

Abstract
Mutations in the proline/tyrosine–nuclear localization signal (PY-NLS) of the Fused in Sarcoma protein (FUS) cause amyotrophic lateral sclerosis (ALS). Here we report the crystal structure of the FUS PY-NLS bound to its nuclear import receptor Karyopherinβ2 (Kapβ2; also known as Transportin). The FUS PY-NLS occupies the structurally invariant C-terminal arch of Kapβ2, tracing a path similar to that of other characterized PY-NLSs. Unlike other PY-NLSs, which generally bind Kapβ2 in fully extended conformations, the FUS peptide is atypical as its central portion forms a 2.5-turn α-helix. The Kapβ2-binding epitopes of the FUS PY-NLS consist of an N-terminal PGKM hydrophobic motif, a central arginine-rich α-helix, and a C-terminal PY motif. ALS mutations are found almost exclusively within these epitopes. Each ALS mutation site makes multiple contacts with Kapβ2 and mutations of these residues decrease binding affinities for Kapβ2 (KD for wild-type FUS PY-NLS is 9.5 nM) up to ninefold. Thermodynamic analyses of ALS mutations in the FUS PY-NLS show that the weakening of FUS-Kapβ2 binding affinity, the degree of cytoplasmic mislocalization, and ALS disease severity are correlated.
Keywords: nucleocytoplasmic transport, Lou Gehrig's disease
The proline/tyrosine–nuclear localization signal (PY-NLS) of RNA-binding protein Fused in Sarcoma (FUS) was first identified in a structure-based bioinformatics approach and shown to bind the import-karyopherin, karyopherinβ2 (Kapβ2) in a Ran-sensitive manner (1). More recently, the signal was shown to direct Kapβ2-mediated nuclear import in cells (2) and to be heavily mutated in ∼5% of familial amyotrophic lateral sclerosis (ALS) (3–5), a progressive and fatal neurodegenerative disorder. ALS mutations in the PY-NLS disrupted nuclear import of FUS, causing its mislocalization and aggregation in the cytoplasm, as evidenced by cytoplasmic FUS inclusions in motor neurons of ALS patients (2, 6–9). The PY-NLS of FUS is also mutated in another neurodegenerative disease, frontotemporal lobar dementia (FTLD), which also is characterized by cytoplasmic mislocalization and aggregation of FUS (10, 11). The pathogenic role of FUS PY-NLS mutants was further confirmed by observations that expression of such proteins in Drosophila, Caenorhabditis elegans, zebrafish, and rats caused neurodegeneration (12–15). Proper FUS localization is important in maintaining neuronal homeostasis, and it is thus important to understand the factors that govern localization of both wild-type and mutant proteins.
PY-NLSs are recognized by the import-karyopherin Kapβ2 (also known as Transportin) for transport through the nuclear pore complex into the nucleus (1, 16–18). These 15- to 100-residue–long sequences are large and diverse and cannot be sufficiently described by a traditional consensus sequence but are instead described by a collection of physical rules that include requirements for intrinsic structural disorder, overall basic character, and a set of sequence motifs. PY-NLS motifs consist of an N-terminal hydrophobic or basic motif and a C-terminal RX2–5PY motif (Fig. 1A). A loose consensus of Φ-G/A/S-Φ-Φ (where Φ is a hydrophobic residue) represents the N-terminal hydrophobic motif whereas the equivalent N-terminal basic motif is a 4- to 20-residue segment that is enriched in basic residues. Kapβ2-bound PY-NLSs show structurally conserved consensus regions that are separated by linkers (19, 20). Extensive mutagenesis and thermodynamic analyses corroborated structural findings that the consensus motifs form three linker-separated binding epitopes (Fig. 1A) (21). Epitope 1 is the hydrophobic or basic N-terminal motif; epitopes 2 and 3 are the arginine residue and the PY dipeptide of the C-terminal RX2–5PY motif, respectively. Each epitope can contribute very differently to total binding energy in different PY-NLSs and signal diversity can be achieved through combinatorial mixing of energetically weak and strong motifs while maintaining affinity appropriate for nuclear import (21).
Fig. 1.

The atypical helical PY-NLS of FUS. (A) Sequences of PY-NLSs of FUS, EWS, and TAF15 and hnRNPs A1, M, and D. The N-terminal hydrophobic (yellow) or basic (blue) motifs that form epitope 1 of the NLS, the central epitope 2 (cyan), and the C-terminal epitope 3 (red) are shown. Residues mutated in ALS are marked with red asterisks. (B) Overall structure of the Kapβ2-FUS PY-NLS complex. The karyopherin is pink and the PY-NLS cyan. Side chains of the FUS PY-NLS is shown in C.
PY-NLSs are very diverse in sequence (21). Within the 15- to 100-residue peptides, obvious sequence conservation is observed only at their C-terminal PY or homologous PΦ (where Φ is a hydrophobic residue) motifs. The FUS PY-NLS has a noticeably conserved RX2–5PY motif at its 521RRERPY526 C terminus, but it is unclear whether the N-terminal Kapβ2-binding epitope of the signal is a hydrophobic (508PGKM511) or a basic (510KMRGEHR518) motif (Fig. 1A) (1, 2). Hence, the effect of the severe P525L ALS mutation in the PY motif was easily predicted but the structural basis of other ALS mutation sites in the FUS PY-NLS, particularly the striking series of five arginine sites, remains unexplained due to degeneracy and complexity of PY-NLS sequences (2, 6). The FUS PY-NLS is remarkably similar to the PY-NLSs of the other two members of the FUS, EWS or EWSR1, and TAF15 (FET) family of RNA-binding proteins that are all mutated in ALS (Fig. 1A) (1, 21). Information on the structural and energetic basis of FUS PY-NLS recognition by Kapβ2 should be directly applicable to EWS and TAF15.
Here we report the crystal structure of Kapβ2 bound to the FUS PY-NLS, which contains conserved N-terminal hydrophobic and C-terminal PY motifs as well as an atypical central arginine-rich α-helix. All ALS mutation sites in the PY-NLS make multiple contacts with Kapβ2. Structural and thermodynamic analyses explain the relative severity of these mutations in disease.
Results and Discussion
We have solved the 2.3-Å crystal structure of Kapβ2 bound to the PY-NLS of FUS (residues 498–526) to understand how the ALS mutations in FUS are involved in Kapβ2 binding and thus nuclear import (Fig. 1 A–C, Fig. S1A, and Table S1). The FUS PY-NLS occupies the PY-NLS binding site in the structurally invariant C-terminal arch of Kapβ2 (22), tracing a path similar to that of other characterized PY-NLSs (Figs. 1 B and C and 2 A and B and Fig. S1A). Unlike other PY-NLSs, which generally bind Kapβ2 in fully extended conformations (1, 19, 20, 23), the FUS peptide is atypical as its central portion forms a 2.5-turn α-helix. The circular dichroism spectrum of the FUS PY-NLS alone showed mostly coiled structure, suggesting that the FUS helix is induced upon Kapβ2 binding (Fig. S1B).
Fig. 2.

Comparative structural analysis of PY-NLSs. (A) Superposition of Kapβ2 residues 297–823 for four different Kap-PY NLS complexes. The FUS PY-NLS is in cyan(PDB ID 4FDD), the hnRNP A1 PY-NLS (PDB ID 2H4M) is in yellow, the hnRNP M PY-NLS (PDB ID 2OT8) is in magenta, and the hnRNP D PY-NLS (PDB ID 2Z5N) is in green. (B) Details of the superposition to compare PY-NLSs from FUS (cyan) and hnRNP A1 (yellow).
Residues 498–506 of the bound FUS peptide are not observed in the electron density. Residues 508PGKM511 form epitope 1 of the NLS and occupy the position of the hydrophobic (FGPM) motif of the PY-NLS of hnRNP A1 (1, 20) (Fig. 2B). These residues make numerous hydrophobic interactions with Kapβ2 residues Trp-730 and Ile-773 (Fig. 3, Bottom). Within this N-terminal hydrophobic motif, FUS residue Lys-510 (mutated to Glu or Arg in ALS) makes hydrophobic interactions with Kapβ2 Trp-730 and salt bridges with Kapβ2 Glu-653 and Asp-693. Further C-terminally, segment 514RGEHRQDRR522 of FUS forms a polarized helix, with all five of its basic residues facing Kapβ2, explaining their periodicity in the sequence (Fig. 3, Middle). Arg-514, His-517, Arg-518, Arg-521, and Arg-522, all mutated in ALS (5), form numerous salt bridges and hydrogen bonds with Kapβ2 residues Asp-509, Asp-543, Asp-550, Glu-588, and Asp-646, which are located on the B helices of HEAT repeats 9–12. Arg-522, the last side chain of the helix, overlaps with arginine residues (or epitopes 2) of the C-terminal RX2–5PY motifs of other PY-NLSs (1, 19, 20), suggesting that epitope 2 in FUS extends beyond from the single-arginine residue to an entire helix (Figs. 1A and 2B and Fig. S2). Here epitope 2 is connected to the N-terminal 508PGKM511 hydrophobic motif (epitope 1) and to the C-terminal 525PY526 motif (epitope 3) by short linkers. Asp-512 and Arg-524 in the two linkers serve as N and C caps, respectively, of the helix (Fig. 3, Top and Middle). Like the N-terminal hydrophobic motif, the structurally conserved PY motif of FUS contacts Kapβ2 (residues Ala-381, Leu-419, Ile-457, and Trp-460) primarily through hydrophobic interactions (Fig. 3, Top). FUS residue Pro-525 in the PY motif, which is mutated in particularly severe juvenile ALS cases (24), makes numerous contacts to these hydrophobic residues of Kapβ2 and also makes intramolecular contacts to the neighboring FUS Tyr-526, possibly mutually positioning each other for additional interactions with Kapβ2.
Fig. 3.
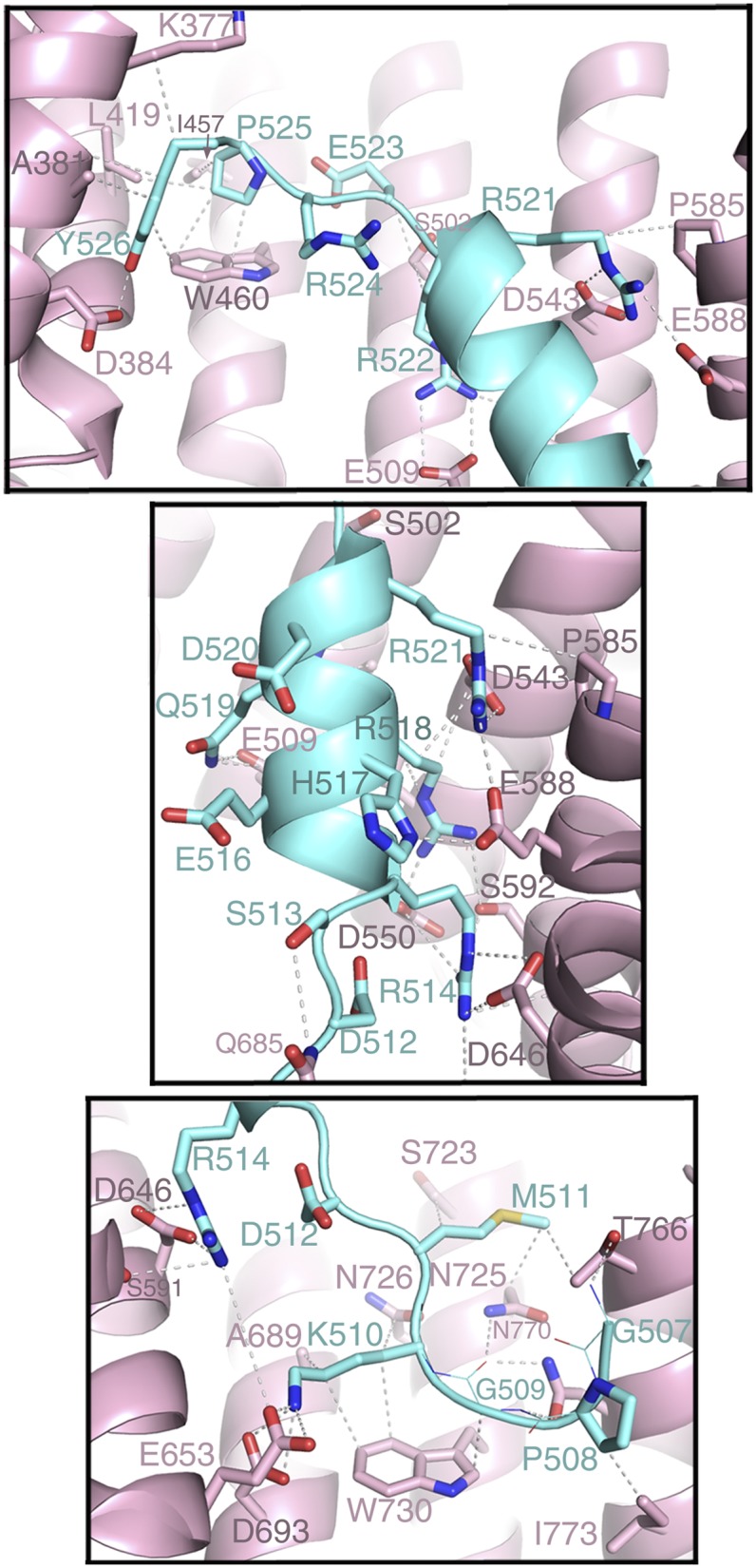
ALS mutation sites make multiple interactions with Kapβ2. The Kap-NLS interface (Kapβ2, pink; FUS PY-NLS, cyan) is shown: Bottom, the C-terminal portion of the PY-NLS spanning residues 523–526; Middle, the central portion of the PY-NLS spanning residues 514–522; and Top, the N-terminal portion of the PY-NLS spanning residues 507–513.
The PY-NLSs of FUS and EWS are highly homologous and are thus expected to adopt similar structures when bound to Kapβ2 (Fig. 1A). We predict that the PY-NLS of the other FET protein, TAF15, also binds Kapβ2 similarly. The 573YGGKM577 segment of TAF15 probably occupies the same position as the 508PGKM511 hydrophobic motif of the FUS PY-NLS. Its 580RNDYRNDQR588 segment probably forms a similar polarized central helix and its C-terminal conserved PY motif is most likely also structurally conserved. EWS and TAF15 were recently suggested to also be mutated in some rare ALS cases (25–28). The few disease mutation sites found thus far are located in the RGG regions that are N-terminal of the PY-NLSs. TAF15 and EWS have not been found to be mutated in FTLD but significant cytoplasmic mislocalization of both proteins was found in FTLD-FUS cases, suggesting impaired Kapβ2-mediated nuclear import even in these wild-type situations (28, 29). We can now explain how all three FET proteins are recognized and imported into the nucleus by Kapβ2.
The FUS PY-NLS is anchored to Kapβ2 through hydrophobic interactions at both ends of the peptide and through electrostatic interactions of its central helix. All PY-NLS side chains that are mutated in ALS either make multiple interactions with Kapβ2 or participate in intramolecular interactions to stabilize the Kapβ2-bound PY-NLS conformation. To determine the energetic contributions of these disease-relevant residues and to map the binding energy distribution along the FUS PY-NLS, we measured dissociation constants (KD) for Kapβ2 binding by isothermal calorimetry (ITC) for a series of alanine and ALS mutants (Fig. 4, Fig. S3, and Table 1). The FUS PY-NLS binds Kapβ2 with high affinity (KD = 9.5 nM). Of the single alanine mutants tested, mutations at Tyr-526, Arg-518, Pro-525, and Arg-522 decreased Kapβ2 binding the most with 70-, 8-, 6-, and 5-fold affinity losses, respectively (Table 1). Mutation of multiple residues to change entire epitopes 1, 2, and 3 of the PY-NLS showed that the central epitope 2 is energetically the strongest [quadruple mutant FUS PY-NLS (R514A, R518A, R521A, R522A) no longer measurably bound Kapβ2], followed by epitope 3 (the P525A, Y526A double mutant decreased affinity 87-fold) and then epitope 1 (quadruple 508PGKM511-AAAA mutant decreased affinity 4-fold). Interestingly, mutations of the nonbasic residues in the epitope 2 helix to generate 514RGGHRGGRR522 did not affect binding affinity, suggesting that introduction of multiple glycines likely did not perturb the helical structure (Table 1). This result suggests that the commonly found RGG motifs might adopt helical structures in PY-NLSs and elsewhere.
Fig. 4.

Decreases in Kapβ2 affinity correlate with cytoplasmic mislocalization. Decreases in Kapβ2 affinity due to alanine and ALS mutations in human FUS PY-NLS (Upper) are correlated with cytoplasmic mislocalization of similar mutations in mouse FUS (Lower). Nucleocytoplasmic index ratios (NCI mut/NCI wt), which describe relative contributions of cytoplasmic intensity of EGFP-FUS, are taken from Kino et al. (6). Residues Glu-523 and Tyr-526 have not been found mutated in ALS and thus are shaded light gray.
Table 1.
ITC analysis of Kapβ2 binding to FUS PY-NLS mutants
| Mutations | KD*, nM | KD, mutant/KD, wild type | |
| WT Kapβ2 | 9.5 ± 1.1 | ||
| Alanine mutants | 508PGKM511_AAAA | 38.00 ± 3.7 | 4 |
| R514A | 16.5 ± 0.1 | 2 | |
| R518A | 77.5 ± 1.0 | 8 | |
| R521A | 27.7 ± 2.6 | 3 | |
| R522A | 52.1 ± 1.4 | 5 | |
| R514A, R518A | 434.8 ± 28.0 | 46 | |
| R521A, R522A | 1,090.9 ± 17.7 | 115 | |
| R514A, R518A, R521A, R522A | ND | >>150† | |
| 514RGGHRGGRR522 | 31.5 ± 18.0 | 3‡ | |
| E523A | 11.9 ± 1.6 | 1 | |
| P525A | 55.6 ± 1.4 | 6 | |
| Y526A | 665.2 ± 14.2 | 70 | |
| P525A, Y526A | 821.9 ± 10.3 | 87 | |
| ALS disease mutants | H517Q | 11.4 ± 0.1 | 1 |
| R521C | 31.0 ± 1.3 | 3 | |
| R524S | 13.5 ± 0.4 | 1 | |
| P525L | 84.8 ± 6.6 | 9 |
ND, not detectable. All experiments (with one exception) were performed three times (±SD).
*Stoichiometry, 0.9–1.1.
†The lowest measurable KD = 1.09 μM in the Kapβ2–FUS-NLS mutant series was used to estimate the KD, mutant/KD, wild type.
‡Exception, where experiments were performed twice.
The weakening of FUS-Kapβ2 binding affinity, the degree of cytoplasmic mislocalization, and ALS disease severity are correlated. ITC analyses of several PY-NLS peptides with ALS mutations showed less than twofold to ninefold decreases in Kapβ2 binding affinity (Fig. 4 and Table 1). Loss of Kapβ2 binding in these mutants correlates with the degree of cytoplasmic mislocalization in cells (2, 6–9). Fig. 4 shows surprisingly tight correlation of the loss of Kapβ2 binding that we measured, with quantitation by Kino et al. of cytoplasmic/nuclear ratios for a large series of FUS mutants relative to the wild-type protein (6). Of the missense ALS mutants, the P525L mutation decreased Kapβ2 affinity the most, by ninefold (Fig. 4 and Table 1). This mutation also caused the most severe mislocalization of FUS to the cytoplasm (6) (Fig. 4) and is found in the most severe juvenile ALS cases (24). Mutation of Arg-522 decreased Kapβ2 affinity by fivefold and showed moderate mislocalization (Fig. 4 and Table 1). Mutations found in mid- to late-onset ALS cases such as the H517Q, R524S mutations and the most prevalent R521C mutation (2, 5, 24) decreased Kapβ2 affinity by 1.2-, 1.4-, and 3-fold, respectively, and caused only mild mislocalization of FUS to the cytoplasm (6).
Interestingly, although many ALS mutant PY-NLSs have reduced binding affinities compared with the wild-type PY-NLS, their affinities for Kapβ2 are still relatively high, in the tens of nanomolar range (for example, KD of the most severe P525L mutant is 85 nM). Such high affinities may be consistent with the initiation of disease later in life. High affinities for Kapβ2 may allow near-normal nuclear function and localization for some FUS mutants or partial mislocalization for others, which are tolerated until the aging cells encounter additional hits such as further impairment of the nuclear transport machinery or other cellular stress that exacerbate FUS mislocalization or trigger aggregation of mislocalized FUS (2, 5, 7, 9). Furthermore, even though ALS mutants bind Kapβ2 rather tightly, effects of decreased affinities compared with those of wild-type FUS may be amplified in cells due to competition with other high-affinity Kapβ2 cargos and with nonspecific competitors (16, 17, 30). Finally, it is also possible that affinities of wild-type and mutant PY-NLS may be attenuated or modulated in full-length FUS as a result of additional Kapβ2-binding sites outside of the PY-NLS or intramolecular interactions within FUS that involve its PY-NLS. Quantitative studies of Kapβ2 binding to the highly aggregation-prone full-length FUS will be important to reveal potential complexities in their interactions.
In summary, Kapβ2 binds the FUS PY-NLS through structurally conserved N-terminal hydrophobic and C-terminal PY motifs as well as an atypical central arginine-rich α-helix. All ALS mutation sites in the signal either make multiple contacts with Kapβ2 or participate in intramolecular interactions to stabilize the Kapβ2-bound PY-NLS conformation. The loss of Kapβ2 affinity upon mutations at ALS sites correlates with both cytoplasmic mislocalization in cells and disease severity, providing further support that Kapβ2 plays a major role in nuclear localization of FUS. Such compartmentalization of FUS is probably also important for proper maintenance of aggregation states of the protein in cells.
Materials and Methods
Protein Expression, Purification, and Complex Formation.
Human Kapβ2 (Uniprot ID U72069) was expressed in pGEX-Tev vector [pGEX-4T3 (GE Healthcare) with a Tev cleavage site] as a GST fusion protein and purified as previously described (1, 19, 31). A truncation version of Kapβ2, which does not interfere with NLS binding, was used for crystallization (residues 337–367 of Kapβ2 were replaced with a GGSGGSG linker) (1, 19). The human FUS PY-NLS (Uniprot ID P35637; residues 498–526) was expressed as a GST fusion protein. GST-FUS PY-NLS was purified by affinity and ion exchange chromatography followed by cleavage of the GST tag using the TEV protease and further purified by gel filtration in buffer containing 20 mM Hepes, pH 7.3, 110 mM potassium acetate, 2 mM DTT, 2 mM magnesium acetate, and 1 mM EGTA with 20% (vol/vol) glycerol. Kapβ2 and the FUS PY-NLS were mixed at molar ratio of 1:5 and the complex was concentrated to 13 mg/mL for crystallization. For ITC analysis, the FUS PY-NLS was subcloned into the pMal-Tev vector [pMal-2c (New England Bio) with a Tev cleavage site] and mutations were generated by site-directed mutagenesis using Quikchange (Stratagene). MBP-FUS PY-NLS proteins were purified as previously described for ITC measurements (1, 19, 31). All proteins were expressed in BL21 (DE3) Escherichia coli cells.
Crystallization, Data Collection, and Structure Determination.
The Kapβ2-FUS PY-NLS complex was crystallized by vapor diffusion using 1 M succinic acid, 0.1 M Hepes, pH 7.0, and 1% (wt/vol) PEG MME 2000 in the reservoir solution. Crystals were flash frozen in liquid nitrogen and 2.3 Å crystallographic data were obtained at temperature 100 K at the beamline 19-ID at the Advanced Photon Source, Argonne National Laboratory (Lemont, IL). Data were processed using HKL2000 (32). The Kapβ2-FUS PY-NLS crystal is in the P21212 space group with cell dimensions of a = 128.720 Å, b = 157.255 Å, and c = 67.543 Å and only one complex is present in the asymmetric unit. Molecular replacement, using the program Phaser (33), was performed with Kapβ2 coordinates from the Kapβ2-hnRNP M-NLS structure (19) (PDB ID 2OT8) as the search model, followed by auto-model building using Buccaneer (34). The model was further improved by manual building in Coot (35). The structure was refined using REFMAC5 (36) and the same test dataset was used throughout the entire refinement process. An Fo − Fc map shows strong electron density for FUS residues 507–526. The refined model shows good stereochemistry with Rfactor = 20.8% and Rfree = 24.5%. A Ramachandran plot for the final model shows that 97.2% of the residues are in most favored and 2.8% in allowed regions. Details are provided in Table S1.The structural model and crystallographic data are deposited in the Protein Data Bank under PDB ID code 4FDD.
Isothermal Titration Calorimetry (ITC).
Binding affinities of the wild-type and mutant FUS PY-NLSs to Kapβ2 were determined using ITC, as previously described (1, 19, 31). Briefly, MBP-FUS PY-NLS proteins and Kapβ2 were dialyzed against buffer containing 20 mM Tris, pH 7.5, 100 mM sodium chloride, 10% (vol/vol) glycerol, and 2 mM β-mercaptoethanol. About 100 μM of wild-type and mutant MBP-FUS PY-NLS proteins was titrated into the sample cell containing 10 μM full-length Kapβ2. All ITC experiments were performed using a MicroCal Omega VP-ITC calorimeter at 20 °C with 35 rounds of 8-μL injections or 56 rounds of 5-μL injections. Data were plotted and analyzed using the single binding-site model of MicroCal Origin software version 7.0.
Circular Dichroism (CD) Spectroscopy.
To determine the secondary structure of the FUS PY-NLS in solution, we performed CD spectroscopy. FUS PY-NLS (20 μM) in buffer containing 20 mM Tris, pH 7.5, 100 mM NaCl, 10% (vol/vol) glycerol, and 2 mM β-mercaptoethanol was scanned at 25 °C from 190 nm to 260 nm with five repeats. The buffer background was subtracted to generate the final spectrum.
Supplementary Material
Acknowledgments
We thank J. Liao, L. Clark, and Q. Sun for advice and help; the University of Texas Southwestern Structural Biology Laboratory for support; and J. Shorter for discussions. This work is funded by National Institutes of Health Grant R01-GM069909 (to Y.M.C.), the Welch Foundation (I-1532 to Y.M.C.), and the University of Texas Southwestern Endowed Scholars Program (Y.M.C.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4FDD).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1207247109/-/DCSupplemental.
References
- 1.Lee BJ, et al. Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell. 2006;126:543–558. doi: 10.1016/j.cell.2006.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dormann D, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwiatkowski TJ, Jr, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 4.Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dormann D, Haass C. TDP-43 and FUS: A nuclear affair. Trends Neurosci. 2011;34:339–348. doi: 10.1016/j.tins.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 6.Kino Y, et al. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res. 2011;39:2781–2798. doi: 10.1093/nar/gkq1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gal J, et al. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging. 2011;32(2323):e2327–e2340. doi: 10.1016/j.neurobiolaging.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol. 2011;69:152–162. doi: 10.1002/ana.22246. [DOI] [PubMed] [Google Scholar]
- 9.Bosco DA, et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–4175. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broustal O, et al. French Clinical and Genetic Research Network on FTD/FTD-MND FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J Alzheimers Dis. 2010;22:765–769. [PubMed] [Google Scholar]
- 11.Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- 12.Kabashi E, et al. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. 2011;7:e1002214. doi: 10.1371/journal.pgen.1002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murakami T, et al. ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum Mol Genet. 2012;21:1–9. doi: 10.1093/hmg/ddr417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang C, et al. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011;7:e1002011. doi: 10.1371/journal.pgen.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanson NA, Jr, Pandey UB. FUS-related proteinopathies: Lessons from animal models. Brain Res. 2012;1462:44–60. doi: 10.1016/j.brainres.2012.01.039. [DOI] [PubMed] [Google Scholar]
- 16.Chook YM, Süel KE. Nuclear import by karyopherin-βs: Recognition and inhibition. Biochim Biophys Acta. 2011;1813:1593–1606. doi: 10.1016/j.bbamcr.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu D, Farmer A, Chook YM. Recognition of nuclear targeting signals by Karyopherin-β proteins. Curr Opin Struct Biol. 2010;20:782–790. doi: 10.1016/j.sbi.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marfori M, et al. Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim Biophys Acta. 2011;1813:1562–1577. doi: 10.1016/j.bbamcr.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 19.Cansizoglu AE, Lee BJ, Zhang ZC, Fontoura BM, Chook YM. Structure-based design of a pathway-specific nuclear import inhibitor. Nat Struct Mol Biol. 2007;14:452–454. doi: 10.1038/nsmb1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imasaki T, et al. Structural basis for substrate recognition and dissociation by human transportin 1. Mol Cell. 2007;28:57–67. doi: 10.1016/j.molcel.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 21.Süel KE, Gu H, Chook YM. Modular organization and combinatorial energetics of proline-tyrosine nuclear localization signals. PLoS Biol. 2008;6:e137. doi: 10.1371/journal.pbio.0060137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cansizoglu AE, Chook YM. Conformational heterogeneity of karyopherin beta2 is segmental. Structure. 2007;15:1431–1441. doi: 10.1016/j.str.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Süel KE, Cansizoglu AE, Chook YM. Atomic resolution structures in nuclear transport. Methods. 2006;39:342–355. doi: 10.1016/j.ymeth.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mackenzie IR, et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: Two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 2011;122:87–98. doi: 10.1007/s00401-011-0838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Couthouis J, et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:2899–2911. doi: 10.1093/hmg/dds116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Couthouis J, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci USA. 2011;108:20881–20890. doi: 10.1073/pnas.1109434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ticozzi N, et al. Genetics of familial Amyotrophic lateral sclerosis. Arch Ital Biol. 2011;149:65–82. doi: 10.4449/aib.v149i1.1262. [DOI] [PubMed] [Google Scholar]
- 28.King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neumann M, et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134:2595–2609. doi: 10.1093/brain/awr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Timney BL, et al. Simple kinetic relationships and nonspecific competition govern nuclear import rates in vivo. J Cell Biol. 2006;175:579–593. doi: 10.1083/jcb.200608141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang ZC, Satterly N, Fontoura BM, Chook YM. Evolutionary development of redundant nuclear localization signals in the mRNA export factor NXF1. Mol Biol Cell. 2011;22:4657–4668. doi: 10.1091/mbc.E11-03-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 34.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.