

Abstract
This chapter describes a workflow for measuring the efficacy of a barcode in identifying species. First, assemble individual sequence databases corresponding to each barcode marker. A controlled collection of taxonomic data is preferable to GenBank data, because GenBank data can be problematic, particularly when comparing barcodes based on more than one marker. To ensure proper controls when evaluating species identification, specimens not having a sequence in every marker database should be discarded. Second, select a computer algorithm for assigning species to barcode sequences. No algorithm has yet improved notably on assigning a specimen to the species of its nearest neighbor within a barcode database. Because global sequence alignments (e.g., with the Needleman–Wunsch algorithm, or some related algorithm) examine entire barcode sequences, they generally produce better species assignments than local sequence alignments (e.g., with BLAST). No neighboring method (e.g., global sequence similarity, global sequence distance, or evolutionary distance based on a global alignment) has yet shown a notable superiority in identifying species. Finally, “the probability of correct identification” (PCI) provides an appropriate measurement of barcode efficacy. The overall PCI for a data set is the average of the species PCIs, taken over all species in the data set. This chapter states explicitly how to calculate PCI, how to estimate its statistical sampling error, and how to use data on PCR failure to set limits on how much improvements in PCR technology can improve species identification.
Keywords: Barcode efficacy in species identification, Probability of correct identification, DNA barcode
1. Introduction
Species are becoming extinct, making conservation of biodiversity a major challenge. The first step to preserving biodiversity is assessment, but there are not enough taxonomists to catalog species throughout the world. DNA barcodes therefore provide the basis of a promising alternative strategy because they require only collection of DNA and not the immediate taxonomic identification of specimens. Although barcodes have many other uses, e.g., identification of novel species, taxonomic classification, and phylogeny, their application to cataloguing biodiversity justifies restricting this chapter to the measurement of a barcode’s efficacy in identifying known species.
In its essence, a barcode is any standardized subset of DNA from a taxonomic specimen (1, 2). The subset may vary, depending on readily recognizable features of a specimen (e.g., is the specimen a vertebrate? a plant? an insect? etc.). If computers could identify the species of a specimen from its barcode, then the barcode would provide a database key for retrieving taxonomic information pertinent to the specimen. A computer catalog of species on Earth then becomes a technical possibility. Early studies indicated that the sequence of cytochrome c oxidase 1 (CO1) gene could correctly identify many species (3), so selection of CO1 as a primary barcode followed naturally (4–10).
Although the selection of a DNA barcode has been natural for some species, it has been problematic for others, particularly plants (11–14) and insects (15, 16). The lack of a clear consensus for a barcode in those species has stimulated interest in the objective, quantitative measurement of the efficacy of a barcode in identifying species. Consensus on an actual barcode for some species remains tentative, but nonetheless, a consensus on measuring barcode efficacy has emerged (14, 15, 17). This chapter summarizes the consensus and indicates how to construct studies to evaluate the relative merits of competing barcodes. For practical methods, the reader is invited to view http://www.ncbi.nlm.nih.gov/CBBresearch/Spouge/html.ncbi/barcode/, a Web site providing information on computer programs pertinent to barcodes. Web pages are supposed to be self-explanatory, so to avoid undue brevity, the second section in this chapter provides some rationale for the computer programs for evaluating barcodes. The third section provides a practical summary of the entire chapter.
2. The Measurement of the Efficacy of Species Identification
To fix our terminology, the term “marker” connotes any contiguous region of DNA (coding or non-coding), whereas the term “barcode” connotes the aggregate of the one or more markers in the “standardized subset of DNA” referred to in the Introduction. Presently, all barcode markers are marker genes like CO1, matK, etc. In slowly evolving organisms like plants, however, intergenic spacers (DNA regions flanked by two genes) are still worthy of consideration as potential markers, because they usually diverge faster than genes, while their ends are still conserved, providing primers for PCR (17, 18). As described below, however, multiple sequence alignments (MSAs) of intergenic markers might complicate the workflow in a barcode database.
To have practical meaning, any measurement of the efficacy of species identification must mirror the performance of a database based on the prospective barcode. In practice, users query the database with a barcode retrieved from a specimen; the database returns the species identification as output, with the assignment “unknown” for any species apparently not yet in the database. Because this chapter restricts itself to discussing the identification of known species, it assumes that each query to the barcode database represents a specimen belonging to a species already in the database.
2.1. The Database
The first step in estimating the efficacy of several prospective barcodes is to assemble the corresponding databases. To ensure the proper controls, specimens not having sequences in every marker database should be eliminated from consideration (14), because if the databases do not contain exactly the same specimens, there might be unappreciated but influential biases. Consider, e.g., a hypothetical experiment that extracts from GenBank all sequences corresponding to two prospective markers, Marker A and Marker B. If Marker A has been the default marker of choice, whereas Marker B has been considered as the last hope for resolving species after Marker A has failed, the GenBank entries for Marker B might be biased toward a subset of particularly difficult specimens. Thus, on GenBank data, Marker B might have fewer correct species assignments than Marker A, even though Marker B is in fact better at resolving species than Marker A. Moreover, relative to a barcode database, GenBank taxonomy is undependable, and undependable taxonomy improperly influences conclusions by occasionally penalizing correct species identification. In addition, GenBank entries do not usually identify individual taxonomic specimens. GenBank data are therefore particularly unsuited to studying barcodes based on more than one marker, because the sequences from different markers cannot be associated with a single specimen. Although studies based on GenBank data have obvious scientific interest, they do not have the same status as a controlled taxonomic study. In summary, the choice of database affects conclusions, so care must be taken that the database reflects the scientific aims of a study.
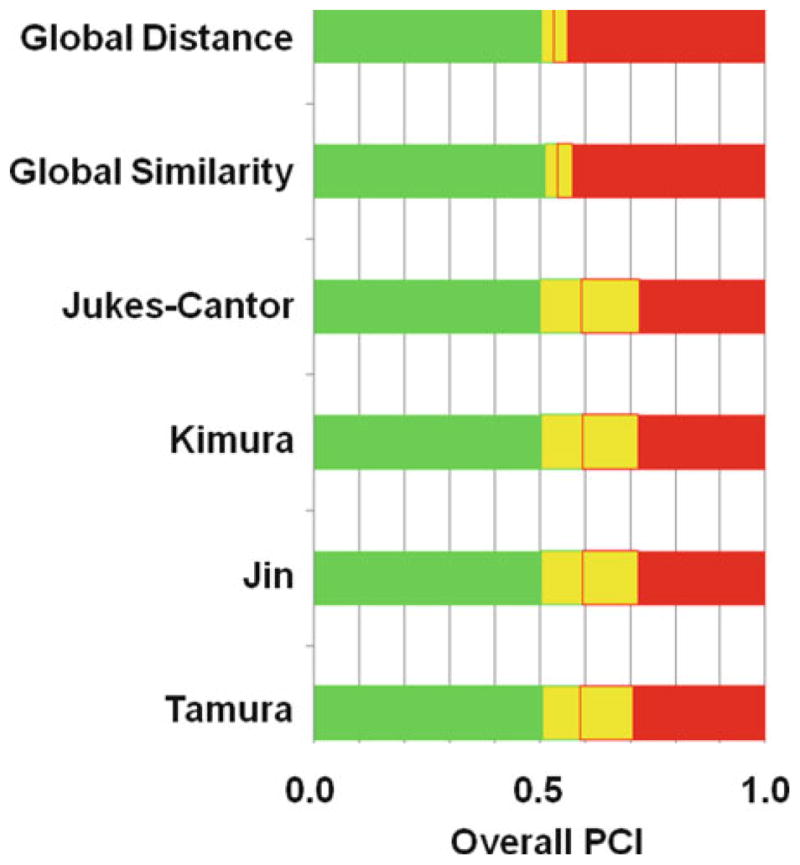
Figure 1 shows some pertinent results for trnH-psbA, a potential barcode marker in plants. By using pairwise alignment and various evolutionary distances in the procedures described below, the best overall probability of correct identification (PCI) in Fig. 1 is about 0.50, which is noticeably lower than the overall PCI of 0.69 from a controlled taxonomic study (14), suggesting that the GenBank entries for trnH-psbA might contain biases, relative to a controlled taxonomic study. The corresponding FASTA sequence file (see the Supplementary Materials) in fact contained genetic crosses (denoted by “x”) and tentative species assignments (denoted by “sp.”, “cf.”, “aff.”), which were obscure, until the Web tools mentioned above found them.
Fig. 1.
2.2. Species Assignment Algorithm
Once an appropriate database has been selected, the computer must assign a species to each barcode query (or declare its failure to assign). The next step, therefore, is to select a computer algorithm for assigning each specimen and its barcode sequence to a species. No algorithm seems to improve noticeably on assigning to a specimen the species of its nearest neighbor within a barcode database (19, 20). Thus, many algorithms begin by estimating a “separation” between the barcode sequences in two specimens. (The term “separation” is preferable to “distance”, which connotes some specific mathematical properties not necessary to barcodes.)
Separation can be based on: (1) sequence alignment similarities, (2) sequence alignment distances, (3) evolutionary distances (which usually require prior alignment of the barcode sequences), or (4) alignment-free distances. Studies have compared different measures of separation, but they are too limited to draw definitive conclusions about which separation provides the best species assignments. There are, however, some distinctly bad measures of separation.
Like any assignment method, species assignment should use all available information. BLAST is a popular sequence comparison tool (21, 22), but as a measure of separation it can mislead, because it compares two sequences with local alignment, which matches and scores only the two most similar subsequences within two sequences (see Fig. 2, which diagrams some of the differences between local and global alignments). Global alignment, which matches the entire length of sequences, is better for measuring the separation of barcode marker sequences. In intergenic markers particularly, BLAST has the possible weakness of matching only small subsequences, because alignments within intergenic spacers often contain large gaps. Short subsequences can exhibit convergent evolution (homoplasy) (23), so on the one hand a BLAST local alignment might make distant species appear spuriously close. On the other hand, a global alignment might resolve the species by highlighting dissimilarities across the whole marker. In the context of barcodes, therefore, a global alignment (e.g., with some close relative of the Needleman–Wunsch Algorithm (24)) is generally preferable to a local alignment (e.g., with the Smith–Waterman Algorithm (25) or BLAST). Other types of alignments exist, but there is little reason to expect them to assign species notably better than global alignment.
Fig. 2.
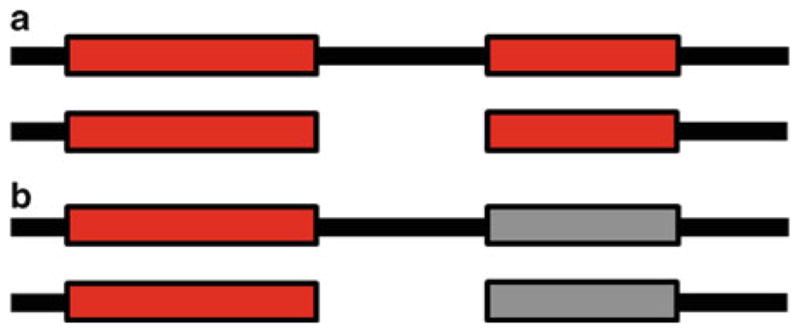
Two types of alignment, global and local. (a) shows a global alignment of two sequences (black lines). Global alignment is an alignment along the complete length of the sequences, so it bridges a gap in the second sequence (white space), to include all pairs of similar subsequences (red rectangles). (b) shows a local alignment of the same two sequences. Local alignment aligns only the pair of most similar subsequences in the sequences, so it does not bridge the gap in the second sequence and does not include the smaller subsequence alignment (now shown in gray). Local alignment can be misleading when identifying species with barcodes because it does not incorporate all available sequence information.
MSAs might be more problematic for intergenic markers than for marker genes like CO1, because intergenic MSAs usually contain many gaps, disrupting the alignment columns representing evolutionary relationships. In practice, the Barcode of Life Database (http://www.boldsystems.org) stores sequences in a global MSA, by using the program HMMer (26) to align sequences before comparing the corresponding barcode marker genes. In fact, many publicly available tools (e.g., MUSCLE (27) or MAFFT (28)) could create barcode MSAs interchangeably with HMMer. The point of using MSAs in a large barcode database, however, is that MSA can be much faster than pairwise sequence alignment. (If there are N barcodes in a database, pairwise alignment requires time proportional to N2.) Although bioinformatics should adapt to the needs of biology and not vice versa, the selection of an intergenic marker as a barcode might exclude MSAs in the workflow of large barcode databases, causing awkward (but probably not insuperable) difficulties.
As separations, the relative merits of global alignment similarity, global alignment distances, or evolutionary distances based on a global alignment have not yet been clearly established, although the differences in species assignment are probably small. Alignment distances and similarities model insertions and deletions in sequences, which are not as well understood as nucleotide substitutions used in evolutionary distances. As a separation, p-distance (the proportion p of alignment pairs containing differing nucleotides) is particularly simple and well-known to taxonomists (20), but in fact no separation based on global alignment has shown any clear superiority in species assignment over the others.
Other species assignment algorithms should be mentioned (29, 30). Many probabilistic algorithms, in particular those producing phylogenetic trees (31, 32), are now a commonplace in taxonomy. Unfortunately, most probabilistic computations are much slower than the nearest neighbor algorithms above. Because they do not noticeably improve identification, they have not found a place in automatic species identification. Alignment-free algorithms are simple and provide faster computation than alignment-based methods (20, 33), but presently, they have not been widely adopted in species identification.
2.3. Probability of Correct Identification
With an appropriate database and species assignment algorithm in hand, a scientist interested in barcode efficacy must measure the algorithm’s success in identifying species. Any reasonable measure of barcode efficacy should reflect the probability that a database based on the prospective barcode identifies a specimen’s species correctly. Consensus has therefore emerged on “the probability of correct identification” (PCI) as the appropriate measurement of barcode efficacy (14, 15, 17). The ambiguities in the definition of PCI accommodate legitimate scientific disagreement about success in species identification, so the concept of PCI actually embraces a broad class of measures.
Consider a particular data set, and assume that PCI can be defined for each species within the data set. The overall PCI for the data set is the average of the species PCIs, taken over all species in the data set. If a few data subsets are particularly important (e.g., angiosperm, basal, and gymnosperm subsets within a plant data set), the PCI for the subsets can be reported separately. In principle, the PCI for each species could be weighted to reflect the species’ importance or the number of specimens representing it in the data set. In practice, however, scientists have not weighted averages when calculating overall PCI. Thus, to calculate the overall PCI of a data set, we now require only a species PCI, a probability to quantify success in identifying each fixed species.
To calculate a species PCI, one can perform a leave-one-out procedure, sometimes called “the jackknife” in statistics (34). Remove each specimen in a species in turn from the database, and consider the separation of the removed specimen from the specimens of the same species remaining in the database. (The leave-one-out procedure cannot sensibly be applied if a species has only a single specimen in the database. Because a singleton species must therefore be omitted from the average in the overall PCI, it usually represents wasted experimental effort. It does, however, provide a “decoy,” which provides a realistic impediment to correct species assignment.)
Scientists legitimately disagree over the definition of “success” in species identification. Some scientists might consider “success” theoretically, as a monophyly, where every specimen in the species is closer to all specimens in the species than to any other specimen (14). On success, the species PCI is 1; on failure, it is 0. Other scientists might consider success more pragmatically, as a correct assignment of the species, where each specimen in the species has as its nearest neighbor(s) only specimens in the species (15). Again, if so, the species PCI is 1; if not, it is 0. The following additional conditions can contribute to success or failure, as desired: ties outside the species for a nearest neighbor, assignment of specimens from other species to the species in question, etc.
Some authors have advanced less stringent criteria for success (e.g., for k > 1, the specimen’s nearest neighbors must contain at least one other specimen from the same species) (33). The species PCI has also been calculated as the fraction of specimens within a species whose nearest neighbor gives the correct assignment (17). Any specific choice might be appropriate in different circumstances, depending on the scientific aim.
Some authors experimented with placing additional conditions on “success” as defined above, e.g., sequence difference (p-distance) thresholds, such as 2% or 3% (15). Detection of unknown species with sequence identity thresholds seems artificial, however (35). The notion of “species” could be redefined by DNA thresholds (1, 2, 36, 37), but such redefinitions generate many conflicts with traditional taxonomy (15).
2.4. PCR Failure
PCI should estimate the success in correctly identifying a known species. Under present technology, species identification with a DNA barcode requires the following criteria:
At least part of the barcode sequence must be present in the specimen.
Laboratory procedures must physically extract it from the specimen.
PCR primers must amplify it.
It must be sequenced.
It must diverge sufficiently, to distinguish species.
It must not diverge excessively, so specimens from a single species remain similar and identifiable.
Thus, PCI must account for PCR failure, if it is to estimate identification success under present technology. Recall that the overall PCI is the average of the PCI for each individual species. The Appendix discusses PCR failure for a barcode based on several markers. For simplicity, this subsection considers here only a bar-code based on a single marker. We revise the species PCI to account for PCR failure, as follows. According to the procedures in the preceding subsection (which ignore PCR failure), let the species have PCI p; and let s be the fraction of specimens from the species with a successful PCR. (Note that s is estimated from all specimens, whereas p is estimated solely from specimens with a successful PCR.) A reasonable procedure might average the “PCR-adjusted species PCI” p′ = ps over all species to produce a “PCR-adjusted overall PCI.” The PCR-adjusted overall PCI faithfully reflects the efficacy of species identification with present technology, whereas the overall PCI (which ignores specimens where PCR failed) reflects the efficacy of species identification with a perfect PCR technology.
Technology reduces PCR failure rates, so arguments have been advanced that PCR failure should be ignored (14). The PCI after any technological advance, however, is bounded below by the PCR-adjusted overall PCI (which reflects present PCR technology); similarly, it is bounded above by the overall PCI (which ignores specimens with failed PCR). The bounds demonstrate that technological advance by itself does not preclude a sober assessment of future prospects. Like any numerical result from a definite procedure with a sensible meaning, the PCR-adjusted overall PCI is useful, and its deliberate omission merely undermines rational discussion about the relative merits of potential barcodes.
2.5. Statistical Sampling Error
The overall PCI is the (unweighted) average of the species PCIs. Let us make a reasonable approximation that species PCIs are mutually independent across all species. Any database is a sample of all possible species, so the overall PCI from the database is an estimate of the “true” overall PCI p. As such, it has a sampling error, calculable with the binomial distribution. Let n be the number of species contributing to the overall PCI. Under mild assumptions (given below), a binomial estimate p̂ is normally distributed with mean p and standard deviation . Thus, the confidence interval contains the true overall PCI p with a confidence determined by z in conjunction with the normal distribution. The larger z is, the broader the interval becomes, and the greater the probability that the interval contains the true value of p. As approximate examples, z = 2 yields an 95% confidence interval; z = 2.6, 99%, etc. (As a useful rule of thumb, the normal approximation holds, if n ≥ 20 and the confidence interval does not include 0.0 or 1.0.) Confidence intervals are worth calculating, because they are often surprisingly broad.
As an aside, the confidence intervals for the overall PCI are crucial to evaluating the relative merits of tentative barcodes, but they have little direct bearing on one’s confidence in the species assignment of a specific specimen, for the following reason. Most taxonomists probably prefer a barcode for which assignment errors are confined to a few species, rather than to have the same errors spread across many species. (If nothing else, alternative strategies might be available for assigning a small number of problematic species.) Overall PCI faithfully reflects taxonomists’ barcode preferences, but the evaluation of a specific species assignment poses a different problem, requiring a different solution.
3. The Summary of the Workflow
Selection of a DNA barcode has been problematic for some species, but there is now a general consensus on the measurement of bar-code efficacy. The procedure for measuring barcode efficacy can be broken into several steps.
First, assemble databases corresponding to the prospective barcodes. The choice of database must be given careful consideration because it can noticeably influence a study’s conclusions. To ensure proper controls, specimens not having a sequence in every marker database should be eliminated from consideration. Because GenBank taxonomy might be undependable, and because most GenBank sequences do not specify a corresponding taxonomic specimen, studies based on GenBank data do not have the same status as a controlled taxonomic study, particularly for barcodes based on more than one marker.
Second, select a computer algorithm for assigning species to barcode sequences. No algorithm seems to improve noticeably on assigning to a specimen the species of its nearest neighbor within a barcode database. A global alignment (e.g., with Needleman–Wunsch algorithm, or some similar algorithm) is recommended, to take advantage of all the information in a barcode sequence. By contrast, BLAST is a local alignment program, which might match only small subsequences within two sequences. Thus, the use of BLAST runs an unnecessary risk when evaluating any prospective barcode, particularly one with an intergenic marker. As long as alignments are in essence global, alignment similarities, alignment distances, and evolutionary distances like p-distance, Kimura 2-Parameter Distance, etc., seem to have approximately equal efficacies in identifying species.
Consensus has emerged on “the probability of correct identification” (PCI) as the appropriate measurement of barcode efficacy. The overall PCI for a data set is the average of the species PCIs, taken over all species in the data set. If a few data subsets are particularly important (e.g., angiosperm, basal, and gymnosperm subsets within a plant data set), the PCI for the subsets can be reported separately.
To calculate a species PCI, remove in turn each specimen in the species from the database, and consider its separation from the remaining specimens (under, e.g., p-distance). Various definitions of identification success within a species are possible: (1) every specimen in the species is closer to all other specimens in the species than to any other specimen; (2) each specimen in the species has another specimen in the species as its nearest neighbor; (3) more stringent versions of the two foregoing definitions, where ties outside the species for a nearest neighbor, or assignment of other species to the species in question, also connote failure; (4) less stringent criteria for success (e.g., for k > 1, the specimen’s nearest k neighbors must contain at least one other specimen from the same species; or (5) probabilistic measures of success, like the fraction of specimens within a species displaying one of the foregoing definitions of success. Scientific purpose makes different definitions of “successful assignment” appropriate to different circumstances.
To estimate success under present technology, PCI must account for PCR failure. Although the case of a barcode with several markers has been relegated to the Appendix, the case of a barcode with only one marker poses no difficulties. Simply estimate the rate of PCR failure within each species by using all specimens, not just the ones with completely successful PCRs. Multiplication of a species PCI by the PCR success rate within the species yields a “PCR-adjusted” species PCI, which can then be averaged over species to yield a PCR-adjusted overall PCI. The overall PCI after technological advance is bounded below by the PCR-adjusted overall PCI; similarly, it is bounded above the overall PCI (which derives from PCR successes only). Thus, present technology bounds prospects for an overall PCI.
A database provides a statistical sample of all possible data. The overall PCI calculated from a database is therefore a statistical estimate of the true overall PCI, and as such, it yields an estimate with a statistical error. The errors are sometimes surprisingly large, and the differences in barcode efficaciousness correspondingly small.
For software relevant to this chapter, see http://www.ncbi.nlm.nih.gov/CBBresearch/Spouge/html.ncbi/barcode/.
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, NLM, NCBI.
Appendix
For a barcode with several markers, each of which can have a failed PCR, specimen identification ultimately relies on the markers with a successful PCR. To quantify the identification process, number the markers {1, 2,…,m}, and consider any subset M of {1, 2,…,m}. For a particular specimen, let the probability that M is the subset of markers with PCR success be denoted by sM, and let the PCI for the barcode based on the marker subset M be pM. A species PCI p can then be calculated from the values of sM and pM (although the calculation depends on the definition of species PCI: see Section 2.3 for various definitions.)
One very reasonable definition of the PCR-adjusted species PCI is the average p = Σ(M) pM sM. For the case of a barcode based on a single marker, e.g., M is a subset of {1}, i.e., the empty set {} or {1}. Because the empty set {} corresponds to a complete absence of information about a specimen, the corresponding PCI is p{} = 0, so p = p{}s{} + p{1}s{1} = p{1}s{1}, which agrees with the formula for the PCR-adjusted PCI in the main text, for a barcode based on a single marker.
Footnotes
For software relevant to this chapter, see http://www.ncbi.nlm.nih.gov/CBBresearch/Spouge/html.ncbi/barcode/
References
- 1.Hebert PD, Cywinska A, Ball SL, Dewaard JR. Biological identifications through DNA barcodes. Proc Biol Sci. 2003;270:313–321. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Floyd R, Abebe E, Papert A, Blaxter M. Molecular barcodes for soil nematode identification. Mol Ecol. 2002;11:839–850. doi: 10.1046/j.1365-294x.2002.01485.x. [DOI] [PubMed] [Google Scholar]
- 3.Hebert PD, Ratnasingham S, Dewaard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci. 2003;270:S96–S99. doi: 10.1098/rsbl.2003.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hajibabaei M, Janzen DM, Burns JM, et al. DNA barcodes distinguish species of tropical lepidoptera. Proc Natl Acad Sci U S A. 2006;103:968–971. doi: 10.1073/pnas.0510466103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hogg ID, Hebert PDN. Biological identification of springtails (hexapoda: Collembola) from the canadian arctic, using mitochondrial DNA barcodes. Can J Zool. 2004;82:749–754. [Google Scholar]
- 6.Lorenz JG, Jackson WE, Beck JC, Hanner R. The problems and promise of DNA barcodes for species diagnosis of primate bio-materials. Philos Trans R Soc Lond B Biol Sci. 2005;360:1869–1877. doi: 10.1098/rstb.2005.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyer CP, Paulay G. DNA barcoding: error rates based on comprehensive sampling. PLoS Biol. 2005;3:e422. doi: 10.1371/journal.pbio.0030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saunders GW. Applying DNA barcoding to red macroalgae: a preliminary appraisal holds promise for future applications. Philos Trans R Soc Lond B Biol Sci. 2005;360:1879–1888. doi: 10.1098/rstb.2005.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith MA, Fisher BL, Hebert PDN. DNA barcoding for effective biodiversity assessment of a hyperdiverse arthropod group: the ants of Madagascar. Philos Trans R Soc Lond B Biol Sci. 2005;360:1825–1834. doi: 10.1098/rstb.2005.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith MA, Woodley NE, Janzen DH, et al. DNA barcodes reveal cryptic host-specificity within the presumed polyphagous members of a genus of parasitoid flies (diptera: Tachinidae) Proc Natl Acad Sci U S A. 2006;103:3657–3662. doi: 10.1073/pnas.0511318103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chase MW, Salamin N, Wilkinson M, et al. Land plants and DNA barcodes: short-term and long-term goals. Philos Trans R Soc Lond B Biol Sci. 2005;360:1889–1895. doi: 10.1098/rstb.2005.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cowan RS, Chase MW, Kress JW, Savolainen V. 300,000 species to identify: problems, progress, and prospects in DNA barcoding of land plants. Taxon. 2006;55:611–616. [Google Scholar]
- 13.Kress WJ, Erickson DL. DNA barcodes: genes, genomics, and bioinformatics. Proc Natl Acad Sci U S A. 2008;105:2761–2762. doi: 10.1073/pnas.0800476105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cbol Plant Working Group. A DNA barcode for land plants. Proc Natl Acad Sci U S A. 2009;106:12794–12797. doi: 10.1073/pnas.0905845106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meier R, Shiyang K, Vaidya G, Ng PK. DNA barcoding and taxonomy in diptera: a tale of high intraspecific variability and low identification success. Syst Biol. 2006;55:715–728. doi: 10.1080/10635150600969864. [DOI] [PubMed] [Google Scholar]
- 16.Huang D, Meier R, Todd PA, Chou LM. Slow mitochondrial coI sequence evolution at the base of the metazoan tree and its implications for DNA barcoding. J Mol Evol. 2008;66:167–174. doi: 10.1007/s00239-008-9069-5. [DOI] [PubMed] [Google Scholar]
- 17.Erickson DL, Spouge JL, Resch A, et al. DNA barcoding in land plants: developing standards to quantify and maximize success. Taxon. 2008;13:1304–1316. [PMC free article] [PubMed] [Google Scholar]
- 18.Kress WJ, Erickson DL. A two-locus global DNA barcode for land plants: the coding rbcl gene complements the non-coding trnhpsba spacer region. PLoS One. 2007;2:e508. doi: 10.1371/journal.pone.0000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Austerlitz F. Comparing phylogenetic and statistical classification methods for DNA barcoding. Paper presented at the second international barcode of life conference; Taipei, Taiwan. 2007. [Google Scholar]
- 20.Little DP, Stevenson DW. A comparison of algorithms for the identification of specimens using DNA barcodes: examples from gymnosperms. Cladistics. 2007;23:1–27. doi: 10.1111/j.1096-0031.2006.00126.x. [DOI] [PubMed] [Google Scholar]
- 21.Altschul SF, Madden TL, Schaffer AA, et al. Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altschul S. Hot papers – bioinformatics – gapped blast and psi-blast: a new generation of protein database search programs by s.F. Altschul, t.L. Madden, a.A. Schaffer, j.H. Zhang, z. Zhang, w. Miller, d.J. Lipman – comments. Scientist. 1999;13:15. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wouters MA, Husain A. Changes in zinc ligation promote remodeling of the active site in the zinc hydrolase superfamily. J Mol Biol. 2001;314:1191–1207. doi: 10.1006/jmbi.2000.5161. [DOI] [PubMed] [Google Scholar]
- 24.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 25.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 26.Eddy SR. Multiple alignment using hidden markov models. Proc Int Conf Intell Syst Mol Biol. 1995;3:114–120. [PubMed] [Google Scholar]
- 27.Edgar RC. Muscle: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katoh K, Misawa K, Kuma K, Miyata T. Mafft: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matz MV, Nielsen R. A likelihood ratio test for species membership based on DNA sequence data. Philos Trans R Soc Lond B Biol Sci. 2005;360:1969–1974. doi: 10.1098/rstb.2005.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen R, Matz M. Statistical approaches for DNA barcoding. Syst Biol. 2006;55:162–169. doi: 10.1080/10635150500431239. [DOI] [PubMed] [Google Scholar]
- 31.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 32.Felsenstein J. Phylogenies from molecular sequences – inference and reliability. Annu Rev Genet. 1988;22:521–565. doi: 10.1146/annurev.ge.22.120188.002513. [DOI] [PubMed] [Google Scholar]
- 33.Kuksa P, Pavlovic V. Efficient alignment-free DNA barcode analytics. BMC Bioinform atics. 2009;10:S9. doi: 10.1186/1471-2105-10-S14-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Efron B, Stein C. The jackknife estimate of variance. Ann Stat. 1981;9:586–596. [Google Scholar]
- 35.Ferguson JWH. On the use of genetic divergence for identifying species. Biol J Linnean Soc. 2002;75:509–516. [Google Scholar]
- 36.Blaxter M, Mann J, Chapman T, et al. Defining operational taxonomic units using DNA barcode data. Philos Trans R Soc Lond B Biol Sci. 2005;360:1935–1943. doi: 10.1098/rstb.2005.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lambert DM, Baker A, Huynen L, et al. Is a large-scale DNA-based inventory of ancient life possible? J Hered. 2005;96(3):279–284. doi: 10.1093/jhered/esi035. [DOI] [PubMed] [Google Scholar]
- 38.Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian protein metabolism. Academic; New York: 1969. pp. 21–123. [Google Scholar]
- 39.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 40.Jin L, Nei M. Limitations of the evolutionary parsimony method of phylogenetic analysis. Mol Biol Evol. 1990;7:82–102. doi: 10.1093/oxfordjournals.molbev.a040588. [DOI] [PubMed] [Google Scholar]
- 41.Tamura K. Model selection in the estimation of the number of nucleotide substitutions. Mol Biol Evol. 1994;11:154–157. doi: 10.1093/oxfordjournals.molbev.a040088. [DOI] [PubMed] [Google Scholar]
- 42.Waterman MS, Smith TF, Beyer WA. Some biological sequence metrics. Adv Math. 1976;20:367–387. [Google Scholar]