

Abstract
Transcript elongation by RNA polymerase II (RNAPII) is accompanied by conserved patterns of histone modification. Whereas histone modifications have established roles in transcription initiation, their functions during elongation are not understood. Mono-ubiquitylation of histone H2B (H2Bub1) plays a key role in coordinating co-transcriptional histone modification by promoting site-specific methylation of histone H3. H2Bub1 also regulates gene expression through an unidentified, methylation-independent mechanism. Here we reveal bidirectional communication between H2Bub1 and Cdk9, the ortholog of metazoan positive transcription elongation factor b (P-TEFb), in the fission yeast Schizosaccharomyces pombe. Chemical and classical genetic analyses indicate that lowering Cdk9 activity or preventing phosphorylation of its substrate, the transcription processivity factor Spt5, reduces H2Bub1 in vivo. Conversely, mutations in the H2Bub1 pathway impair Cdk9 recruitment to chromatin and decrease Spt5 phosphorylation. Moreover, an Spt5 phosphorylation-site mutation, combined with deletion of the histone H3 Lys4 methyltransferase Set1, phenocopies morphologic and growth defects due to H2Bub1 loss, suggesting independent, partially redundant roles for Cdk9 and Set1 downstream of H2Bub1. Surprisingly, mutation of the histone H2B ubiquitin-acceptor residue relaxes the Cdk9 activity requirement in vivo, and cdk9 mutations suppress cell-morphology defects in H2Bub1-deficient strains. Genome-wide analyses by chromatin immunoprecipitation also demonstrate opposing effects of Cdk9 and H2Bub1 on distribution of transcribing RNAPII. Therefore, whereas mutual dependence of H2Bub1 and Spt5 phosphorylation indicates positive feedback, mutual suppression by cdk9 and H2Bub1-pathway mutations suggests antagonistic functions that must be kept in balance to regulate elongation. Loss of H2Bub1 disrupts that balance and leads to deranged gene expression and aberrant cell morphologies, revealing a novel function of a conserved, co-transcriptional histone modification.
Author Summary
Modification of histone proteins is an important transcriptional regulatory mechanism in eukaryotic cells. Although various histone modifications are found primarily within the coding regions of transcribed genes, how they influence transcription elongation remains unclear. Among these modifications is mono-ubiquitylation of histone H2B (H2Bub1), which is needed for co-transcriptional methylation of histone H3 at specific sites. Here we show that H2Bub1 and Cdk9, the kinase component of positive transcription elongation factor b (P-TEFb), are jointly regulated by a positive feedback loop: Cdk9 activity is needed for co-transcriptional H2Bub1, and H2Bub1 in turn stimulates Cdk9 activity toward one of its major substrates, the conserved elongation factor Spt5. We provide genetic evidence that the combined action of H2Bub1 on Spt5 phosphorylation and histone methylation accounts for the gene-regulatory effects of this modification. Surprisingly, our genetic and genome-wide studies indicate that P-TEFb and H2Bub1 act in opposition on elongating RNA polymerase. We suggest that the positive feedback linking P-TEFb and H2Bub1 helps to maintain a balance between their opposing actions. These results highlight a novel regulatory role for a conserved histone modification during transcription elongation.
Introduction
The elongation phase of transcription is a point of regulation for many genes transcribed by RNAPII in eukaryotes, and control of elongation is critical for coupling of transcription to downstream steps in gene expression [1], [2]. Whereas the regulation of transcription at the initiation step has been studied extensively, many of the mechanisms governing elongation in vivo remain to be elucidated. Transcription is accompanied by post-translational modification of nucleosomal histones in a highly conserved pattern, stereotypical features of which include methylation of histone H3 Lys4 (H3K4me) and acetylation of histones H3 and H4 at 5′ ends, methylation of histone H3 Lys36 (H3K36me) towards 3′ ends, and mono-ubiquitylation of histone H2B at a conserved site in the carboxyl-terminus (H2Bub1) throughout coding regions of genes [3]. Conservation of this pattern suggests an important role in coordinating gene expression, but the precise functions of individual modifications in elongation control are poorly understood.
H2Bub1, which appears to play a central role in the interplay between chromatin and the RNAPII elongation complex, is catalyzed in budding yeast by the ubiquitin-conjugating enzyme Rad6 and the E3 ubiquitin ligase Bre1 [4]. Formation of H2Bub1 on transcribed chromatin also requires PAF, a conserved complex with multiple functions during elongation [5]. H2Bub1 is required for co-transcriptional generation of H3K4me by the methyltransferase Set1 in yeast, and contributes to global H3K4me levels in metazoans [6]–[9]. In vitro, H2Bub1 directly stimulates activity of Set1 towards a reconstituted chromatin substrate [10], [11].
H2Bub1 also acts independently of histone methylation; our work in the fission yeast S. pombe revealed that the Set1-independent pathway is important for normal cell growth and morphology, and for elongation by RNAPII at select target genes [12]. Similar findings have now been reported in S. cerevisiae and mammalian cells [13]–[15], but the mechanism of methylation-independent effects of H2Bub1 is unknown.
Co-transcriptional histone modifications are regulated by a conserved subset of cyclin-dependent kinases (CDKs) associated with the transcription machinery [16]. In metazoans these include Cdk7, a component of the initiation factor TFIIH, and Cdk9, catalytic subunit of positive transcription elongation factor b (P-TEFb). Cdk7, Cdk9 and their yeast orthologs phosphorylate multiple proteins important for elongation, including the Rpb1 subunit of RNAPII, at Ser2, Ser5 and Ser7 positions within the repeated YSPTSPS motif of its carboxyl-terminal domain (CTD) [17]–[19]; and the Spt5 subunit of a conserved elongation factor, known in metazoans as DRB-sensitivity inducing factor (DSIF) [20], [21]. Those phosphorylations control recruitment of pre-mRNA-processing and chromatin-modifying enzymes [17], [22], [23].In metazoans, moreover, Cdk9 activity overcomes promoter-proximal pausing imposed by unphosphorylated DSIF and a negative elongation factor (NELF) [20], [24], [25]. Pausing is thought to act both as a quality control over gene expression, by facilitating recruitment of mRNA-processing factors [26], [27]; and as a rate-limiting determinant of expression for subsets of stringently regulated genes [28], [29].
P-TEFb also regulates histone modification. In mammalian cells, levels of H2Bub1, H3K4me, and H3K36me decrease after depletion of Cdk9 by RNA interference (RNAi), or treatment with flavopiridol, an inhibitor of Cdk9 and related kinases [30], [31]. Bur1, the essential ortholog of Cdk9 in budding yeast, is required for co-transcriptional H3K36me and consequent action of the Rpd3S histone deacetylase complex to prevent transcription initiation within coding regions [32]–[34]. Similarly, co-transcriptional H2Bub1 depends on Bur1 [35], [36] and the carboxyl-terminal region of Spt5, which contains sites phosphorylated by Bur1 [37], [38]. Budding yeast contains another, non-essential CDK, Ctk1, which is the major Rpb1-Ser2 kinase in vivo and is required for H3K36me [39], [40]. Although Ctk1 was proposed to be a yeast-specific P-TEFb paralog [41], putative orthologs of Ctk1 have recently been identified in human and Drosophila cells [42], [43], suggesting faithful conservation of the entire CTD kinase network.
CDKs are themselves regulated by histone modification. In mammals, P-TEFb binds the bromodomain protein Brd4, whose recruitment to promoter-proximal sites is stimulated by acetylation of histone H4 [44], [45]. Phosphorylation of histone H3 also favors recruitment of P-TEFb [46], [47]. On the other hand, certain histone marks work in opposition to specific CDKs during elongation. For example, in budding yeast, lethality of a BUR1 deletion is suppressed by deletion of the H3K36 methyltransferase Set2 [34], [48]; and H2B de-ubiquitylation by Ubp8—a component of the SAGA complex—is required for Ctk1 recruitment and Ser2 phosphorylation at some genes [49].
In fission yeast, the P-TEFb ortholog is the essential Cdk9/Pch1 complex. In vitro, Cdk9 phosphorylates the Rpb1 CTD at both Ser5 and Ser2 [50]; inhibition of Cdk9 in vivo reduced the apparent stoichiometry of Rpb1 phosphorylation but did not cause selective loss of either Ser5 or Ser2 signals [51]. Cdk9 also phosphorylates the CTD of S. pombe Spt5 [52]; spt5 mutations that prevent this phosphorylation cause slow growth, defects in transcript elongation and, when combined with partial truncations of the Rpb1 CTD, synthetic lethality [53]. Genetic epistasis suggests, moreover, that the Spt5 CTD contains an exclusive, albeit nonessential, target of Cdk9 in vivo [51]. The potential roles of CDKs and their substrates in regulating histone modifications in fission yeast have not been explored.
Here we uncover a mutual dependence of H2Bub1 and P-TEFb function in S. pombe: mutations that impair phosphorylation of Spt5 by Cdk9 diminish levels of H2Bub1 and, reciprocally, mutants unable to generate H2Bub1 have reductions in chromatin-associated Cdk9 and phosphorylated Spt5. Ablation of the preferred phosphoacceptor site in Spt5 combined with deletion of set1+ phenocopies morphological defects of htb1-K119R mutants, suggesting that Cdk9 and Set1 govern separate, partially redundant pathways downstream of H2Bub1. Despite the dependencies between H2Bub1 and Spt5 phosphorylation, mutations that impair Cdk9 function suppress abnormal cell morphologies and reverse an RNAPII distribution defect, both due to loss of H2Bub1. Conversely, growth of htb1-K119R cells is resistant to selective Cdk9 inhibition, relative to that of htb1+ cells. Taken together, biochemical and genetic results suggest that P-TEFb and H2Bub1 oppose one another to regulate RNAPII elongation, but that proper balance between the two is ensured via a positive feedback loop, in which phosphorylation by Cdk9 stimulates H2Bub1 and vice versa.
Results
H2Bub1 influences global RNAPII distribution in S. pombe
Our previous results suggested that H2Bub1 promotes expression of a subset of genes [12]. To ask whether H2Bub1 might act more generally in transcription, we combined ChIP with hybridization of recovered DNA sequences to microarrays that cover the entire S. pombe genome at 200 base-pair intervals (ChIP-chip). We first mapped genome-wide distribution of H2Bub1 in wild-type cells with an antibody that specifically recognizes the ubiquitylated form of histone H2B [[54], Figure S1A]. A FLAG-epitope tag fused to the carboxyl-terminus of histone H2B allowed us to normalize for total H2B occupancy by parallel ChIP-chip with anti-FLAG antibody. The FLAG-tagged H2B was ubiquitylated to a similar extent as the native protein ([12], Figure S1B). To correlate the H2Bub1 pattern with transcription, we also performed ChIP-chip with an RNAPII-specific antibody. We grouped genes into quartiles according to their total levels of RNAPII enrichment and plotted average distributions of H2Bub1 within each group (Figure 1A). This analysis confirmed the presence of H2Bub1 throughout coding regions of transcribed genes in S. pombe, similar to its distribution in S. cerevisiae and metazoans [13], [55]–[57]. Furthermore, H2Bub1 enrichment was correlated with that of RNAPII throughout the genome (r = 0.58).
Figure 1. Loss of H2Bub1 globally alters distribution of RNAPII in gene coding regions.

(A) Average distribution of H2Bub1 at 540 S. pombe genes, as determined by ChIP-chip. Genes were grouped according to total levels of RNAPII enrichment (see key at top). The grey box in the “average gene” representation at bottom denotes the gene coding region; 5′ and 3′ untranslated regions are denoted by thin black lines. The arrow denotes the transcription start site. (B) Average distribution of RNAPII at 540 S. pombe genes, as determined by ChIP-chip in a wild-type strain (JTB62-1). Genes were grouped according to total levels of RNAPII enrichment. (C) As in (B), determined in an htb1-K119R mutant strain (JTB67-1). Gene groupings were created using wild-type RNAPII enrichment values. (D) Average distributions of differences between mutant and wild-type RNAPII enrichment grouped according to RNAPII enrichment in wild-type cells. The key below the graph illustrates the statistical significance of the differences for each group at 50 positions along the average gene. The rows of the key are color-coded according to the graph. Open squares denote p>0.01; light shading denotes 0.01>p>10exp-5; dark shading denotes p<10exp-5 (one-sample t-tests; μ0 = 0). Note that there is only light shading for the last row (corresponding to the blue curve).
Because of this genome-wide association, we sought to determine the impact of H2Bub1 loss on RNAPII distribution within genes, by ChIP-chip in wild-type and htb1-K119R mutant cells. This analysis revealed alterations in RNAPII occupancy within gene coding regions in the htb1-K119R mutant: a global, downstream shift in average RNAPII density, reflecting both a decrease in occupancy within the 5′ halves of genes and a more pronounced increase near the 3′ ends (Figure 1B and 1C). These changes occurred in all classes of genes, but varied quantitatively depending on their overall levels of RNAPII cross-linking (Figure 1D; Dataset S1). Genes highly enriched for RNAPII (Figure 1D; red line) had a significant (P<10exp-5) decrease in RNAPII 5′-end occupancy in the absence of H2Bub1, whereas the increase at 3′ ends failed to reach significance (P>0.01). In the other three classes of genes the increases in RNAPII occupancy at the 3′ end were the most significant changes (Figure 1D). Therefore, H2Bub1 globally affects RNAPII distribution in S. pombe genes.
Previous reports have shown that H2Bub1 affects nucleosome stability [9], [14], [58], [59]. We performed ChIP-chip with an antibody that recognizes histone H3 to determine whether the changes in RNAPII distribution caused by H2Bub1 loss were correlated with changes in nucleosome occupancy. The average histone H3 occupancy in gene coding regions did not differ significantly between wild-type and htb1-K119R cells (Figure S2; Datasets S2 and S3). These data suggest that the observed changes in RNAPII occupancy are unlikely to be caused solely by altered nucleosome distribution.
To ascertain whether genes affected by the htb1-K119R mutation were associated with particular cellular functions, we searched for Gene Ontology (GO) terms that were significantly enriched among the 500 genes with the largest increases or decreases in RNAPII occupancy in the htb1-K119R mutant. The genes exhibiting loss of RNAPII occupancy were enriched for ribosomal protein genes (P = 3.58exp-26, 17.7%), consistent with a role for H2Bub1 in promoting RNAPII occupancy at heavily transcribed genes.
The ChIP-chip analysis revealed that RNAPII density was reduced throughout coding regions of 25 of 40 genes whose expression decreased by ≥2-fold in our previous microarray analysis of gene expression [12](Table S1). Furthermore, this group of genes is significantly enriched among the highest RNAPII-occupancy class that we defined by ChIP-chip (P = 7.96exp-5). Thus, the ChIP-chip results and our previous gene-expression data argue that loss of H2Bub1 has a particularly strong negative impact on steady-state levels of poly(A+) mRNA produced from highly transcribed genes. However, the global redistribution of RNAPII in htb1-K119R cells (Figure 1D) suggests that H2Bub1 might affect gene expression more broadly through additional, post-transcriptional mechanisms.
P-TEFb activity promotes H2Bub1 in fission yeast
Loss of H2Bub1 caused changes in cell morphology and RNAPII distribution that resemble those produced by inactivation of Rpb1 CTD kinases or loss of Rpb1-CTD Ser2 phosphorylation [12], [51], [60], [61]. We therefore investigated possible interactions between H2Bub1 and three CDKs implicated in transcription: the TFIIH-associated Cdk7 ortholog Mcs6 [61], Cdk9, and the Ctk1 ortholog Lsk1 [62]. First, we determined the effect on H2Bub1 of selective inhibition of Mcs6 or Cdk9, made possible by mutating the “gatekeeper” residue in each kinase to Gly, to render the enzyme sensitive to bulky adenine analogs that do not affect wild-type kinases [63]. We previously replaced mcs6+, cdk9+ and lsk1+ with alleles encoding analog-sensitive (AS) mutant versions, each of which was sensitive to the inhibitory analog 3-MB-PP1 [51]. Selective inhibition of Cdk9, by addition of 20 µM 3-MB-PP1 to cdk9as cells, caused diminution of H2Bub1 signals in whole-cell extracts (Figure 2A). Cdk9 inhibition also decreased levels of H3K4 di- and trimethylation (H3K4me2 and H3K4me3, respectively) and H3K36 trimethylation (H3K36me3) (Figure 2A and data not shown)—modifications likewise associated with actively transcribed genes [3]. Inhibition of the TFIIH-associated kinase in mcs6as cells had small or no effects on H2Bub1, H3K4me2, H3K4me3 or H3K36me3. These results suggest a predominant role for Cdk9 in co-transcriptional histone modification, and a possibly exclusive requirement in generating H2Bub1.
Figure 2. H2Bub1 depends on Cdk9 activity and Spt5 phosphorylation.
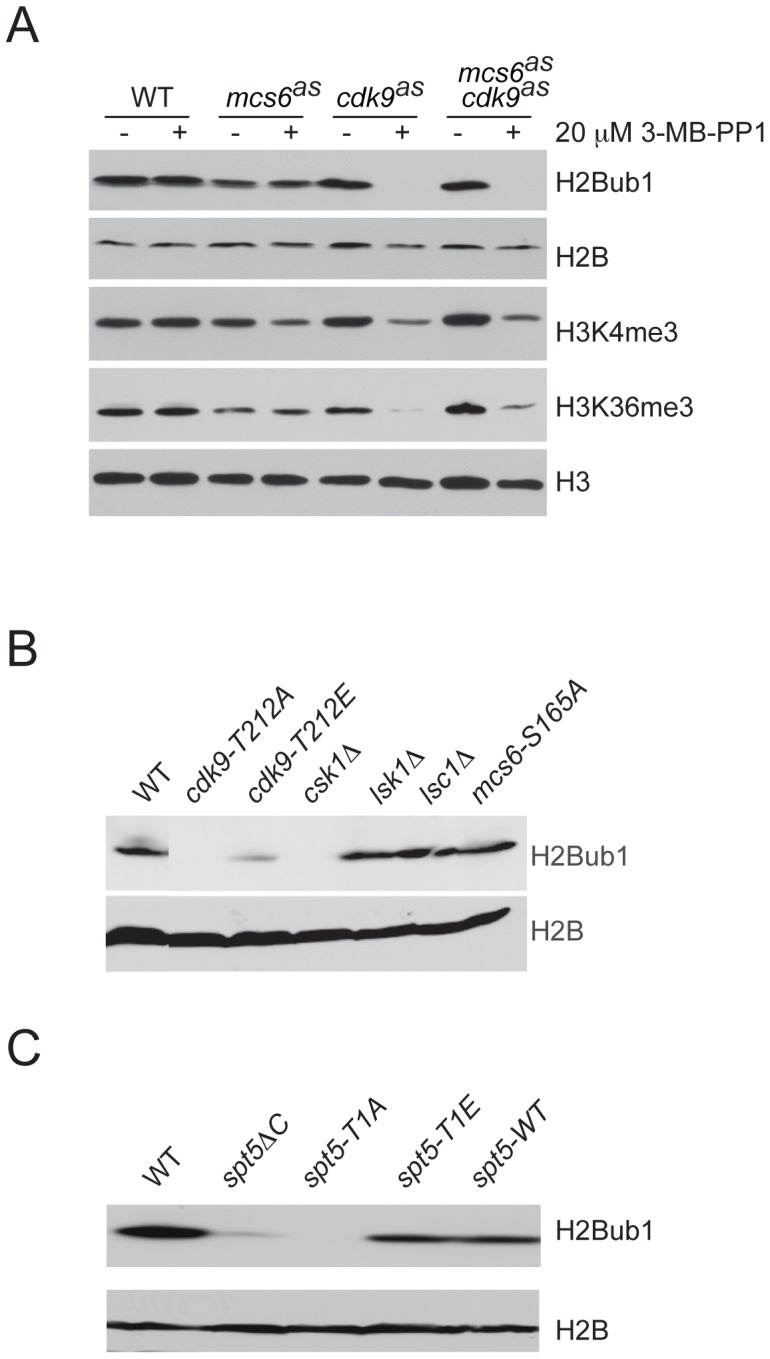
(A) Immunoblots of whole-cell extracts from wild-type (wt) (JS78) or AS mutant strains (LV7, LV77, LV42), as indicated, grown in the absence (−) or presence (+) of 20 µM 3-MB-PP1, added 20 min prior to harvest. Antibodies are indicated at right. (B) Immunoblots of extracts from indicated strains probed for total histone H2B and H2Bub1, as indicated at right. “T212A” and “T212E” denote strains cdk9-T212A (HD7-24) and cdk9-T212E (HG127). (C) Immunoblots of extracts from wild-type (JS78) or indicated spt5 mutant strains.
Bur1 in budding yeast and Cdk9 in mammalian cells are proposed to stimulate H2Bub1 by favoring recruitment of PAF, which can in turn associate directly with the H2B ubiquitylation machinery [10], [37], [38], [64]. Inhibition of Cdk9as decreased association of both the PAF component Rtf1 and the E2 ubiquitin-conjugating enzyme Rhp6 with transcribed chromatin (Figure S3A and S3B), suggesting faithful conservation of the P-TEFb-H2Bub1 pathway in S. pombe.
H2Bub1 depends on activated Cdk9 and phosphorylation of Spt5
To further probe the dependence of H2Bub1 on P-TEFb, we tested effects of other mutations that perturb Cdk9 function (Figure 2B). Two mutant strains in which Cdk9 activity is reduced by lack of phosphorylation at Thr212 of the activation segment (T loop)—cdk9-T212A and csk1Δ, which is missing the CDK-activating kinase (CAK) responsible for that phosphorylation [50]—had nearly undetectable levels of H2Bub1. In contrast, a cdk9-T212E mutation, which substitutes a Glu residue for Thr212 to mimic constitutive phosphorylation [65], had only a small negative effect on H2Bub1. Loss of H2Bub1 occurred uniquely in mutants with impaired Cdk9 function; neither mcs6-S165A, a T-loop mutation that renders Mcs6 refractory to CAK [66], [67], nor deletions of lsk1+ or lsc1+ [which encodes the cyclin partner of Lsk1 [68]], affected bulk H2Bub1 levels (Figure 2B). Therefore, among S. pombe CDKs implicated in transcript elongation, Cdk9 is uniquely required for H2Bub1.
Impaired Cdk9 function could lead to H2Bub1 loss by decreasing phosphorylation of the Spt5 CTD nonapeptide repeat TPAWNSGSK at Thr1, a site modified by Cdk9 in vitro [52]. To test this we measured H2Bub1 levels in a series of spt5 truncation mutants: spt5ΔC, in which the entire CTD is deleted; or variants containing seven repeats, either of wild-type sequence (spt5-WT), or with every Thr1 position mutated to Ala (spt5-T1A) or Glu (spt5-T1E) [53]. H2Bub1 decreased in spt5ΔC and spt5-T1A mutants, but not in spt5-WT or an spt5-T1E mutant that mimics constitutive phosphorylation (Figure 2C). We conclude that Cdk9 activity and Spt5-CTD phosphorylation are required for H2Bub1 in vivo.
Cdk9 activity is required for Spt5-Thr1 phosphorylation in vivo
To investigate this connection, and to assess relative contributions of different CDKs to Spt5 phosphorylation in vivo, we generated an antibody specific for the Spt5 CTD phosphorylated on the Thr1 residue (anti-Spt5-T1P), which recognized Spt5 only after it had been phosphorylated by Cdk9 in vitro (Figure S4A). In whole-cell extracts, the phosphorylated Spt5 signal was undetectable after treatment of cdk9as cells with 20 µM 3-MB-PP1, which had little or no effect on Spt5 phosphorylation in wild-type, mcs6as or lsk1as cells, and did not alter Spt5 expression levels in any of the strains (Figure 3A). Spt5-Thr1 phosphorylation was nearly abolished in cdk9as cells at 3-MB-PP1 doses as low as 300 nM (Figure S4B). In contrast, the IC50 for inhibition of cdk9as cell growth was ∼10–15 µM, and Rpb1-CTD Ser2 phosphorylation was relatively resistant to inhibition of Cdk9 alone [51]. There was no additive sensitivity of Spt5-P to the analog in mcs6as cdk9as cells, nor any loss of Spt5-P signal in mcs6as cells treated with 3-MB-PP1 doses up to 40 µM (Figure S4B, S4C). We conclude that Cdk9 is the major, and possibly sole, kinase responsible for phosphorylation of Spt5-Thr1 in vivo, and that levels of Spt5-P are sensitive even to sublethal reductions in Cdk9 activity.
Figure 3. H2Bub1 independently stimulates Cdk9-mediated Spt5 phosphorylation and Set1-dependent H3K4 methylation.

(A) Immunoblots of extracts from strains carrying spt5-myc with or without an as kinase allele as indicated (CS111, CS112, CS155, CS159, LV125 and LV167, respectively). Cultures were grown in the absence (−) or presence (+) of 20 µM 3-MB-PP1, added 20 min prior to harvest. Antibody reactivities are indicated at right. (B) Immunoblots of extracts from indicated strains (JS78, JTB67-1, JTB62-1, JTB331, HD7-24 and JTB297, respectively), probed for phosphorylated (Spt5-P) or total Spt5. (C) Fluorescent images of DAPI/calcofluor-stained cells from indicated strains (spt5-WT, JTB350, spt5-T1A, JTB352, spt5-T1E, JTB354, respectively). (D) Quantification of abnormal septation in strains of indicated genotypes (JTB67-1, spt5-WT, JTB350, spt5-T1A, JTB352, spt5-T1E, JTB354, JTB418 and JTB428, respectively). Error bars represent standard deviations from 2 independent experiments; at least 200 cells were counted in each. (E) Immunoblots of extracts from indicated strains (JTB204, JTB80-2, and JTB331, respectively), probed for phosphorylated (Spt5-P) or total Spt5. (F) Immunoblots of extracts from indicated strains (JTB204, JTB80-2, JTB67-1, spt5-WT, spt5-T1A and spt5-T1E, respectively), probed for H3K4me3 or total H3.
A role for Spt5 phosphorylation downstream of H2Bub1
There is a precedent for H2Bub1 regulating the function of a CTD kinase [49]. We wondered whether Cdk9 and the H2Bub1 machinery could be reciprocally regulated, such that the H2Bub1-defective mutants htb1-K119R and brl2Δ (which lacks the Bre1 ortholog required for H2Bub1 [12]) would have global alterations in Spt5-P. Indeed, Spt5-P was decreased relative to total Spt5 (detected with antibodies specific for wild-type Spt5-CTD irrespective of phosphorylation state; Figure S4D) in both htb1-K119R and brl2Δ strains. Conversely, Spt5-P was slightly increased in cells lacking the ubiquitin-specific protease encoded by ubp8+ (Figure 3B). (Cells lacking a second putative H2B de-ubiquitylating enzyme, encoded by the UBP10 homolog ubp16+ [69], had no change in levels of H2Bub1 or Spt5-P [data not shown]). These data suggest a positive feedback loop connecting Cdk9 and the H2Bub1 machinery in vivo.
Previous reports have documented cooperative action of H2Bub1 and the FACT complex, a conserved elongation factor that controls nucleosome dynamics during RNAPII elongation [14], [70], [71]. This prompted us to ask whether FACT might also influence Spt5-P levels in vivo. There was no reduction in levels of Spt5-P, relative to total Spt5, in a strain deleted for pob3+, which encodes a subunit of FACT (Figure S5A). This argues that promotion of Spt5-P reflects a FACT-independent function of H2Bub1.
Another consequence of H2Bub1 loss is reduction in H3K4me—a chromatin modification strongly implicated in gene activation. We showed previously, however, that loss of H2Bub1 caused more severe phenotypes than did ablation of H3K4me due to deletion of set1+. Those phenotypes include increased frequency of cells with division septa in asynchronous populations, and the appearance of cells with abnormal septation patterns (multiple septa separating two nuclei and unseparated chains of cells in which septa had formed) [12]. To test whether impaired Spt5-Thr1 phosphorylation might account for the more severe phenotypes caused by H2Bub1 loss, relative to those due to absence of H3K4me, we constructed an spt5-T1A set1Δ double mutant; the combination, which abolished two protein modifications shown to depend on H2Bub1, phenocopied the increased and aberrant septation produced by H2Bub1 loss (Figure 3C, 3D). Unlike the htb1-K119R and brl2Δ mutants, a set1Δ mutant showed no reduction in Spt5 phosphorylation (Figure 3E). Conversely, the spt5-T1A mutant retained detectable levels of H3K4me3 despite reduced H2Bub1 (Figure 3F). Therefore, H2Bub1 promotes Spt5-P and H3K4me by independent pathways. Importantly, the spt5-T1A mutation by itself did not cause morphological defects, even though it lowered H2Bub1 levels. We attribute this to the residual Set1 function present in the spt5-T1A strain (Figure 3F). A similar double mutant carrying spt5-T1E was morphologically normal, as was an spt5-T1A set2Δ strain, confirming the specificity of the genetic interaction (Figure 3C, 3D). (The pob3Δ mutation had no effect on cell morphology by itself, and did not modify phenotypes caused by htb1-K119R, suggesting that these effects are also FACT-independent [Figure S5B].) Together, these results suggest that Cdk9 and Set1 operate downstream of H2Bub1 to modify components of the transcription machinery and chromatin, respectively, and thereby regulate transcript elongation.
Next, to investigate the basis for reduced Spt5-P in H2Bub1-deficient cells, we measured Spt5-P occupancy at specific genes in the htb1-K119R mutant by ChIP. Control experiments confirmed that the Spt5-P antibody was suitable for immunoprecipitation and ChIP (Figure S6A, S6B). As predicted by immunoblot results, treatment of cdk9as spt5-myc cells with 3-MB-PP1 abolished the Spt5-P ChIP signal without affecting total Spt5 recruitment.
We then compared cross-linking of Spt5-P to chromatin in htb1+ and htb1-K119R cells, at the Cdk9-sensitive eng1+ and Cdk9-insensitive aro1+ genes [51] (Figure 4A and 4B); and at two loci (nup189+ and SPBC354.10+) that, according to our RNAPII ChIP-chip analysis, displayed a 3′ shift in RNAPII distribution in the absence of H2Bub1 (Figure S7A, S7B). A non-transcribed sequence served as a negative control (Figure S8). Although Spt5-P was broadly enriched within gene coding regions, as we observed for H2Bub1, the patterns of Spt5-P and H2Bub1 crosslinking across eng1+ and aro1+ differed, suggesting that the interdependence between these modifications is influenced by other, potentially locus-specific factors (Figure 4A, 4B, 4I, and 4J). Nevertheless, levels of Spt5-P were reduced to varying degrees on all four genes in the htb1-K119R mutant (Figure 4A and 4B, Figure S7A and S7B). Total Spt5 crosslinking was unchanged in htb1-K119R relative to wild-type cells, and the effect on Spt5-P was most evident near the 5′ ends of genes we tested, suggesting that H2Bub1 promotes co-transcriptional phosphorylation (but not recruitment) of Spt5 during the transition from initiation to elongation (Figure 4C–4F; Figure S7C–S7F).
Figure 4. H2Bub1 enhances Cdk9 recruitment and Spt5 phosphorylation at transcribed genes.

(A,B) Spt5 phosphorylation was measured by ChIP using anti-Spt5-P in spt5-myc (MS265; black bars) and spt5-myc htb1-K119R (LV239; gray bars) strains and quantified at the indicated genes by qPCR. Enrichment is plotted as a percentage of input signal for each primer pair. Positions of PCR primer pairs within coding regions are indicated schematically at top. (C,D) Spt5-myc occupancy was measured by ChIP as in A and B. (E,F) Spt5-P enrichment normalized to total Spt5-myc occupancy. (G,H) Cdk9-myc occupancy was measured by ChIP in cdk9-myc (MS264; black bars) and cdk9-myc htb1-K119R (KL259; gray bars) strains. (I,J) H2Bub1 enrichment was measured by ChIP and normalized to H2B-FLAG occupancy. Error bars denote standard deviations from 3 independent experiments. Asterisks denote a significant difference between wild-type and mutant (“*” p<0.04, “**” p<0.02; unpaired t-test).
We hypothesized that the reduction in Spt5-P could be due to a defect in recruitment of Cdk9 to chromatin in the htb1-K119R mutant. To test this possibility we compared Cdk9 crosslinking to eng1+, aro1+, nup189+ and SPBC354.10+ by ChIP in htb1+ cdk9-myc and htb1-K119R cdk9-myc strains (Figure 4G and 4H, Figure S7G and S7H). This analysis revealed that Cdk9 recruitment was impaired at all four loci in htb1-K119R cells, consistent with the reduction in Spt5-P signals. Diminished Spt5-P and Cdk9 crosslinking to these loci was not an indirect consequence of reduced RNAPII recruitment, because RNAPII crosslinking and mRNA levels were not reduced at three of the four genes examined (Figure S9). We conclude that lack of H2Bub1 specifically impedes Cdk9 recruitment to transcribed chromatin, leading to impaired phosphorylation of Spt5 within gene coding regions.
Mutual genetic suppression by P-TEFb and H2Bub1 pathway mutations
Biochemical analyses revealed a reciprocal relationship between Spt5-P and H2Bub1, suggestive of a positive feedback loop. The synthetic phenotype of spt5-T1A set1Δ double mutants, moreover, indicated that the Cdk9-Spt5 axis was one of two partially redundant gene-regulatory pathways downstream of H2Bub1. To detect and characterize direct genetic interactions between P-TEFb and H2Bub1, we tested sensitivity of a cdk9as htb1-K119R double mutant strain to 3-MB-PP1. Unexpectedly, cells bearing a cdk9as allele were less sensitive to growth inhibition by 3-MB-PP1 in an htb1-K119R, compared to an htb1+, background (Figure 5A). In contrast, the htb1-K119R mutation exacerbated 3-MB-PP1-sensitivity of mcs6as and lsk1as cells (data not shown). Therefore, lack of H2Bub1 specifically reduced dependency on active Cdk9, consistent with the two pathways acting antagonistically. Absence of H2Bub1 did not fully bypass the requirement for Cdk9 activity, however, because higher doses of 3-MB-PP1 were still capable of arresting proliferation of cdk9as htb1-K119R cells.
Figure 5. Mutual phenotypic suppression in cdk9 htb1-K119R double mutants.

(A) For indicated strains (JS78, LV7, JTB62-1, JTB67-1, MS249, LV193), growth in increasing [3-MB-PP1] is plotted as a percentage of growth in the absence of 3-MB-PP1. Error bars denote standard deviations from 3 independent experiments. (B) Images of DAPI- and calcofluor-stained htb1-K119R (JTB67-1) and cdk9as htb1-K119R (LV193) cells grown in the absence (top) or presence (middle) of 10 µM 3-MB-PP1 for 7 hr, or after inhibitor washout and return to growth (bottom). (C) Fluorescent images of DAPI/calcofluor-stained wild-type (JS78), cdk9-T212A (HD7-24), brl2Δ (JTB331), brl2Δ cdk9-T212A (JTB335), ubp8Δ (JTB297), ubp8Δ cdk9-T212A (JTB336), htb1-K119R (JTB67-1), cdk9-T212A htb1-K119R (LV252) cdk9-T212E htb1-K119R (LV256), and mcs6-S165A htb1-K119R (LV254) cells. (D) Quantification of abnormal septation patterns in strains of indicated genotypes (JTB62-1, JTB67-1, JTB325, JTB326, JTB331, JTB335, JTB377, JTB333 respectively). Error bars represent standard deviations from 2 independent experiments; at least 200 cells were counted in each. (E) Flocculation of indicated htb1-FLAG strains (JTB62-1, JTB67-1, JTB325, JTB326) was quantified as described in Materials and Methods. Error bars denote standard deviations from 2 independent experiments.
In the absence of inhibitory analogs, cdk9as htb1-K119R cells displayed hyperseptated and branched morphologies characteristic of H2Bub1-defective mutants. After treatment with 10 µM 3-MB-PP1 for 7 hr, however, cell morphology reverted to normal (Figure 5B). This effect was reversible; after washout of 3-MB-PP1 and return to drug-free medium, the “reverted” cdk9as htb1-K119R cells re-acquired an aberrant, hyperseptated morphology.
To confirm that impairment of Cdk9 function could suppress phenotypes caused by the absence of H2Bub1, we combined a cdk9-T212A mutation, which prevents activation of Cdk9 by a CAK but allows cell viability [50], with htb1-K119R or brl2Δ. As was the case with chemical inhibition of Cdk9, cdk9-T212A rescued morphologic phenotypes of H2Bub1-defective mutants (Figure 5C, 5D). Importantly, there was no suppression of htb1-K119R or brl2Δ by lsk1 loss-of-function alleles (Figure 5D and data not shown), establishing the specificity of the interaction between Cdk9 and H2Bub1. Similarly, neither cdk9-T212E nor mcs6-S165A could correct the aberrant morphologies of htb1-K119R cells (Figure 5C). Reversal of septation phenotypes by cdk9-T212A was accompanied by loss of the flocculation observed in htb1-K119R single mutants; ∼15% of wild-type or cdk9-T212A htb1-K119R double mutant cells settled out of liquid cultures after 1 hr, compared to ∼60% of cdk9+ htb1-K119R cells (Figure 5E), consistent with suppression of htb1-K119R by reduction of Cdk9 activity.
We next asked if aberrant morphologies due to htb1-K119R could be suppressed by mutations in the known Cdk9 substrates: Spt5 and Rpb1 [51]. By itself, spt5-T1A did not modify htb1-K119R phenotypes (Figure 6A), implicating another Cdk9 target (or targets) in the observed suppression. We attempted to verify this by combining cdk9as with spt5-T1A in an htb1-K119R background, to allow selective inhibition of Cdk9 in the absence of Spt5-Thr1 phosphorylation. Unexpectedly, the triple-mutant cells grown in the absence of inhibitory analogs had nearly normal morphology—a septation index close to that of the wild-type strain, <5% multiseptated cells and no chained cells (Figure 6B). In vitro, the activity of Cdk9as was reduced ∼3-fold relative to that of wild-type Cdk9, even in the absence of drugs (Figure S10). This is likely to be due to decreased affinity for ATP—a known consequence of the gatekeeper mutation in other AS kinases [72], [73]. The data suggest that loss of Spt5-Thr1 phosphorylation, combined with the partial reduction of Cdk9 activity, suppressed morphological abnormalities caused by loss of H2Bub1.
Figure 6. Cdk9 activity towards multiple substrates is required for abnormal morphologies of H2Bub1-deficient cells.

(A) Quantification of abnormal septation patterns in the indicated htb1-FLAG strains (JTB378, JTB351, JTB379, JTB353, JTB380, JTB355). Error bars represent standard deviations from 2 independent experiments; at least 200 cells were counted in each. (B) As in (A) for the indicated cdk9as strains (KL289, KL291, KL293). Cells were grown in the presence of either DMSO or 3-MB-PP1 as described in Figure 5B. (C) As in (A) for the indicated htb1-FLAG strains (MS260, MS256, MS261, MS257, MS272, MS259). (D) Flocculation of indicated strains (JS78, JTB67-1, JTB351, JTB353, KL291, KL293, MS259) was quantified as described in Materials and Methods. (E) Growth rates in liquid rich medium (YES) were measured for strains analyzed in (D).
These results reveal multiple, distinct roles for Spt5-Thr1 phosphorylation in mediating regulatory interactions between H2Bub1 and Cdk9. When Cdk9 activity levels are normal, Spt5-P serves, in combination with Set1, to promote H2Bub1 functions in maintaining normal cell morphology (Figure 3C, 3D). When H2Bub1 is absent, however, Spt5-P, together with at least one other Cdk9-dependent pathway, contributes to aberrant and excessive septation (Figure 6B). A similar distinction between specific elimination of Spt5-P and a general reduction in Cdk9 activity is apparent in a set1Δ background, in which spt5-T1A resulted in cell-morphology defects but cdk9-T212A had no effect (Figure 3D). Thus, the positive feedback between H2Bub1 and Cdk9, involving a single Cdk9 target (Spt5), is genetically distinguishable from the antagonism, which involves multiple targets (Spt5 and one or more others).
Cdk9 also contributes to phosphorylation of Ser2 and Ser5 of RNAPII CTD repeats [50], [51], [74]. An rpb1-S2A mutation that replaced Ser2 in all repeats with Ala in the context of a truncated but still functional CTD [53] failed to suppress htb1-K119R in either a cdk9+ or cdk9as background (Figure 6C). Combined loss of both Spt5-Thr1 and Rpb1-Ser2 in a cdk9+ background also did not suppress htb1-K119R septation phenotypes. Similarly, in a cdk9as background, spt5-T1A but not rpb1-S2A suppressed flocculation due to an htb1-K119R mutation (Figure 6D). Therefore, diminished Cdk9 activity must rescue phenotypes caused by H2Bub1 loss through reduced phosphorylation of Spt5-Thr1 and another target (or targets) besides Rpb1-Ser2.
The suppression of hyperseptation and flocculation was not simply a consequence of reduced growth rate, because hyperseptated, flocculating cdk9+ spt5-T1A htb1-K119R cells and suppressed cdk9as spt5-T1A htb1-K119R cells grew at similar rates in liquid culture. Moreover, the non-suppressed, cdk9as rpb1-S2A htb1-K119R strain grew more slowly than did the suppressed strain (Figure 6E). These data indicate that suppression of htb1-K119R is a specific consequence of reduced Cdk9 activity.
RNAPII redistribution due to htb1-K119R depends on Cdk9 activity
Our results thus far indicated that loss of H2Bub1 partially alleviated the cellular requirement for Cdk9 activity and that, conversely, selective impairment of Cdk9 function suppressed phenotypes of H2Bub1-defective mutants. This mutual suppression led us to hypothesize that P-TEFb and H2Bub1 might act antagonistically to control transcript elongation. We tested such an interaction by asking whether a reduction in Cdk9 activity could reverse the altered RNAPII distribution observed in an htb1-K119R mutant. We analyzed genome-wide RNAPII crosslinking by ChIP-chip in cdk9-T212A and cdk9-T212A htb1-K119R strains (Figure 7; Datasets S4 and S5). The cdk9-T212A mutation caused a global redistribution of RNAPII occupancy that was essentially opposite to that observed in the htb1-K119R strain: an increase in RNAPII density in the 5′ halves of genes, and a decrease toward the 3′ ends of genes that was particularly significant at highly transcribed loci (compare Figure 7C and Figure 1D). Impaired Cdk9 activity also led to reversal of the RNAPII 3′ shift caused by htb1-K119R (Figure 7F). Global RNAPII occupancy profiles in cdk9-T212A htb1+ and cdk9-T212A htb1-K119R mutants were nearly identical, suggesting that, when Cdk9 activity was reduced, H2Bub1 had little or no effect on RNAPII dynamics at the majority of genes (Figure 7A–7E). Loss of H2Bub1 reduced RNAPII occupancy within the 5′ halves of genes even in the context of reduced Cdk9 activity, however, suggesting that this effect is partially Cdk9-independent (Figure 7D, 7E). Nonetheless, full Cdk9 activity is required for the increase in RNAPII density at the 3′ ends of genes in the absence of H2Bub1 (Figure 7E). These data argue that P-TEFb and H2Bub1 antagonize one another to govern the distribution of elongating RNAPII within genes (Figure 8).
Figure 7. Opposing effects of H2Bub1 and Cdk9 activity on RNAPII distribution revealed by ChIP–chip.

(A) Average distribution of RNAPII at 540 S. pombe genes, as determined by ChIP-chip in a cdk9-T212A strain (JTB325). Genes were grouped according to total levels of RNAPII enrichment. (B) As in (A) for cdk9-T212A htb1-K119R (JTB326). (C) Average distributions of differences between cdk9-T212A (JTB325) and wild-type (JTB62-1) RNAPII enrichment grouped according to RNAPII enrichment in wild-type cells. (D) As in (C) for differences between cdk9-T212A htb1-K119R and wild-type RNAPII enrichment. (E) As in (C) for differences between cdk9-T212A htb1-K119R and cdk9-T212A RNAPII enrichment. (F) As in (C) for differences between cdk9-T212A htb1-K119R and htb1-K119R RNAPII enrichment. The keys below C-F illustrate the statistical significance of the differences for each group at 50 positions along the average gene. The rows of the key are color-coded according to the graph. Open squares denote p>0.01; light shading denotes 0.01>p>10exp-5; dark shading denotes p<10exp-5 (one-sample t-tests; μ0 = 0). Note that there is only light shading for the last row (corresponding to the blue curve).
Figure 8. Model depicting positive and negative interactions between P-TEFb and H2Bub1 during transcript elongation (see text for details).

Discussion
Here we combined chemical genetics and genomics to reveal reciprocal relationships between a CDK and a co-transcriptional histone modification in fission yeast. First, we showed interdependence of Cdk9-mediated Spt5 phosphorylation and H2Bub1—evidence for co-regulation governed by positive feedback. Importantly, we found that this feedback loop and the histone methyltransferase Set1 support independent functions of H2Bub1 in vivo. Second, we uncovered genetic interactions between mutations that compromise either Cdk9 activity or H2Bub1, which indicate opposing functions. To explain this paradox we propose a homeostatic mechanism that ensures optimal balance of P-TEFb and H2Bub1 functions during elongation.
A positive feedback loop linking Spt5-P and H2Bub1
A key Cdk9 substrate in the H2Bub1 pathway is Spt5, an ancient component of the transcription machinery that promotes RNAP processivity [75]–[77]. Eukaryotic Spt5 orthologs contain carboxyl-terminal sites phosphorylated by P-TEFb; S. pombe Spt5 has a contiguous array of nonapeptide repeats, which are phosphorylated at Thr1 by Cdk9 in vitro [52]. We showed that Cdk9 activity is necessary for phosphorylation of Spt5-Thr1 in vivo, consistent with genetic epistasis between Spt5-CTD truncation and selective Cdk9 inhibition [51]. Furthermore, reduction of Cdk9 activity or mutation of Spt5-Thr1 to Ala reduced H2Bub1. In budding yeast, Spt5 phosphorylation promotes H2Bub1 through recruitment of the PAF complex [37], [38], and Spt5 also influences H2Bub1 in mammalian cells [31], [78], suggesting a conserved signaling pathway.
Cdk9-dependent phosphorylation of the Rpb1 CTD has been implicated in formation of H2Bub1 in mammalian cells [30], [79]. The sites phosphorylated by Cdk9 within the CTD repeat—Ser2 and Ser5—are essential for cell viability, however, precluding a definitive proof of their involvement in this pathway. Because Ser2 is not essential in S. pombe [53], [68], we have been able to test its contribution to H2Bub1 more directly; the rpb1-S2A mutation had no effect on H2Bub1 levels (data not shown). Understanding the relative contributions of the Rpb1 CTD and other Cdk9 substrates in directing H2Bub1 will probably require better tools for study of metazoan P-TEFb and its targets in vivo.
The identification of Spt5-Thr1 phosphorylation as a specific and sensitive indicator of Cdk9 activity allowed us to uncover another, unexpected relationship between P-TEFb and H2Bub1 in vivo: Cdk9-mediated phosphorylation of Spt5 depends on H2Bub1. ChIP analysis suggested a possible explanation—decreased recruitment of Cdk9 to H2Bub1-deficient chromatin. In mammalian cells, basal association of P-TEFb with the HIV-1 promoter decreased upon depletion of the Bre1 ortholog RNF20, consistent with a conserved role for H2Bub1 in P-TEFb recruitment [80]. Based on our data and results in budding yeast, we propose that a positive feedback loop connects Cdk9 activity and H2Bub1 through the sequence: 1) phosphorylation of Spt5 by Cdk9; 2) PAF recruitment by Spt5-P; 3) recruitment and/or activation of the Rhp6 (S. pombe ortholog of Rad6)-Brl1/2 complex by PAF, to generate H2Bub1; and 4) Cdk9 recruitment, leading to reiteration of the cycle (Figure 8). Although the mechanism by which H2Bub1 facilitates Cdk9 recruitment remains to be determined, there is precedent in metazoans for P-TEFb association with modified histones [44], [45].
Two independent protein modification pathways downstream of H2Bub1
Our results indicate that H3K4me and Spt5-P are independently stimulated by H2Bub1 in vivo. Importantly, elimination of either modification alone did not cause septation phenotypes, but their combined ablation phenocopied the aberrant cell morphologies produced by H2Bub1 loss. Therefore, we have identified a molecular intermediate—Spt5-P—in the H3K4me-independent pathway downstream of H2Bub1 (Figure 8). A challenge for the future will be to ascertain how Set1 cooperates with Cdk9, acting through Spt5-Thr1, to regulate gene expression.
Cdk9-H2Bub1 antagonism during RNAPII elongation
The mutually reinforcing relationship between Cdk9 activity and H2Bub1 seemed to predict the two would act in concert during transcription. Instead, genetic analysis indicated they work in opposition: loss of H2Bub1 partially relieved the requirement for Cdk9 activity in vivo, and reduction in Cdk9 activity suppressed cell-morphology phenotypes of H2Bub1-deficient mutants. Simultaneous mutation of two known sites of Cdk9-dependent phosphorylation (Spt5-Thr1 and Rpb1-Ser2) did not suppress htb1-K119R, implicating at least one other Cdk9 target in opposing H2Bub1 function. Consistent with the genetic interaction, Cdk9 and H2Bub1 exerted inverse effects on global RNAPII distribution: whereas RNAPII density shifted toward the 3′ ends of genes in an htb1-K119R mutant, lowering Cdk9 activity produced the opposite effect in an htb1+ background, and prevented the 3′ shift in the double-mutant strain.
The antagonism we observe between Cdk9 activity and H2Bub1 is incomplete—loss of H2Bub1 did not entirely bypass the essential function of Cdk9, and led to altered RNAPII occupancy in the 5′ portions of genes even when Cdk9 activity was reduced. Conversely, although the presence of cdk9-T212A eliminated aberrant cell morphologies associated with htb1-K119R, the growth rate of double mutant cells was slower than that of either single mutant (data not shown). These results imply that other pathways can modulate the functions of Cdk9 and H2Bub1 in transcript elongation.
Our results strengthen the notion that P-TEFb activity is required to overcome repressive effects of histone modifications, first suggested by work in budding yeast. In S. cerevisiae, the instructive genetic interactions occurred between BUR1 and the H3K36 methyltransferase Set2; lethality of a bur1 null mutation was suppressed by deletion of SET2 [34], [48]. In fission yeast, however, set2 deletion did not suppress growth arrest due to inhibition of Cdk9as (L.V. and R.P.F., unpublished observations). Interestingly, bur1Δ was also weakly suppressed by deletion of RAD6 [48], implying that a regulatory interaction between P-TEFb and H2Bub1 might be conserved in budding yeast.
In mammalian cells, the Bre1/Brl2 ortholog RNF20 selectively represses transcription of genes implicated in tumorigenesis; its knockdown reduces H2Bub1, enhances the transcriptional response to growth factors and promotes cellular transformation [13]. These effects were recently shown to depend on the transcription elongation factor TFIIS [81]. This extends the parallel between mammals and fission yeast, by suggesting that H2Bub1 selectively modulates gene expression in both settings by restraining transcript elongation, albeit through different factors—TFIIS or P-TEFb, respectively. Given the established dependence of H2Bub1 on CDK9 activity in mammalian cells [30], [31], it will now be important to ask if metazoan P-TEFb function is also reciprocally influenced by H2Bub1.
Evidence for a chromatin-based checkpoint in RNAPII elongation
How can we reconcile biochemical evidence for mutual dependence of Cdk9 activity and H2Bub1 with genetic evidence for their antagonism? We propose a homeostatic mechanism whereby Cdk9-H2Bub1 interdependence ensures a balance between their opposing functions. In this scenario, Cdk9 promotes H2Bub1 to regulate its own effects on elongation (Figure 8). In an htb1-K119R mutant, the balance is disrupted, resulting in Cdk9-dependent accumulation of RNAPII in 3′ regions of genes. We suggest this pattern reflects unchecked elongation that is poorly responsive to mRNA processing signals, and may imply enhancement of the RNAPII pausing normally associated with mRNA 3′ end processing [82], [83]. H2Bub1 has been linked to various aspects of RNA processing in budding yeast and in mammalian cells, including splicing, 3′ end formation, and nuclear export [30], [57], [84]–[86]. In fission yeast, Cdk9 has been implicated in mRNA 5′-end formation, through its role in recruiting a capping enzyme to transcribed chromatin [87]. Still to be determined are the full range of H2Bub1-dependent functions that depend on P-TEFb activity and the mechanism(s) by which H2Bub1 and Cdk9 influence each other.
In cell division, faithful genome duplication and segregation are ensured by checkpoints—extrinsic signaling pathways that enforce dependency between intrinsically independent events [88]. Transcription and mRNA-processing could be intrinsically coupled through chromatin structure, because removal of nucleosomal barriers is likely to be a limiting step for elongation [89]. Here we have uncovered a role for a covalent chromatin modification, H2Bub1, in regulating elongation through an extrinsic pathway involving Cdk9 and Spt5. We propose a checkpoint-like function of H2Bub1, to set thresholds of Cdk9 activity and Spt5-P required for elongation and mRNA maturation, with the balance between opposing functions of Cdk9 and H2Bub1 maintained by virtue of their interdependence. The absence of H2Bub1 (and consequent decreases in Spt5-P and H3K4me) might hinder mRNA processing events normally coupled to Cdk9-mediated phosphorylations, without fully alleviating the Cdk9-dependence of elongation; “rescue” of these defects by reducing Cdk9 activity would be analogous to rescue of cell-cycle checkpoint mutants by drugs or mutations that slow another process.
Materials and Methods
S. pombe strains and media
Strains used in this study are listed in Table S2. Cells were grown in YE medium containing 250 mg/L each of adenine, leucine, histidine, and uracil (YES). Strains were constructed by standard genetic techniques [90].
Immunological methods
To detect Spt5 phosphorylation by immunoblotting, S. pombe whole-cell extracts were prepared as previously described [37], [91]. To detect chromatin modifications, cells were lysed in trichloroacetic acid, as described [92]. The Spt5-P antibody was raised against the peptide acetyl-NSGNK[pT]PAWNVGNK[pT]PAWNSC-amide injected into rabbits, and purified from whole serum after depletion with the same peptide in unphosphorylated form immobilized on resin, by adsorption to and elution from resin-bound acetyl-AWNSGSK[pT]PAWNSGSC-amide by 21st Century Biochemicals (Marlboro, MA). Other antibodies are listed in Text S1.
Chromatin immunoprecipitation and microarray analysis
ChIP was carried out as described previously [12], [93]. Amplification, labeling, and hybridization of control and immunoprecipitated DNA samples for ChIP-chip were carried out as described previously and normalized data are included as Datasets S1, S2, S3, S4, S5 [94]. Further details are included in Text S1. Sequences of primers used for qPCR analysis are available upon request.
Microscopy
S. pombe cells were fixed and stained with diamino-phenylindole (DAPI) and calcofluor as described previously [51]. Cells were viewed using a Leica DM5000b microscope and photographed with a CCD camera. Images were processed using Volocity software.
Chemical genetic methods
Cultures were treated with 3-MB-PP1 at indicated doses, or with vehicle (DMSO), for indicated times at 30°C prior to extract preparation. Dose-response to 3-MB-PP1 was determined as previously described [51]. To analyze effects of 3-MB-PP1 on cell morphologies, cells were grown at 30°C to OD600∼0.1 in YES and treated with 10 µM 3-MB-PP1 or DMSO for 7 hr, then fixed and stained for microscopy immediately or after return to growth in drug-free YES for 7 hr.
Flocculation assays
Assays were carried out as described previously [95].
Supporting Information
Microarray data for ChIP-chip performed with a H2Bub1 antibody (wild-type) and a RNAPII antibody (wild-type and htb1-K119R).
(ZIP)
Microarray data for ChIP-chip performed with a histone H3 antibody (wild-type).
(ZIP)
Microarray data for ChIP-chip performed with a histone H3 antibody (htb1-K119R).
(ZIP)
Microarray data for ChIP-chip performed with a RNAPII antibody (cdk9-T212A).
(ZIP)
BED file containing microarray data for ChIP-chip performed with a RNAPII antibody (cdk9-T212A htb1-K119R).
(ZIP)
Specificity of H2Bub1 antibody for ChIP in S. pombe. (A) H2Bub1 occupancy was measured by ChIP in wild-type (JTB62-1) and htb1-K119R (JTB67-1) strains and quantified at the inactive hsp16 + gene and constitutive act1 + gene. Values were normalized to H2B-FLAG occupancy measured in parallel to control for differences in nucleosome density. Positions of primer pairs used for qPCR are indicated in schematic at top. (B) Immunoblots of whole-cell extracts from untagged (JTB204), htb1-FLAG (JTB62-1), and htb1-K119R-FLAG (JTB67-1) strains using the indicated antibodies.
(PDF)
Loss of H2Bub1 does not significantly alter nucleosome density in S. pombe. (A) Average distribution of histone H3 at 540 S. pombe genes, as determined by ChIP-chip in a wild-type strain (JTB62-1). Genes were grouped according to total levels of RNAPII enrichment (see key at top). The grey box in the “average gene” representation at bottom denotes the gene coding region; 5′ and 3′ untranslated regions are denoted by thin black lines. The arrow denotes the transcription start site. (B) As in (A), determined in an htb1-K119R mutant strain (JTB67-1). Gene groupings were created using wild-type RNAPII enrichment values. (C) Average distributions of differences between mutant and wild-type histone H3 enrichment grouped according to RNAPII enrichment in wild-type cells. The key below the graph illustrates the statistical significance of the differences for each group at 50 positions along the average gene. The rows of the key are color-coded according to the graph. Open squares denote p>0.01; light shading denotes 0.01>p>10exp-5 (one-sample t-tests; μ0 = 0).
(PDF)
Chromatin association of Rhp6 and the PAF protein Rtf1 requires Cdk9 activity. (A) Rhp6-TAP occupancy was measured by ChIP in the indicated strains and quantified at the eng1+ gene by qPCR. Enrichment is plotted as percentage of the input signal for each primer pair. Positions of PCR primer pairs are indicated in the schematic at top. (B) Rtf1-TAP occupancy was measured by ChIP in the indicated strains and quantified at the act1+ gene by qPCR. Enrichment is plotted as percentage of the input signal for each primer pair. Positions of PCR primer pairs are indicated in the schematic at top. In all cases, treatment of the cdk9as strains was with either DMSO (-) or 20 µM 3-MB-PP1 for 3 hr prior to harvesting. Error bars denote standard deviations from three independent experiments.
(PDF)
Validation of Spt5 as an exclusive Cdk9 target using a phospho-specific Spt5 antibody. (A) Kinase reactions containing recombinant GST-Spt5 substrate and indicated combinations of purified, recombinant Cdk9/Pch1 and Csk1 were analyzed by immunoblotting with: affinity-purified antibody specific for phosphorylated Spt5 (Spt5-P); crude serum specific for wild-type Spt5 CTD repeats, regardless of phosphorylation state (Spt5-CTD; see Text S1); or anti-GST antibody (GST). (B,C) Immunoblots of whole-cell extracts from indicated spt5-myc strains after treatment with increasing concentrations of 3-MB-PP1. Antibodies are indicated at right. (D) Both phospho-isoform- and pan-specific total Spt5 antibodies are specific for the wild-type nonapeptide repeat sequence of the Spt5 CTD. Whole-cell extracts from the indicated strains were probed with affinity-purified antibody (top, “Spt5-P”) or crude serum (bottom, “Spt5”) from a rabbit immunized with an Spt5-CTD phosphopeptide (see Materials and Methods). Spt5 antibodies discriminate against nonapeptides with substitutions at the Thr1 position, but not against unphosphorylated, wild-type Spt5-CTD (see Figure 3A), illustrating why phospho-site mutant proteins cannot be used as controls for specificity of phosphospecific antibodies.
(PDF)
The FACT subunit Pob3 has functions distinct from those of H2Bub1. (A) Immunoblots of whole-cell extracts from wild-type (JTB204), brl2Δ (JTB331), cdk9-T212A (HD7-24), and pob3Δ (JTB281-1) strains using the indicated antibodies. (B) Quantification of abnormal septation in strains of indicated genotypes (JTB317 and JTB318, respectively). Error bars represent standard deviations from 2 independent experiments; at least 200 cells were counted in each.
(PDF)
Anti-Spt5-P specifically immunoprecipitates Spt5-P from wild-type and spt5-myc strains. (A) Spt5-P immunoprecipitation from whole-cell extracts. Extracts from indicated strains (bottom) were incubated with or without anti-Spt5-P (top), and immunoprecipitates were analyzed by immunoblotting with antibodies indicated at right. The cdk9as strain was treated prior to extract preparation with either DMSO or 5 µM 3-MB-PP1, as indicated at bottom, for 20 min. For each treatment we analyzed 10% of input extract (“input”), 10% of supernatant after precipitation (“sup”) and 50% of immunoprecipitated material (“IP”). Antibodies are indicated at right. (B) ChIP of Spt5-P and Spt5-myc at eng1 +. Patterns of Spt5-P occupancy on eng1+ are similar in wild-type (“WT,” top; JS78) and spt5-myc (middle; CS111) strains. Distribution of total Spt5 on eng1+ was measured in the spt5-myc strain (bottom). (C) Spt5-myc (top) and Spt5-P (middle) occupancies were measured by ChIP in a cdk9as spt5-myc strain and quantified at indicated positions within eng1 + by qPCR. Cells were treated prior to lysis with either DMSO or 5 µM 3-MB-PP1 for 20 min. Enrichment is plotted as percentage of input signal for each primer pair. The ratios of Spt5-P/Spt-myc at each position are plotted at bottom.
(PDF)
Impact of H2Bub1 on Spt5 phosphorylation and Cdk9 recruitment at nup189+ and SPB354.10+. (A,B) Spt5 phosphorylation was measured by ChIP using anti-Spt5-P in spt5-myc (MS265; black bars) and spt5-myc htb1-K119R (LV239; gray bars) strains and quantified at the indicated genes by qPCR. In all graphs of this figure the enrichment is plotted as percentage of the input signal for each primer pair, positions of PCR primer pairs within coding regions are indicated in the schematic at top, error bars denote standard deviations from 3 independent experiments and asterisks denote a significant difference between wild-type and mutant (“*” p<0.04, “**” p<0.02; unpaired t-test). (C,D) Spt5-myc occupancy was measured by ChIP as in C and D. (E,F) Spt5-P enrichment normalized to total Spt5-myc occupancy. (G,H) Cdk9-myc occupancy was measured by ChIP in cdk9-myc (MS264; black bars) and cdk9-myc htb1-K119R (KL259; gray bars) strains.
(PDF)
ChIP of Spt5-P, Spt5-myc, and Cdk9-myc at an intergenic sequence. (A) Occupancy of total Spt5, phospho-Spt5 and Cdk9 was measured by ChIP using anti-myc or anti-Spt5-P in wild-type strains (MS265 for Spt5-myc and Spt5-P IP; MS264 for Cdk9-myc IP), followed by qPCR using primers covering the 5′ end of eng1 (eng1-1) and an intergenic region as control (control). In all graphs of this figure the enrichment is plotted as percentage of the input signal for each primer pair and error bars denote standard deviations from 3 independent experiments. (B) Same experiment as in (A) in htb1-K119R strains (LV239 for Spt5-myc and Spt5-P IP; KL259 for Cdk9-myc IP).
(PDF)
Impact of H2Bub1 on RNAPII occupancy and mRNA expression at individual genes. (A-D) RNAPII occupancy was measured by ChIP in wild-type (JTB62-1; black bars) and htb1-K119R (JTB67-1; gray bars) strains and quantified at the indicated genes by qPCR. Enrichment is plotted as percentage of the input signal for each primer pair, and positions of PCR primer pairs within coding regions are indicated in the schematic at top of each graph. (E) Levels of mRNA from the indicated genes were quantified by qRT-PCR and normalized to act1+. Throughout this figure error bars denote standard deviations from 3 independent experiments and asterisks denote a significant difference between wild-type and mutant (“*” p<0.04, “**” p<0.02; unpaired t-test).
(PDF)
Kinase activity of Cdk9 and Cdk9as complexes purified from S. pombe. Lysates from pcm1-myc (CS145) or cdk9as pcm1-myc (CS165) strains were incubated either with anti-myc-bound protein G beads (α-myc-beads) to co-immunoprecipitate Cdk9 bound to Pcm1-myc or with protein G alone (mock). Input lysate amounts in each immunoprecipitation were varied as indicated. Immunoprecipitates were analyzed by immunoblotting (top panels) and for Rpb1-CTD kinase activity (middle panels). Phosphorylation signals were visualized by autoradiography and quantified with a phosphorimager (bottom).
(PDF)
RNAPII distributions at H2Bub1-stimulated genes in the htb1-K119R strain.
(DOC)
S. pombe strains used in this study.
(DOC)
Supplemental materials and methods.
(DOC)
Acknowledgments
We thank N. Minsky and M. Oren for providing the H2Bub1 antibody for ChIP-chip experiments. We thank A. Verreault, S. Drouin, and members of the Fisher and Tanny labs for helpful discussions. We acknowledge J. Vogel for use of microscopes.
Footnotes
The authors have declared that no competing interests exist.
This work was supported by fellowships to KML from NIH (www.nih.gov/) (T32 CA78207) and to LV from the Caja Madrid Foundation (www.fundacioncajamadrid.es/); by NIH grants GM076021 to RPF, EB001987 to KMS, and GM052470 to BS; by Canadian Institutes for Health Research (CIHR)(www.cihr-irsc.gc.ca/e) grant MOP-97890 to JCT; and by start-up funds awarded to JCT by the Sackler Program in Epigenetics. JCT is supported by a fellowship from FRQS (20274)(www.frsq.gouv.qc.ca). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Fuda NJ, Ardehali MB, Lis JT. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature. 2009;461:186–192. doi: 10.1038/nature08449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perales R, Bentley D. “Cotranscriptionality”: the transcription elongation complex as a nexus for nuclear transactions. Mol Cell. 2009;36:178–191. doi: 10.1016/j.molcel.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–599. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 4.Osley MA. H2B ubiquitylation: the end is in sight. Biochim Biophys Acta. 2004;1677:74–78. doi: 10.1016/j.bbaexp.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 5.Jaehning JA. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Biophys Acta. 2010;1799:379–388. doi: 10.1016/j.bbagrm.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bray S, Musisi H, Bienz M. Bre1 is required for Notch signaling and histone modification. Dev Cell. 2005;8:279–286. doi: 10.1016/j.devcel.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 7.Kim J, Hake SB, Roeder RG. The human homolog of yeast BRE1 functions as a transcriptional coactivator through direct activator interactions. Mol Cell. 2005;20:759–770. doi: 10.1016/j.molcel.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 8.Zhu B, Zheng Y, Pham AD, Mandal SS, Erdjument-Bromage H, et al. Monoubiquitination of human histone H2B: the factors involved and their roles in HOX gene regulation. Mol Cell. 2005;20:601–611. doi: 10.1016/j.molcel.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 9.Chandrasekharan MB, Huang F, Sun ZW. Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation. Epigenetics. 2010;5:460–468. doi: 10.4161/epi.5.6.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J, Guermah M, McGinty RK, Lee JS, Tang Z, et al. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell. 2009;137:459–471. doi: 10.1016/j.cell.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Racine A, Page V, Nagy S, Grabowski D, Tanny JC. Histone H2B ubiquitylation promotes activity of the intact Set1 histone methyltransferase complex in fission yeast. J Biol Chem. 2012;287:19040–19047. doi: 10.1074/jbc.M112.356253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanny JC, Erdjument-Bromage H, Tempst P, Allis CD. Ubiquitylation of histone H2B controls RNA polymerase II transcription elongation independently of histone H3 methylation. Genes Dev. 2007;21:835–847. doi: 10.1101/gad.1516207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shema E, Tirosh I, Aylon Y, Huang J, Ye C, et al. The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev. 2008;22:2664–2676. doi: 10.1101/gad.1703008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleming AB, Kao CF, Hillyer C, Pikaart M, Osley MA. H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol Cell. 2008;31:57–66. doi: 10.1016/j.molcel.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 15.Shukla A, Bhaumik SR. H2B-K123 ubiquitination stimulates RNAPII elongation independent of H3-K4 methylation. Biochem Biophys Res Commun. 2007;359:214–220. doi: 10.1016/j.bbrc.2007.05.105. [DOI] [PubMed] [Google Scholar]
- 16.Fuchs SM, Laribee RN, Strahl BD. Protein modifications in transcription elongation. Biochim Biophys Acta. 2009;1789:26–36. doi: 10.1016/j.bbagrm.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phatnani HP, Greenleaf AL. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006;20:2922–2936. doi: 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 18.Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, et al. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol Cell. 2009;34:387–393. doi: 10.1016/j.molcel.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, et al. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol. 2009;29:5455–5464. doi: 10.1128/MCB.00637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wada T, Takagi T, Yamaguchi Y, Watanabe D, Handa H. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. Embo J. 1998;17:7395–7403. doi: 10.1093/emboj/17.24.7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wada T, Takagi T, Yamaguchi Y, Ferdous A, Imai T, et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998;12:343–356. doi: 10.1101/gad.12.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komarnitsky P, Cho EJ, Buratowski S. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 2000;14:2452–2460. doi: 10.1101/gad.824700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schroeder SC, Schwer B, Shuman S, Bentley D. Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev. 2000;14:2435–2440. doi: 10.1101/gad.836300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, et al. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell. 2006;21:227–237. doi: 10.1016/j.molcel.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 25.Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, et al. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell. 1999;97:41–51. doi: 10.1016/s0092-8674(00)80713-8. [DOI] [PubMed] [Google Scholar]
- 26.Core LJ, Lis JT. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science. 2008;319:1791–1792. doi: 10.1126/science.1150843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glover-Cutter K, Kim S, Espinosa J, Bentley DL. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat Struct Mol Biol. 2008;15:71–78. doi: 10.1038/nsmb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilchrist DA, Dos Santos G, Fargo DC, Xie B, Gao Y, et al. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell. 2010;143:540–551. doi: 10.1016/j.cell.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Min IM, Waterfall JJ, Core LJ, Munroe RJ, Schimenti J, et al. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev. 2011;25:742–754. doi: 10.1101/gad.2005511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pirngruber J, Shchebet A, Schreiber L, Shema E, Minsky N, et al. CDK9 directs H2B monoubiquitination and controls replication-dependent histone mRNA 3′-end processing. EMBO Rep. 2009;10:894–900. doi: 10.1038/embor.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pirngruber J, Shchebet A, Johnsen SA. Insights into the function of the human P-TEFb component CDK9 in the regulation of chromatin modifications and co-transcriptional mRNA processing. Cell Cycle. 2009;8:3636–3642. doi: 10.4161/cc.8.22.9890. [DOI] [PubMed] [Google Scholar]
- 32.Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 33.Drouin S, Laramee L, Jacques PE, Forest A, Bergeron M, et al. DSIF and RNA polymerase II CTD phosphorylation coordinate the recruitment of Rpd3S to actively transcribed genes. PLoS Genet. 2010;6:e1001173. doi: 10.1371/journal.pgen.1001173. doi: 10.1371/journal.pgen.1001173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu Y, Sutton A, Sternglanz R, Prelich G. The BUR1 cyclin-dependent protein kinase is required for the normal pattern of histone methylation by SET2. Mol Cell Biol. 2006;26:3029–3038. doi: 10.1128/MCB.26.8.3029-3038.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laribee RN, Krogan NJ, Xiao T, Shibata Y, Hughes TR, et al. BUR kinase selectively regulates H3 K4 trimethylation and H2B ubiquitylation through recruitment of the PAF elongation complex. Curr Biol. 2005;15:1487–1493. doi: 10.1016/j.cub.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 36.Wood A, Schneider J, Dover J, Johnston M, Shilatifard A. The Bur1/Bur2 complex is required for histone H2B monoubiquitination by Rad6/Bre1 and histone methylation by COMPASS. Mol Cell. 2005;20:589–599. doi: 10.1016/j.molcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Warfield L, Zhang C, Luo J, Allen J, et al. Phosphorylation of the transcription elongation factor Spt5 by yeast Bur1 kinase stimulates recruitment of the PAF complex. Mol Cell Biol. 2009;29:4852–4863. doi: 10.1128/MCB.00609-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou K, Kuo WH, Fillingham J, Greenblatt JF. Control of transcriptional elongation and cotranscriptional histone modification by the yeast BUR kinase substrate Spt5. Proc Natl Acad Sci U S A. 2009;106:6956–6961. doi: 10.1073/pnas.0806302106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiu H, Hu C, Hinnebusch AG. Phosphorylation of the Pol II CTD by KIN28 enhances BUR1/BUR2 recruitment and Ser2 CTD phosphorylation near promoters. Mol Cell. 2009;33:752–762. doi: 10.1016/j.molcel.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao T, Hall H, Kizer KO, Shibata Y, Hall MC, et al. Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev. 2003;17:654–663. doi: 10.1101/gad.1055503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wood A, Shilatifard A. Bur1/Bur2 and the Ctk complex in yeast: the split personality of mammalian P-TEFb. Cell Cycle. 2006;5:1066–1068. doi: 10.4161/cc.5.10.2769. [DOI] [PubMed] [Google Scholar]
- 42.Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, et al. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;24:2303–2316. doi: 10.1101/gad.1968210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, et al. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011;25:2158–2172. doi: 10.1101/gad.16962311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, et al. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 45.Yang Z, Yik JH, Chen R, He N, Jang MK, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 46.Zippo A, Serafini R, Rocchigiani M, Pennacchini S, Krepelova A, et al. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138:1122–1136. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 47.Karam CS, Kellner WA, Takenaka N, Clemmons AW, Corces VG. 14-3-3 mediates histone cross-talk during transcription elongation in Drosophila. PLoS Genet. 2010;6:e1000975. doi: 10.1371/journal.pgen.1000975. doi: 10.1371/journal.pgen.1000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 49.Wyce A, Xiao T, Whelan KA, Kosman C, Walter W, et al. H2B ubiquitylation acts as a barrier to Ctk1 nucleosomal recruitment prior to removal by Ubp8 within a SAGA-related complex. Mol Cell. 2007;27:275–288. doi: 10.1016/j.molcel.2007.01.035. [DOI] [PubMed] [Google Scholar]
- 50.Pei Y, Du H, Singer J, St Amour C, Granitto S, et al. Cyclin-dependent kinase 9 (Cdk9) of fission yeast is activated by the CDK-activating kinase Csk1, overlaps functionally with the TFIIH-associated kinase Mcs6, and associates with the mRNA cap methyltransferase Pcm1 in vivo. Mol Cell Biol. 2006;26:777–788. doi: 10.1128/MCB.26.3.777-788.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Viladevall L, St Amour CV, Rosebrock A, Schneider S, Zhang C, et al. TFIIH and P-TEFb coordinate transcription with capping enzyme recruitment at specific genes in fission yeast. Mol Cell. 2009;33:738–751. doi: 10.1016/j.molcel.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pei Y, Shuman S. Characterization of the Schizosaccharomyces pombe Cdk9/Pch1 protein kinase: Spt5 phosphorylation, autophosphorylation, and mutational analysis. J Biol Chem. 2003;278:43346–43356. doi: 10.1074/jbc.M307319200. [DOI] [PubMed] [Google Scholar]
- 53.Schneider S, Pei Y, Shuman S, Schwer B. Separable functions of the fission yeast Spt5 carboxyl-terminal domain (CTD) in capping enzyme binding and transcription elongation overlap with those of the RNA polymerase II CTD. Mol Cell Biol. 2010;30:2353–2364. doi: 10.1128/MCB.00116-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Minsky N, Shema E, Field Y, Schuster M, Segal E, et al. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat Cell Biol. 2008;10:483–488. doi: 10.1038/ncb1712. [DOI] [PubMed] [Google Scholar]
- 55.Schulze JM, Jackson J, Nakanishi S, Gardner JM, Hentrich T, et al. Linking cell cycle to histone modifications: SBF and H2B monoubiquitination machinery and cell-cycle regulation of H3K79 dimethylation. Mol Cell. 2009;35:626–641. doi: 10.1016/j.molcel.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jung I, Kim SK, Kim M, Han YM, Kim YS, et al. H2B monoubiquitylation is a 5′-enriched active transcription mark and correlates with exon-intron structure in human cells. Genome Res. 2012;22:1026–1035. doi: 10.1101/gr.120634.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, et al. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat Chem Biol. 2011;7:113–119. doi: 10.1038/nchembio.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Batta K, Zhang Z, Yen K, Goffman DB, Pugh BF. Genome-wide function of H2B ubiquitylation in promoter and genic regions. Genes Dev. 2011;25:2254–2265. doi: 10.1101/gad.177238.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coudreuse D, van Bakel H, Dewez M, Soutourina J, Parnell T, et al. A gene-specific requirement of RNA polymerase II CTD phosphorylation for sexual differentiation in S. pombe. Curr Biol. 2010;20:1053–1064. doi: 10.1016/j.cub.2010.04.054. [DOI] [PubMed] [Google Scholar]
- 61.Lee KM, Miklos I, Du H, Watt S, Szilagyi Z, et al. Impairment of the TFIIH-associated CDK-activating kinase selectively affects cell cycle-regulated gene expression in fission yeast. Mol Biol Cell. 2005;16:2734–2745. doi: 10.1091/mbc.E04-11-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karagiannis J, Bimbo A, Rajagopalan S, Liu J, Balasubramanian MK. The nuclear kinase Lsk1p positively regulates the septation initiation network and promotes the successful completion of cytokinesis in response to perturbation of the actomyosin ring in Schizosaccharomyces pombe. Mol Biol Cell. 2005;16:358–371. doi: 10.1091/mbc.E04-06-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knight ZA, Shokat KM. Chemical genetics: where genetics and pharmacology meet. Cell. 2007;128:425–430. doi: 10.1016/j.cell.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 64.Kim J, Roeder RG. Direct Bre1-Paf1 Complex Interactions and RING Finger-independent Bre1-Rad6 Interactions Mediate Histone H2B Ubiquitylation in Yeast. J Biol Chem. 2009;284:20582–20592. doi: 10.1074/jbc.M109.017442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gerber HB, Pikman Y, Fisher RP. The CDK-activating kinase (CAK) Csk1 is required for normal levels of homologous recombination and resistance to DNA damage in fission yeast. PLoS ONE. 2008;3:e1492. doi: 10.1371/journal.pone.0001492. doi: 10.1371/journal.pone.0001492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hermand D, Pihlak A, Westerling T, Damagnez V, Vandenhaute J, et al. Fission yeast Csk1 is a CAK-activating kinase (CAKAK). Embo J. 1998;17:7230–7238. doi: 10.1093/emboj/17.24.7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee KM, Saiz JE, Barton WA, Fisher RP. Cdc2 activation in fission yeast depends on Mcs6 and Csk1, two partially redundant Cdk-activating kinases CAKs). Curr Biol. 1999;9:441–444. doi: 10.1016/s0960-9822(99)80194-8. [DOI] [PubMed] [Google Scholar]
- 68.Karagiannis J, Balasubramanian MK. A cyclin-dependent kinase that promotes cytokinesis through modulating phosphorylation of the carboxy terminal domain of the RNA Pol II Rpb1p sub-unit. PLoS ONE. 2007;2:e433. doi: 10.1371/journal.pone.0000433. doi: 10.1371/journal.pone.0000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weake VM, Workman JL. Histone ubiquitination: triggering gene activity. Mol Cell. 2008;29:653–663. doi: 10.1016/j.molcel.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 70.Pavri R, Zhu B, Li G, Trojer P, Mandal S, et al. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell. 2006;125:703–717. doi: 10.1016/j.cell.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 71.Kari V, Shchebet A, Neumann H, Johnsen SA. The H2B ubiquitin ligase RNF40 cooperates with SUPT16H to induce dynamic changes in chromatin structure during DNA double-strand break repair. Cell Cycle. 2011;10:3495–3504. doi: 10.4161/cc.10.20.17769. [DOI] [PubMed] [Google Scholar]
- 72.Kraybill BC, Elkin LL, Blethrow JD, Morgan DO, Shokat KM. Inhibitor scaffolds as new allele specific kinase substrates. J Am Chem Soc. 2002;124:12118–12128. doi: 10.1021/ja0264798. [DOI] [PubMed] [Google Scholar]
- 73.Larochelle S, Batliner J, Gamble MJ, Barboza NM, Kraybill BC, et al. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat Struct Mol Biol. 2006;13:55–62. doi: 10.1038/nsmb1028. [DOI] [PubMed] [Google Scholar]
- 74.Guiguen A, Soutourina J, Dewez M, Tafforeau L, Dieu M, et al. Recruitment of P-TEFb (Cdk9-Pch1) to chromatin by the cap-methyl transferase Pcm1 in fission yeast. EMBO J. 2007;26:1552–1559. doi: 10.1038/sj.emboj.7601627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grohmann D, Nagy J, Chakraborty A, Klose D, Fielden D, et al. The initiation factor tfe and the elongation factor Spt4/5 compete for the RNAP clamp during transcription initiation and elongation. Mol Cell. 2011;43:263–274. doi: 10.1016/j.molcel.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martinez-Rucobo FW, Sainsbury S, Cheung AC, Cramer P. Architecture of the RNA polymerase-Spt4/5 complex and basis of universal transcription processivity. Embo J. 2011;30:1302–1310. doi: 10.1038/emboj.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klein BJ, Bose D, Baker KJ, Yusoff ZM, Zhang X, et al. RNA polymerase and transcription elongation factor Spt4/5 complex structure. Proc Natl Acad Sci U S A. 2011;108:546–550. doi: 10.1073/pnas.1013828108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen Y, Yamaguchi Y, Tsugeno Y, Yamamoto J, Yamada T, et al. DSIF, the Paf1 complex, and Tat-SF1 have nonredundant, cooperative roles in RNA polymerase II elongation. Genes Dev. 2009;23:2765–2777. doi: 10.1101/gad.1834709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang F, Yu X. WAC, a functional partner of RNF20/40, regulates histone H2B ubiquitination and gene transcription. Mol Cell. 2011;41:384–397. doi: 10.1016/j.molcel.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bres V, Yoshida T, Pickle L, Jones KA. SKIP interacts with c-Myc and Menin to promote HIV-1 Tat transactivation. Mol Cell. 2009;36:75–87. doi: 10.1016/j.molcel.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shema E, Kim J, Roeder RG, Oren M. RNF20 Inhibits TFIIS-Facilitated Transcriptional Elongation to Suppress Pro-oncogenic Gene Expression. Mol Cell. 2011;42:477–488. doi: 10.1016/j.molcel.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Birse CE, Lee BA, Hansen K, Proudfoot NJ. Transcriptional termination signals for RNA polymerase II in fission yeast. EMBO J. 1997;16:3633–3643. doi: 10.1093/emboj/16.12.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boireau S, Maiuri P, Basyuk E, de la Mata M, Knezevich A, et al. The transcriptional cycle of HIV-1 in real-time and live cells. J Cell Biol. 2007;179:291–304. doi: 10.1083/jcb.200706018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shieh GS, Pan CH, Wu JH, Sun YJ, Wang CC, et al. H2B ubiquitylation is part of chromatin architecture that marks exon-intron structure in budding yeast. BMC Genomics. 2011;12:627. doi: 10.1186/1471-2164-12-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vitaliano-Prunier A, Babour A, Herissant L, Apponi L, Margaritis T, et al. H2B ubiquitylation controls the formation of export-competent mRNP. Mol Cell. 2012;45:132–139. doi: 10.1016/j.molcel.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tomson BN, Davis CP, Warner MH, Arndt KM. Identification of a role for histone H2B ubiquitylation in noncoding RNA 3′-end formation through mutational analysis of Rtf1 in Saccharomyces cerevisiae. Genetics. 2011;188:273–289. doi: 10.1534/genetics.111.128645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.St Amour CV, Sansó M, Bösken CA, Lee KM, Larochelle S, et al. Separate domains of fission yeast Cdk9 (P-TEFb) are required for capping enzyme recruitment and primed (Ser7-phosphorylated) Rpb1 carboxyl-terminal domain substrate recognition. Mol Cell Biol. 2012;32:2372–2383. doi: 10.1128/MCB.06657-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 89.Workman JL. Nucleosome displacement in transcription. Genes Dev. 2006;20:2009–2017. doi: 10.1101/gad.1435706. [DOI] [PubMed] [Google Scholar]
- 90.Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- 91.Sansó M, Gogol M, Ayté J, Seidel C, Hidalgo E. Transcription factors Pcr1 and Atf1 have distinct roles in stress- and Sty1-dependent gene regulation. Eukaryot Cell. 2008;7:826–835. doi: 10.1128/EC.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kao CF, Osley MA. In vivo assays to study histone ubiquitylation. Methods. 2003;31:59–66. doi: 10.1016/s1046-2023(03)00088-4. [DOI] [PubMed] [Google Scholar]
- 93.Sansó M, Vargas-Perez I, Quintales L, Antequera F, Ayté J, et al. Gcn5 facilitates Pol II progression, rather than recruitment to nucleosome-depleted stress promoters, in Schizosaccharomyces pombe. Nucleic Acids Res. 2011;39:6369–6379. doi: 10.1093/nar/gkr255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell. 2007;27:393–405. doi: 10.1016/j.molcel.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 95.Samuelsen CO, Baraznenok V, Khorosjutina O, Spahr H, Kieselbach T, et al. TRAP230/ARC240 and TRAP240/ARC250 Mediator subunits are functionally conserved through evolution. Proc Natl Acad Sci U S A. 2003;100:6422–6427. doi: 10.1073/pnas.1030497100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Microarray data for ChIP-chip performed with a H2Bub1 antibody (wild-type) and a RNAPII antibody (wild-type and htb1-K119R).
(ZIP)
Microarray data for ChIP-chip performed with a histone H3 antibody (wild-type).
(ZIP)
Microarray data for ChIP-chip performed with a histone H3 antibody (htb1-K119R).
(ZIP)
Microarray data for ChIP-chip performed with a RNAPII antibody (cdk9-T212A).
(ZIP)
BED file containing microarray data for ChIP-chip performed with a RNAPII antibody (cdk9-T212A htb1-K119R).
(ZIP)
Specificity of H2Bub1 antibody for ChIP in S. pombe. (A) H2Bub1 occupancy was measured by ChIP in wild-type (JTB62-1) and htb1-K119R (JTB67-1) strains and quantified at the inactive hsp16 + gene and constitutive act1 + gene. Values were normalized to H2B-FLAG occupancy measured in parallel to control for differences in nucleosome density. Positions of primer pairs used for qPCR are indicated in schematic at top. (B) Immunoblots of whole-cell extracts from untagged (JTB204), htb1-FLAG (JTB62-1), and htb1-K119R-FLAG (JTB67-1) strains using the indicated antibodies.
(PDF)
Loss of H2Bub1 does not significantly alter nucleosome density in S. pombe. (A) Average distribution of histone H3 at 540 S. pombe genes, as determined by ChIP-chip in a wild-type strain (JTB62-1). Genes were grouped according to total levels of RNAPII enrichment (see key at top). The grey box in the “average gene” representation at bottom denotes the gene coding region; 5′ and 3′ untranslated regions are denoted by thin black lines. The arrow denotes the transcription start site. (B) As in (A), determined in an htb1-K119R mutant strain (JTB67-1). Gene groupings were created using wild-type RNAPII enrichment values. (C) Average distributions of differences between mutant and wild-type histone H3 enrichment grouped according to RNAPII enrichment in wild-type cells. The key below the graph illustrates the statistical significance of the differences for each group at 50 positions along the average gene. The rows of the key are color-coded according to the graph. Open squares denote p>0.01; light shading denotes 0.01>p>10exp-5 (one-sample t-tests; μ0 = 0).
(PDF)
Chromatin association of Rhp6 and the PAF protein Rtf1 requires Cdk9 activity. (A) Rhp6-TAP occupancy was measured by ChIP in the indicated strains and quantified at the eng1+ gene by qPCR. Enrichment is plotted as percentage of the input signal for each primer pair. Positions of PCR primer pairs are indicated in the schematic at top. (B) Rtf1-TAP occupancy was measured by ChIP in the indicated strains and quantified at the act1+ gene by qPCR. Enrichment is plotted as percentage of the input signal for each primer pair. Positions of PCR primer pairs are indicated in the schematic at top. In all cases, treatment of the cdk9as strains was with either DMSO (-) or 20 µM 3-MB-PP1 for 3 hr prior to harvesting. Error bars denote standard deviations from three independent experiments.
(PDF)
Validation of Spt5 as an exclusive Cdk9 target using a phospho-specific Spt5 antibody. (A) Kinase reactions containing recombinant GST-Spt5 substrate and indicated combinations of purified, recombinant Cdk9/Pch1 and Csk1 were analyzed by immunoblotting with: affinity-purified antibody specific for phosphorylated Spt5 (Spt5-P); crude serum specific for wild-type Spt5 CTD repeats, regardless of phosphorylation state (Spt5-CTD; see Text S1); or anti-GST antibody (GST). (B,C) Immunoblots of whole-cell extracts from indicated spt5-myc strains after treatment with increasing concentrations of 3-MB-PP1. Antibodies are indicated at right. (D) Both phospho-isoform- and pan-specific total Spt5 antibodies are specific for the wild-type nonapeptide repeat sequence of the Spt5 CTD. Whole-cell extracts from the indicated strains were probed with affinity-purified antibody (top, “Spt5-P”) or crude serum (bottom, “Spt5”) from a rabbit immunized with an Spt5-CTD phosphopeptide (see Materials and Methods). Spt5 antibodies discriminate against nonapeptides with substitutions at the Thr1 position, but not against unphosphorylated, wild-type Spt5-CTD (see Figure 3A), illustrating why phospho-site mutant proteins cannot be used as controls for specificity of phosphospecific antibodies.
(PDF)
The FACT subunit Pob3 has functions distinct from those of H2Bub1. (A) Immunoblots of whole-cell extracts from wild-type (JTB204), brl2Δ (JTB331), cdk9-T212A (HD7-24), and pob3Δ (JTB281-1) strains using the indicated antibodies. (B) Quantification of abnormal septation in strains of indicated genotypes (JTB317 and JTB318, respectively). Error bars represent standard deviations from 2 independent experiments; at least 200 cells were counted in each.
(PDF)
Anti-Spt5-P specifically immunoprecipitates Spt5-P from wild-type and spt5-myc strains. (A) Spt5-P immunoprecipitation from whole-cell extracts. Extracts from indicated strains (bottom) were incubated with or without anti-Spt5-P (top), and immunoprecipitates were analyzed by immunoblotting with antibodies indicated at right. The cdk9as strain was treated prior to extract preparation with either DMSO or 5 µM 3-MB-PP1, as indicated at bottom, for 20 min. For each treatment we analyzed 10% of input extract (“input”), 10% of supernatant after precipitation (“sup”) and 50% of immunoprecipitated material (“IP”). Antibodies are indicated at right. (B) ChIP of Spt5-P and Spt5-myc at eng1 +. Patterns of Spt5-P occupancy on eng1+ are similar in wild-type (“WT,” top; JS78) and spt5-myc (middle; CS111) strains. Distribution of total Spt5 on eng1+ was measured in the spt5-myc strain (bottom). (C) Spt5-myc (top) and Spt5-P (middle) occupancies were measured by ChIP in a cdk9as spt5-myc strain and quantified at indicated positions within eng1 + by qPCR. Cells were treated prior to lysis with either DMSO or 5 µM 3-MB-PP1 for 20 min. Enrichment is plotted as percentage of input signal for each primer pair. The ratios of Spt5-P/Spt-myc at each position are plotted at bottom.
(PDF)
Impact of H2Bub1 on Spt5 phosphorylation and Cdk9 recruitment at nup189+ and SPB354.10+. (A,B) Spt5 phosphorylation was measured by ChIP using anti-Spt5-P in spt5-myc (MS265; black bars) and spt5-myc htb1-K119R (LV239; gray bars) strains and quantified at the indicated genes by qPCR. In all graphs of this figure the enrichment is plotted as percentage of the input signal for each primer pair, positions of PCR primer pairs within coding regions are indicated in the schematic at top, error bars denote standard deviations from 3 independent experiments and asterisks denote a significant difference between wild-type and mutant (“*” p<0.04, “**” p<0.02; unpaired t-test). (C,D) Spt5-myc occupancy was measured by ChIP as in C and D. (E,F) Spt5-P enrichment normalized to total Spt5-myc occupancy. (G,H) Cdk9-myc occupancy was measured by ChIP in cdk9-myc (MS264; black bars) and cdk9-myc htb1-K119R (KL259; gray bars) strains.
(PDF)
ChIP of Spt5-P, Spt5-myc, and Cdk9-myc at an intergenic sequence. (A) Occupancy of total Spt5, phospho-Spt5 and Cdk9 was measured by ChIP using anti-myc or anti-Spt5-P in wild-type strains (MS265 for Spt5-myc and Spt5-P IP; MS264 for Cdk9-myc IP), followed by qPCR using primers covering the 5′ end of eng1 (eng1-1) and an intergenic region as control (control). In all graphs of this figure the enrichment is plotted as percentage of the input signal for each primer pair and error bars denote standard deviations from 3 independent experiments. (B) Same experiment as in (A) in htb1-K119R strains (LV239 for Spt5-myc and Spt5-P IP; KL259 for Cdk9-myc IP).
(PDF)
Impact of H2Bub1 on RNAPII occupancy and mRNA expression at individual genes. (A-D) RNAPII occupancy was measured by ChIP in wild-type (JTB62-1; black bars) and htb1-K119R (JTB67-1; gray bars) strains and quantified at the indicated genes by qPCR. Enrichment is plotted as percentage of the input signal for each primer pair, and positions of PCR primer pairs within coding regions are indicated in the schematic at top of each graph. (E) Levels of mRNA from the indicated genes were quantified by qRT-PCR and normalized to act1+. Throughout this figure error bars denote standard deviations from 3 independent experiments and asterisks denote a significant difference between wild-type and mutant (“*” p<0.04, “**” p<0.02; unpaired t-test).
(PDF)
Kinase activity of Cdk9 and Cdk9as complexes purified from S. pombe. Lysates from pcm1-myc (CS145) or cdk9as pcm1-myc (CS165) strains were incubated either with anti-myc-bound protein G beads (α-myc-beads) to co-immunoprecipitate Cdk9 bound to Pcm1-myc or with protein G alone (mock). Input lysate amounts in each immunoprecipitation were varied as indicated. Immunoprecipitates were analyzed by immunoblotting (top panels) and for Rpb1-CTD kinase activity (middle panels). Phosphorylation signals were visualized by autoradiography and quantified with a phosphorimager (bottom).
(PDF)
RNAPII distributions at H2Bub1-stimulated genes in the htb1-K119R strain.
(DOC)
S. pombe strains used in this study.
(DOC)
Supplemental materials and methods.
(DOC)