. 1991;12:435–445. [Google Scholar]
- 29.Kangas E, Tidor B. Optimizing electrostatic affinity in ligand–receptor binding: Theory, computation, and ligand properties. J Chem Phys. 1998;109:7522–7545. [Google Scholar]
- 30.Gilson MK, Sharp KA, Honig BH. Calculating the electrostatic potential of molecules in solution: Method and error assessment. J Comput Chem. 1988;9:327–335. [Google Scholar]
- 31.Irwin JJ, Shoichet BK. ZINC: A free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desmet J, Maeyer MD, Hazes B, Lasters I. The dead-end elimination theorem and its use in protein side-chain positioning. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 33.Leach AR, Lemon AP. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm. Proteins. 1998;33:227–239. doi: 10.1002/(sici)1097-0134(19981101)33:2<227::aid-prot7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 34.Matayoshi ED, Wang GT, Krafft GA, Erickson J. Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science. 1990;247:954–958. doi: 10.1126/science.2106161. [DOI] [PubMed] [Google Scholar]
- 35.Wu TD, Schiffer CA, Gonzales MJ, Taylor J, Kantor R, Chou S, Israelski D, Zolopa AR, Fessel WJ, Shafer RW. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J Virol. 2003;77:4836–4847. doi: 10.1128/JVI.77.8.4836-4847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nalam MNL, Ali A, Altman MD, Reddy GSKK, Chellappan S, Kairys V, Ozen A, Cao H, Gilson MK, Tidor B, Rana TM, Schiffer CA. Evaluating the substrate-envelope hypothesis: Structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J Virol. 2010;84:5368–5378. doi: 10.1128/JVI.02531-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freire E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discovery Today. 2008;13:869–874. doi: 10.1016/j.drudis2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King NM, Melnick L, Prabu-Jeyabalan M, Nalivaika EA, Yang SS, Gao Y, Nie X, Zepp C, Heefner DL, Schiffer CA. Lack of synergy for inhibitors targeting a multi-drug-resistant HIV-1 protease. Protein Sci. 2002;11:418–429. doi: 10.1110/ps.25502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greco WR, Hakala MT. Evaluation of methods for estimating the dissociation constant of tight binding enzyme inhibitors. J Biol Chem. 1979;254:12104–12109. [PubMed] [Google Scholar]
- 41.Petropoulos CJ, Parkin NT, Limoli KL, Lie YS, Wrin T, Huang W, Tian H, Smith D, Winslow GA, Capon DJ, Whitcomb JM. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2000;44:920–928. doi: 10.1128/aac.44.4.920-928.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W, Charles W, Carter Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.Collaborative Computational Project Number 4. The CCP4suite: Programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. [Google Scholar]
- 44.Navaza J. AMoRe: An automated package for molecular replacement. Acta Crystallogr. 1994;A50:157–163. [Google Scholar]
- 45.Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. How does a symmetric dimer recognize an asymmetric substrate? A substrate complex of HIV-1 protease. J Mol Biol. 2000;301:1207–1220. doi: 10.1006/jmbi.2000.4018. [DOI] [PubMed] [Google Scholar]
- 46.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 47.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 48.Schomaker V, Trueblood KN. On the rigid-body motion of molecules in crystals. Acta Crystallogr. 1968;B24:63–76. [Google Scholar]
- 49.Kuriyan J, Weis WI. Rigid protein motion as a model for crystallographic temperature factors. Proc Natl Acad Sci USA. 1991;88:2773–2777. doi: 10.1073/pnas.88.7.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tickle IJ, Moss DS. IUCr99 Computing School. IUCr; London: 1999. Modeling rigid-body thermal motion in macromolecular crystal structure refinement. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.