

Ribonucleotide reductases (RNRs) catalyze the conversion of nucleotides to deoxynucleotides in all organisms and thus play a central role in nucleic acid metabolism (1–3). Recently, substantive progress has been made in elucidation of the structure (4, 5), function (6), and regulation (7) of reductases. In addition, recently obtained sequences of reductases from archaea and deeply rooted eubacteria (8–10) have allowed speculation on the role of these enzymes in evolution. As outlined below, as many questions have been raised as answered, guaranteeing that reductases will be of interest to investigate experimentally for some time to come.
One of the most amazing revelations concerning RNRs is that despite their central role in metabolism, the essential metal cofactors have not been evolutionarily conserved. The RNRs have been divided into classes on the basis of these observations (Fig. 1). The class I and class II RNRs, requiring a diferric–tyrosyl radical (11) and adenosylcobalamin (AdoCbl) (12, 13), respectively, have been most extensively studied. From these studies and more recent studies on the intriguing RNR utilizing an essential glycyl radical cofactor (class III) found only in obligate anaerobes (14), a general mechanistic paradigm has evolved (15, 16) (Fig. 1). Whether the class IV RNR, containing a putative manganese cluster–tyrosyl radical cofactor (17), is in accord with this paradigm remains to be established.
Figure 1.
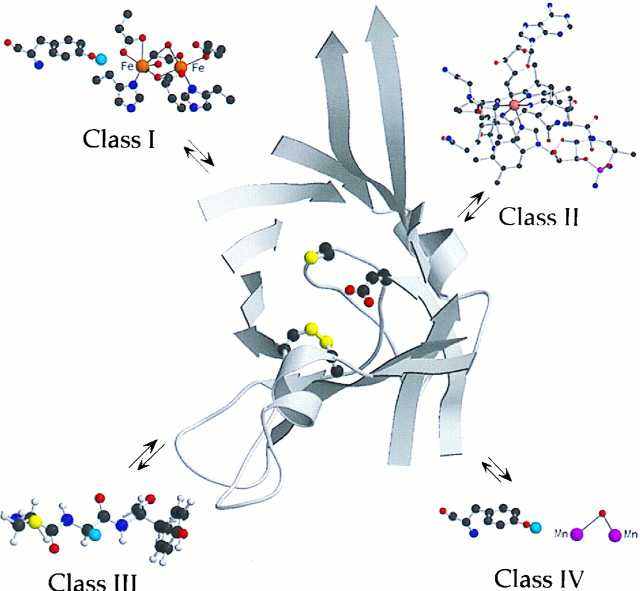
The active site of the Escherichia coli R1 subunit, adapted from Uhlin and Eklund (4). Cys-439, the site of the putative thiyl radical, is at the tip of the central loop of the barrel with Glu-441, the conserved glutamate adjacent to this residue. In addition, a disulfide is shown between Cys-225 and Cys-419, the site of delivery of the reducing equivalents to make deoxynucleotide. Each class of RNR thus far characterized has a different cofactor: the class I RNR has a diferric–tyrosyl radical (blue balls represent the orbital with the unpaired electron); class II utilizes AdoCbl; class III utilizes S-adenosylmethionine and an iron–sulfur cluster to generate a glycyl radical; and class IV is proposed to have a Mn cofactor adjacent to a tyrosyl radical. The cofactors’ function is proposed to be the generation of the thiyl radical, which will be located, regardless of the class of RNR, in an active site with secondary and tertiary structure similar to that observed in class I R1. The figure was generated by using molscript (34) and raster3d (35).
The model is that, despite the diversity of cofactors and the enzymes’ primary and quaternary structures, the mechanism of nucleotide reduction, involving complex and exquisitely controlled radical-dependent redox chemistry, has been conserved (16). The function of the diverse metallo-cofactors, as first proposed in the 1960s, is to initiate some type of radical-dependent reduction process. This model has grown more sophisticated and complex, because of the availability of data from physical organic studies on these enzymes. In addition, multiple sequences from the class I, II, and III RNRs and the resulting accessibility of large amounts of these proteins have facilitated examination in detail of the function of the cofactors (Fig. 1). These studies collectively have allowed further definition of the role of each cofactor as a generator of an essential thiyl radical that initiates the nucleotide reduction process by 3′-hydrogen abstraction from the nucleotide (6, 18, 19).
Support for this postulate has recently been provided from studies on both the structure and function of class I and class II RNRs. Eklund and his collaborators have reported the first structure of each of the two subunits of the E. coli class I RNR: R1 (4, 7) and R2 (20, 21). R2 contains the diferric–tyrosyl radical cofactor and R1 contains the active site, including the essential cysteine designated to become a thiyl radical. A cartoon of the active site of this RNR (R1) is also shown in Fig. 1, where the three required cysteines (the thiyl radical and the two cysteines delivering the reducing equivalents) are housed in a 10-stranded α,β-barrel with a loop penetrating the center of this structure containing at its tip the precursor to the thiyl radical. The remarkable congruency in the chemistry observed with the active site mutants of class I and II RNRs and a variety of mechanism-based inhibitors of class I, II, and III RNRs (22) strongly suggest that the active site of the class I enzyme will serve as a paradigm for the active site structure that will be found in the other classes of RNRs. Recent rapid freeze quench EPR spectroscopy has provided the first direct evidence with class II enzymes that the metallo-cofactor generates, in a kinetically competent fashion, a thiyl radical that is directly involved in nucleotide reduction (6). The chemical similarities in all classes of RNRs thus suggest that this will be a common theme (Fig. 1).
Support for the importance of thiyl radicals in catalysis is provided for class I RNRs indirectly by sequence studies of RNRs from Thermoplasma acidophila by Tauer and Benner (8) and the more recent studies on other organisms of Riera et al. (9) and Jordan et al. (10). These reductases use AdoCbl as a cofactor (class II), but, strikingly, their active sites are homologous to those of the class I RNRs. Recent rapid freeze quench EPR studies on the T. acidophila RNR (23) suggest that it also generates a thiyl radical in a kinetically competent fashion. These recent sequence data in conjunction with physical methods thus support the proposal that the metallo-cofactors generate thiyl radicals in a nucleotide-binding domain that will share common secondary and tertiary structural features.
Further support for the model that all classes of RNRs share a common secondary and tertiary nucleotide reduction domain has been proposed on the basis of the observation of gross similarities in the complex regulation of the selectivity of the nucleotide reduction process in the class I, II, and III RNRs (2, 24). The appropriate deoxynucleotide (dNTP) governs which nucleotide is reduced, that is, the specificity of nucleotide reduction. The regulation argument has thus been used as an evolutionary determinant (24). However, the structure of E. coli R1 (composed of two identical protomers) suggests that the specificity site is at the R1 dimer interface. Furthermore, the studies to date suggest that the Lactobacillus leichmannii and T. acidophilus RNRs are monomers. Thus it is unlikely that the structural similarities in regulation have been evolutionarily conserved, unless the active unit of the class I RNRs is actually a protomer of R1, or that as yet undetected dimers are the catalytically active species in the L. leichmannii and T. acidophilus RNRs.
The recent structure of R1 has better defined the site that governs specificity of reduction, and in this class I RNR a second allosteric site that governs turnover rate (7). This second site does not appear to have been evolutionarily conserved. This initial structural glimpse has provided a starting point for thinking about the amazing complexity of the specificity of the reduction process essential for the fidelity of DNA replication.
An added layer of regulatory complexity is apparent from recent studies in Saccharomyces cerevisiae that have revealed two class I RNRs (both R1 and R2 subunits) with a high degree of sequence homology, proposed to be involved in replication and repair (25–27).† One of these RNRs is dramatically inducible by DNA-damaging agents and is clearly involved in a complex signal transduction cascade in some way controlling the cell cycle (28). Whether two such RNRs exist in humans remains to be established. These questions are central to understanding nucleic acid metabolism They take on a new light given the recent approval of the Food and Drug Administration of gemcitabine (29). This nucleoside, subsequent to its phosphorylation to the diphosphate, is a stoichiometric inactivator of reductase. Decreased dNDP pools are thought to lead to decreased dNTP pools, a situation that facilitates the incorporation of gemcitabine 5′-triphosphate into nucleic acid by DNA polymerase, where it functions as a chain terminator. This synergism is thought to provide an explanation for the observed cytotoxicity and therapeutic efficacy of the drug (ref. 30; W. A. van der Donk, G. Yu, L. Perez, R. J. Sanchez, and J.S., unpublished work).
Studies in yeast and recent studies in mammalian cell culture (31–33) raise additional provocative questions central to understanding nucleic acid metabolism. Is it possible that assembly and disassembly of the diferric–tyrosyl radical cofactor is intimately involved in regulation of the nucleotide reduction process? This proposition would require a link between nucleic acid and energy metabolism as one electron is required for reduction of the essential tyrosyl radical and one electron is required for its regeneration from diferrous R2. Recent studies in yeast defining the gene products responsible for iron and copper homeostasis raise the intriguing possibility that this question will be answerable.
Thus the past five years have solidified in a gratifying way our understanding of the mechanism of nucleotide reduction and the prominent role of the metallo-cofactors in this process. They have also raised thought-provoking questions for the class I RNRs regarding the regulation of nucleotide reduction in replication and repair in vivo at the level of allosteric feedback and cofactor formation and destruction, as well as the prospect of reductase compartmentalization. Thus the future studies on this exciting system appear to be as intriguing as those of the past.
Acknowledgments
Thanks to all of the Stubbe group members who have worked on this project, thanks to Joe Kappock for preparation of Fig. 1, and thanks to Novartis for support of the Stubbe group. Our work on ribonucleotide has been generously supported over the years by National Institutes of Health Grant GM29595.
Footnotes
The companions to this commentary are published on pages 53, 475, and 13487 of volume 94.
Amazingly, the second R2, with 47% sequence identity in comparison to the mammalian R2, lacks several residues essential for iron cluster formation, yet this gene product appears to be essential (25).
References
- 1.Sjoberg B M. Struct Bonding (Berlin) 1997;88:139–173. [Google Scholar]
- 2.Reichard P. Science. 1993;260:1773–1777. doi: 10.1126/science.8511586. [DOI] [PubMed] [Google Scholar]
- 3.Stubbe J, van der Donk W A. Chem Biol. 1995;2:793–801. doi: 10.1016/1074-5521(95)90084-5. [DOI] [PubMed] [Google Scholar]
- 4.Uhlin U, Eklund H. Nature (London) 1994;370:533–539. doi: 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- 5.Uhlin U, Eklund H. J Mol Biol. 1996;262:358–369. doi: 10.1006/jmbi.1996.0519. [DOI] [PubMed] [Google Scholar]
- 6.Licht S L, Gerfen G J, Stubbe J. Science. 1996;271:477–481. doi: 10.1126/science.271.5248.477. [DOI] [PubMed] [Google Scholar]
- 7.Ericksson M, Uhlin U, Ramaswamy S, Ekberg M, Regnstrom K, Sjoberg B M, Eklund H. Structure. 1997;5:1078–1092. doi: 10.1016/s0969-2126(97)00259-1. [DOI] [PubMed] [Google Scholar]
- 8.Tauer A, Benner S A. Proc Natl Acad Sci USA. 1997;94:53–58. doi: 10.1073/pnas.94.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riera J, Robb F T, Weiss R, Fontecave M. Proc Natl Acad Sci USA. 1997;94:475–478. doi: 10.1073/pnas.94.2.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jordon A, Torrents E, Jeanthon C, Eliasson R, Hellman U, Wenstedt C, Barbe J, Gibert I, Reichard P. Proc Natl Acad Sci USA. 1997;94:13487–13492. doi: 10.1073/pnas.94.25.13487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sjöberg B-M, Reichard P, Gräslund A, Ehrenberg A. J Biol Chem. 1977;252:536–541. [PubMed] [Google Scholar]
- 12.Beck W S, Goulian M, Larsson A, Reichard P. J Biol Chem. 1966;241:2177–2179. [PubMed] [Google Scholar]
- 13.Blakley R L. J Biol Chem. 1965;240:2173–2180. [PubMed] [Google Scholar]
- 14.Reichard P. J Biol Chem. 1993;268:8383–8386. [PubMed] [Google Scholar]
- 15.Ashley G W, Harris G, Stubbe J. J Biol Chem. 1986;261:3958–3964. [PubMed] [Google Scholar]
- 16.Stubbe J. J Biol Chem. 1990;265:5329–5332. [PubMed] [Google Scholar]
- 17.Auling G, Follmann H. In: Manganese-Dependent Ribonucleotide Reduction and Overproduction of Nucleotides in Coryneform Bacteria. Sigel H, Sigel A, editors. Vol. 30. New York: Dekker; 1994. [Google Scholar]
- 18.Gerfen, G. J., van der Donk, W. A., Yu, G., Farrar, C., Griffin, R. G., Stubbe, J., McCarthy, J. R., Matthews, D. P. & Jarvi, E. T. (1998) J. Am. Chem. Soc., in press.
- 19.Persson A L, Eriksson M, Katterle F, Potsch S, Sahlin M, Sjoberg B M. J Biol Chem. 1997;272:31533–31541. doi: 10.1074/jbc.272.50.31533. [DOI] [PubMed] [Google Scholar]
- 20.Nordlund P, Eklund H. J Mol Biol. 1993;232:123–164. doi: 10.1006/jmbi.1993.1374. [DOI] [PubMed] [Google Scholar]
- 21.Nordlund P, Sjöberg B-M, Eklund H. Nature (London) 1990;345:593–598. doi: 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- 22.Stubbe J. Adv Enzymol Relat Areas Mol Biol. 1990;63:349–417. doi: 10.1002/9780470123096.ch6. [DOI] [PubMed] [Google Scholar]
- 23.Wu J J. Ph.D. thesis. Cambridge: Massachusetts Institute of Technology; 1998. [Google Scholar]
- 24.Reichard P. Trends Biochem Sci. 1997;22:81–85. doi: 10.1016/s0968-0004(97)01003-7. [DOI] [PubMed] [Google Scholar]
- 25.Huang M, Elledge S J. Mol Cell Biol. 1997;17:6105–6113. doi: 10.1128/mcb.17.10.6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elledge S J, Zhou Z, Allen J B. Trends Biochem Sci. 1992;17:119–123. doi: 10.1016/0968-0004(92)90249-9. [DOI] [PubMed] [Google Scholar]
- 27.Wang P J, Chabes A, Casagrande R, Tian X C, Thelander L, Huffaker T C. Mol Cell Biol. 1997;17:6114–6121. doi: 10.1128/mcb.17.10.6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen J B, Zhou Z, Siede W, Friedberg E C, Elledge S J. Genes Dev. 1994;8:2416–2428. doi: 10.1101/gad.8.20.2401. [DOI] [PubMed] [Google Scholar]
- 29.Hertel L W, Kroin J S, Grossman C S, Grindey G B, Dorr A F, Storniolo A M V, Plunkett W, Gandhi V, Huang P. ACS Symp Ser. 1996;639:265–278. [Google Scholar]
- 30.Plunkett W, Huang P, Xu Y-Z, Heinemann V, Grunewald R, Gandhi V. Semin Oncol. 1995;22:3–10. [PubMed] [Google Scholar]
- 31.Brischwein K, Engelcke M, Riedinger J J, Probst H. Eur J Biochem. 1997;244:286–293. doi: 10.1111/j.1432-1033.1997.00286.x. [DOI] [PubMed] [Google Scholar]
- 32.Fan H, Villegas C, Wright J A. Proc Natl Acad Sci USA. 1996;93:14036–14040. doi: 10.1073/pnas.93.24.14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Filatov D, Bjorklund S, Johnson E, Thelander L. J Biol Chem. 1996;271:23698–23704. doi: 10.1074/jbc.271.39.23698. [DOI] [PubMed] [Google Scholar]
- 34.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 35.Merritt E A, Bacon D J. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]