

Background: Glucocorticoid receptor (GR) translational isoforms with different lengths in the intrinsically disordered (ID) N-terminal domain (NTD) have different activities.
Results: The folded conformations of the NTDs are thermodynamically globular-like proteins with stabilities that correlate with transcriptional activity.
Conclusion: Stability of the ID NTD is a functional regulator of GR.
Significance: Activity modulation through ID stability tuning is a new regulatory paradigm.
Keywords: Allosteric Regulation, Intrinsically Disordered Proteins, Protein Folding, Signaling, Steroid Hormone Receptor, Ensemble, Thermodynamic Coupling
Abstract
Intrinsically disordered (ID) sequence segments are abundant in cell signaling proteins and transcription factors. Because ID regions commonly fold as part of their intracellular function, it is crucial to understand the folded states as well as the transitions between the unfolded and folded states. Specifically, it is important to determine 1) whether large ID segments contain different thermodynamically and/or functionally distinct regions, 2) whether any ID regions fold upon activation, 3) the degree of coupling between the different ID regions, and 4) whether the stability of ID domains is a determinant of function. In this study, we thermodynamically characterized the full-length ID N-terminal domain (NTD) of human glucocorticoid receptor (GR) and two of its naturally occurring translational isoforms. The protective osmolyte trimethylamine N-oxide (TMAO) was used to induce folding transitions. Each of the three NTD isoforms was found to undergo a cooperative folding transition that is thermodynamically indistinguishable (based on m-values) from that of a globular protein of similar size. The extrapolated stabilities for the NTD isoforms showed clear correlation with the known activities of their corresponding GR translational isoforms. The data reveal that the full-length NTD can be viewed as having at least two thermodynamically coupled regions, a functional region, which is indispensable for GR transcriptional activity, and a regulatory region, the length of which serves to regulate the stability of NTD and thus the activity of GR. These results suggest a new functional paradigm whereby steroid hormone receptors in particular and ID proteins in general can have multiple functionally distinct ID regions that interact and modulate the stability of important functional sites.
Introduction
The classic view of protein structure-function relationships has been challenged by the increasing number of proteins, particularly cell signaling proteins and transcription factors, found to contain intrinsically disordered (ID)2 regions (1–4). Many of these ID regions have binding sites for protein partners, and folding coupled with binding often occurs when these regions encounter such partners (1, 4–7). Thus, in some cases, the conditionally folded states (which may be transient) are presumed to be among the functional states of ID proteins. Mutation, truncation, and translocation of ID regions have been implicated in a variety of diseases (5, 8, 9). However, the mechanisms by which these ID regions regulate protein functions are largely unknown. Osmolyte-induced folding of some ID proteins has been reported, and the induced folded states were determined to be functionally relevant (10, 11). These results pave the way for more quantitative studies on the role of folding and stability of these ID regions in mediating function. However, the folding cooperativity is not well characterized (12) nor is the degree to which folded states of ID proteins resemble their folded globular counterparts. Although it is well known that amino acid composition of ID proteins differs significantly from that of globular proteins (3, 4), the level at which these differences are manifested is not known. Are the folded states of ID sequence qualitatively different from globular proteins, or can the folded, native states of ID proteins be understood in terms of the same thermodynamic principles that describe globular proteins?
To address these questions, we carried out thermodynamic characterization of the ID N-terminal domains (NTDs) of three human glucocorticoid receptor (GR/Nr3c1) translational isoforms. GR is a hormone-dependent nuclear transcription factor in the steroid hormone receptor family that contains three modular domains: the ID NTD (GR 1–420), the DNA binding domain (GR 421–486), and the ligand binding domain (GR 528–777). The ID NTDs for steroid hormone receptors are extremely important for transcription regulation, serving as hubs to recruit co-regulators that form the final transcription complex (13–15). Within the human GR NTD, mutational mapping has identified a subregion, termed the activation function-1 (AF1) region, which comprises residues 77–262 and is essential for the full transcriptional activity of the receptor (16). AF1 regions are found in all steroid hormone receptors but differ widely in size and primary sequence (15). Although constructs that lack NTD residues outside of the AF1 region are able to stimulate transcription in the context of simplified promoter-reporter systems (17), several pathogenic mutations have been mapped to these non-AF1 regions (18), indicating that sequences outside AF1 do indeed play a role in maintaining or tuning function. This has also been demonstrated recently with the identification of several human GR NTD translational isoforms differing only in the lengths of their ID NTDs (19) (see Fig. 1). These isoforms have varying activities, different tissue distributions, unique gene regulation sets and have been shown to be derived from a single GR mRNA through well known translational regulatory mechanisms (19). With the exception of the GR isoforms that truncate the entire AF1 region and thus presumably ablate co-activator binding, all other isoforms are active and possess different potencies in transcription regulation (19). Especially intriguing is the observation (discussed below) that the deletion of as few as eight amino acids results in an almost 2-fold increase in activity (19).
FIGURE 1.

Schematic representation of the GR NTD sequence showing the positions of different GR translational isoform start sites (19). The N-terminal domains of the A (starting at position 1), C2 (starting at position 90), and C3 (starting at position 98) isoforms correspond to the constructs GR A-NTD, GR C2-NTD and GR C3-NTD, respectively (shown within squares). The transactivation domain AF1 (GR 77–262) is shown in gray.
Here we investigate the thermodynamic basis for these results by examining the ID NTDs of three representative human GR translational isoforms: GR A-NTD, GR C2-NTD, and GR C3-NTD, which correspond to GR 1–420, GR 90–420, and GR 98–420 in the full-length GR, respectively (Fig. 1). To determine the stability of the folded conformations of these ID proteins, the naturally occurring protective osmolyte trimethylamine N-oxide (TMAO) was used to induce their unfolded to folded transitions. Protective organic osmolytes are small molecules in cells that function to stabilize and protect intracellular proteins against commonly occurring denaturing environmental stresses (20, 21). TMAO has been demonstrated to force thermodynamically unstable proteins to fold and regain high functional activities (10, 22, 23) and thus is an ideal compound to study folding/unfolding reactions.
In summary, for each isoform, we found a TMAO-induced cooperative folding transition to an apparent globular protein-like folded conformation. Our results demonstrate that the GR NTD contains at least two thermodynamically coupled but functionally distinct regions, one regulatory (R) and the other functional (F). The use of alternative translation start sites in cells to vary the length of the R region results in GR translational isoforms of varying stability and activity. We show that this activity is correlated to the stability of each isoform, i.e. to its inherent propensity to fold.
EXPERIMENTAL PROCEDURES
Protein Expression, Purification, and Storage
Plasmids to express GR A-NTD, GR C2-NTD, GR C3-NTD, and GR 1–97 with 10 tandem histidines tagged on both the N and C termini were optimized for Escherichia coli expression, synthesized by DNA 2.0 (Menlo Park, CA), and inserted into the pJ411 bacterial expression vector under T7 promoter control. BL21(DE3)pLysS competent cells (Novagen) transformed with expression plasmids were plated on LB plates with 30 μg/ml kanamycin and incubated at 37 °C for 24 h. Single colonies were picked, inoculated into 50 ml of LB medium with 30 μg/ml kanamycin, and grown overnight at 30 °C with shaking at 250 rpm. The following morning, 10 ml of each overnight culture was transferred to 500 ml of LB medium and grown at 37 °C to an A600 of 0.8. The temperature was adjusted to 15 °C, and after 1 h, the culture was induced by 1 mm isopropyl β-d-thiogalactopyranoside (American Bioanalytical). After 18 h at 15 °C with shaking at 250 rpm, the E. coli cells were collected by centrifugation for 15 min at 5000 rpm at 4 °C, and the pellets were washed with cold PBS (137 mm NaCl, 7 mm Na2HPO4, 3 mm KCl, 1.4 mm KH2PO4) and recentrifuged. To the cell pellets from a 2-liter culture was added 50 ml of denaturing lysis buffer (6 m guanidine hydrochloride, 100 mm monosodium phosphate, 10 mm Tris, 20 mm imidazole, pH 8.0) plus one Complete EDTA-free protease inhibitor mixture tablet (Roche Applied Science). Cells were lysed by flash freezing twice in a dry ice and ethanol mixture for 20 min and then thawed for 10 min in a 42 °C water bath. The cell lysate was centrifuged at 30,000 × g at 4 °C for 1 h. The supernatant from 4 liters of induced cells was loaded by gravity flow onto an FPLC column packed with 20 ml of nickel-nitrilotriacetic acid Superflow resin (Qiagen) pre-equilibrated with the denaturing lysis buffer. After thorough washing the column with the denaturing lysis buffer at a flow rate of 2 ml/min, protein was renatured on the column by flowing through native lysis buffer (100 mm monosodium phosphate, 10 mm Tris, 20 mm imidazole, pH 8.0). Afterward, native lysis buffer containing 200 mm imidazole was applied to the column at 2 ml/min to wash out contaminants and degradation products with His tag only on one terminus. Finally, target protein was eluted from the column by native lysis buffer containing 500 mm imidazole. Protein purity was checked by SDS-PAGE. The purified target protein was dialyzed against storage buffer (10 mm HEPES, 80 mm NaCl, 1 mm EDTA, 10% glycerol, pH 7.6). Protein concentration was then determined by A280 according to the Edelhoch method (24). Each 500-μl protein aliquot in a 1.5-ml Protein Lobind Tube (Eppendorf) was then flash frozen in an ethanol-dry ice bath and stored at −80 °C.
Preparation of TMAO Buffers
Trimethylamine N-oxide dihydrate (98% pure; Acros Organics) was dissolved in 100 mm Tris, 200 mm NaCl, 50 mm arginine buffer to make the 0, 0.5, 1, 1.5, 2, and 2.5 m TMAO buffers. The pH was adjusted to 7.4 for each buffer separately. To absorb any impurities, activated carbon (12–20 mesh; Sigma-Aldrich) was added to each buffer solution and stirred for 30 min while protected from light. The buffer was then filtered (0.22-μm filter; Millipore), divided into aliquot fractions, and stored at −80 °C. TMAO buffers with similar concentrations were mixed to obtain the target TMAO concentrations used in the fluorescence measurements, thus minimizing the pH changes that could occur from mixing two TMAO buffers with a large concentration difference. To aid protein solubility (25), arginine was included in the TMAO buffers to a concentration of 50 mm; this efficiently prevented protein aggregation.
Circular Dichroism Spectra
Far UV circular dichroism spectra for GR A-NTD, C2-NTD, C3-NTD, and GR 1–97 were recorded at 22 °C with an Aviv CD spectrometer (Model 215) from 250 to 190 nm with a bandwidth of 1.0 nm and scan step of 1 nm in a 0.1-cm quartz cell. All spectra were recorded in PBS buffer and corrected for the contribution of respective buffers. Each spectrum shown is an average of three spectra. Molar ellipticities at 200 and 222 nm for each construct were used in this study to compare with the available values for the ID proteins (26) and those from the Protein Circular Dichroism Data Bank (27).
TMAO-induced Protein Folding Transitions Monitored by Tryptophan Emission Fluorescence Intensity
Fluorescence emission spectra for each purified recombinant GR NTD construct were measured using an Aviv ATF 105 fluorometer (Aviv Biomedical) in TMAO buffers of varying concentrations. Freshly prepared dithiothreitol 1 m stock solution was added to TMAO buffer to a final concentration of 1 mm. Protein at 0.5 μm in a volume of 150 μl of TMAO buffer was allowed to rest in a “submicro” fluorometer cell at 22 °C (Santa Cells) for 5 min to allow for temperature stabilization and protein conformation equilibrium to be reached. Emission spectra were then recorded with excitation at 295 nm. All spectra were corrected for the contribution of buffer. Fluorescence emission intensities at 338 nm were recorded as a function of TMAO concentration and normalized to the intensity at 0 m TMAO concentration. The resulting sigmoid curve was fitted to a two-state cooperative folding transition using the linear extrapolation method to determine the stability (ΔGU→F) and m-value for each construct (28). The linear extrapolation method has been shown to be valid for fitting the data of protecting osmolyte-induced folding of ID proteins (29).
Protease Protection Assay
Protease digestions of purified GR A-NTD, GR C2-NTD, and GR C3-NTD proteins at 1 mg/ml were performed with sequencing grade trypsin (Promega) at 22 °C. Digestions were performed at a protein:trypsin mass ratio of 1000:1 in 10 mm HEPES, 80 mm NaCl, 1 mm EDTA, 10% glycerol buffer, pH 7.6 for 0, 5, 10, and 30 min. Digestions were quenched by mixing the protein and trypsin mixture with 6× Laemmli sample buffer and boiling at 100 °C for 10 min. A 40-μg sample at each time point was separated on 4–15% Tris-HCl gel (Bio-Rad) with SDS-Tris-glycine gel running buffer.
Assessment of Side Chain and Backbone Contributions to the m-value for TMAO-induced Folding
The m-values were calculated based on the group transfer free energy model (30) using experimentally measured values for the backbone and the side chain contributions (12, 31). Briefly, the m-value considers the contribution of each residue using the difference in exposed surface area to a 1.4-Å rolling sphere in the native state (crystal structure) relative to the denatured state (average surface area from a library of Gly-X-Gly rotamers),
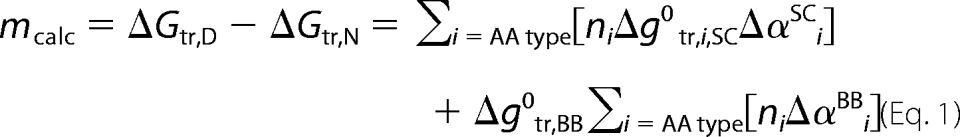 |
where ni is the total number of groups of type i present in the protein, Δg0tr is the free energy of transfer of the side chain (SC) or backbone (BB) from aqueous solution to 1 m TMAO, and Δαi is the fractional change in solvent accessibility from the native to denatured state (12, 31). Crystal structures with diverse folds chosen for analysis were RNase T1, P protein, staphylococcal nuclease, sperm whale myoglobin, carbonic anhydrase, adenylate kinase, triose-phosphate isomerase, porphobilinogen deaminase, mitogen-activated protein kinase, phosphoglycerate dehydrogenase, and seryl-tRNA synthetase (Protein Data Bank codes are in supplemental Table S1). Only the A-chain was considered for calculations in the asymmetric unit, and any bound metals and/or ligands were removed. For sensitivity analyses, every amino acid was changed (i.e. so that there is 0% sequence identity between mutant and WT sequences) based on the expected probabilities from the BLOSUM62 matrix (32) and a similar substitution matrix for order to disorder mutations (33). 100,000 in silico mutant sequences for each protein were generated, and the m-values were recalculated using the same fractional change in solvent accessibility (supplemental Table S1).
RESULTS
Naturally Occurring Osmolyte TMAO Can Induce Cooperative Folding Transitions in the ID NTDs of Different GR Translational Isoforms
Tryptophan emission spectra were measured for the GR A-NTD, GR C2-NTD, and GR C3-NTD constructs in buffers with different TMAO concentrations using an excitation wavelength of 295 nm. (Note that all three constructs contain two Trp residues, corresponding to positions 213 and 364 as numbered in the full-length GR A-NTD). As shown in Fig. 2 (inset), in the absence of TMAO, the maximum emission wavelength is 348 nm for the GR A-NTD construct. The same was also found for GR C2-NTD and GR C3-NTD constructs (data not shown), indicating that the Trp residues are solvent-exposed in the ID proteins. In the presence of 2.2 m TMAO, however, the emission wavelength maximum shows a significant blue shift (338 versus 348 nm), indicating that TMAO induces a conformation wherein the Trp side chains are excluded from solvent. The transition from the unfolded to the folded forms of each construct was followed by monitoring the fluorescence intensities at 338 nm as a function of TMAO concentration. As shown in Fig. 2, a clear sigmoidal transition is seen for each construct, suggestive of a TMAO-induced folding transition. These data were fit to a two-state model using the linear extrapolation method, which assumes that the stability of the folded state varies linearly with TMAO concentration (28). From the fits, the free energy of the folded state (relative to the unfolded state), ΔG0U→F, was determined for each construct (Table 1). The m-values, which represent the linear denaturant dependence of the apparent stabilities (i.e. the slopes of the free energy changes), were also determined from the fits (Table 1). The significance of the m-values is discussed in more detail below.
FIGURE 2.

TMAO-induced conformational transitions of GR A-NTD (open squares), GR C2-NTD (open circles), and GR C3-NTD (open triangles) monitored by tryptophan emission fluorescence intensity at 338 nm with excitation at 295 nm. Fluorescence emission intensity at each TMAO concentration was normalized to that at 0 m TMAO concentration. The folding experiments were performed at 22 °C with a protein concentration of 0.5 μm. Nonlinear least square fits to two-state transitions using the linear extrapolation method were carried out for each construct (solid line). The best fit thermodynamic parameters are shown in Table 1. Inset, GR A-NTD fluorescence emission spectrum with excitation at 295 nm in 0 m TMAO with λmax at 348 nm (solid line) and in 2.2 m TMAO with λmax at 338 nm (dashed line).
TABLE 1.
Stabilities and m-values for characterized GR N-terminal domain constructs
| Construct | ΔG0U→F | m-value |
|---|---|---|
| kcal/mol | kcal/(mol × m) | |
| GR A-NTD | 10.1 ± 0.5 | 8.8 ± 0.4 |
| GR C2-NTD | 8.9 ± 0.4 | 6.9 ± 0.4 |
| GR C3-NTD | 7.6 ± 0.3 | 6.7 ± 0.3 |
The quality of the individual fits to the data as well as the independence of the fitted parameters from the emission wavelength (data not shown) indicate that the transitions are well approximated by a two-state model and that the ΔG0U→F obtained for each construct represents a reasonable estimate of the thermodynamic stability difference between the unfolded state and a TMAO-induced folded state. The importance of this conclusion becomes clear when the apparent stabilities for the different constructs are compared. For example, the change in the apparent folding/unfolding free energy, ΔG0U→F, from 10.1 to 7.6 kcal/mol upon truncation of residues 1–97 indicates that removing the first 97 residues actually stabilizes the folded state of the protein. This result is in stark contrast to what might be expected when amino acids are deleted from a protein. Namely, deletion of amino acids generally destabilizes the folded states of proteins. However, as discussed below, this result portends an important organizational relationship between the extreme N terminus and the core region of NTD and suggests a regulation strategy that may be unique to ID proteins.
Protease Protection Assay Validates the Rank Order of Stability Estimates from TMAO Folding of the GR A-NTD, GR C2-NTD, and GR C3-NTD Constructs
The stability measurements obtained from TMAO-induced folding provide an estimate of the extrapolated stability of the folded state. To determine whether the apparent free energies from Table 1 are reporting on the relative stabilities of the unfolded proteins in the absence of TMAO, limited trypsin digestions of GR A-NTD, GR C2-NTD, and GR C3-NTD were performed (Fig. 3). A hallmark feature of ID proteins in vitro is their high intrinsic sensitivity to protease digestion (34). Thus, as expected, all three constructs are highly sensitive to trypsin. After 5 min with a protein:trypsin mass ratio of 1000:1 at 22 °C, significant degradation could be observed for all three constructs with complete digestion being reached at ∼30 min. Comparing the trypsin sensitivity among the three isoforms in the absence of TMAO, however, reveals that GR A-NTD is the most sensitive, whereas GR C3-NTD is the least sensitive, a result that is consistent with the rank order of stabilities obtained from the TMAO folding experiments (Table 1): GR C3-NTD is 1.3 kcal/mol more stable than GR C2-NTD and 2.5 kcal/mol more stable than GR A-NTD. In short, both the apparent free energies and the sensitivities to trypsin digestion for GR A-NTD, GR C2-NTD, and GR C3-NTD indicate that truncation of successive increments of amino acids from the extreme N terminus of GR incrementally stabilizes the folded state of the remaining NTD residues and that the stabilization adequately reflects the impact of the truncation on the propensity to form structure under native conditions.
FIGURE 3.

Protease protection assays for GR A-NTD, GR C2-NTD, and GR C3-NTD. Trypsin digestions were performed at a protein (1 mg/ml):trypsin mass ratio of 1000:1 at 22 °C in 10 mm HEPES, 80 mm NaCl, 1 mm EDTA, 10% glycerol buffer, pH 7.6 for 0, 5, 10, and 30 min, respectively. After quenching, 45-μg samples from each time point were separated on 4–15% Tris-HCl gels with SDS-Tris-glycine gel running buffer.
The Folded States of GR NTDs Induced by TMAO Are Thermodynamically Similar to Globular Proteins of Similar Length
The apparent two-state nature of the fits in Fig. 2 provides additional information. Specifically, the m-values for the three GR NTDs obtained from the fits (Table 1) can be quantitatively compared with m-values obtained for other proteins. According to the linear extrapolation model (28), the stability of the protein at any osmolyte concentration, ΔGU→F(CTMAO), can be described by the linear expression
where ΔG0U→F is the intercept of the line and corresponds to the stability in the absence of osmolyte and m (i.e. the m-value) is the slope of the osmolyte dependence. Importantly, for transitions that are known to be two-state, the m-value has been shown to be proportional to the amount of surface area that is buried upon folding the protein, which correlates with the protein sequence length (31). For denaturant-induced unfolding (i.e. urea or guanidine hydrochloride), the linear relationship between m-value and protein chain length has been established for known globular proteins (35). Shown in Fig. 4A are the renaturation dependent TMAO m-values as a function of chain length for a series of globular proteins that have been destabilized and refolded with TMAO. Shown are Barstar (36), RCAM-T1 (11), P protein (37), and Nank 1–7 (38). For comparison, m-values for GR A-NTD, GR C2-NTD, and GR C3-NTD from Table 1 are also shown. Of note is that the data for all three GR NTD isoforms are well described by the same line that relates the folded globular proteins to their m-values. In fact, all seven data points could be fit to a line with a correlation coefficient of R2 = 0.97. The importance of this result is 2-fold. First, it supports the conclusion that each of the various isoforms (i.e. GR A-NTD, GR C2-NTD, and GR C3-NTD) undergoes folding transitions wherein the bulk of the surface area burial is occurring in a cooperative two-state manner with increasing TMAO concentration, thus supporting our original inference. Second, the fact that the GR NTD constructs and the known globular proteins could be fit to the same linear relationship indicates that TMAO affects the folded and unfolded states of ID and globular proteins to a similar degree on a per amino acid basis (12) even if the molecular basis of these effects are not known (see Fig. 6 and “Discussion”). This surprising result strongly suggests that the full-length NTD and the two truncated translational isoforms are able to adopt native folds that are thermodynamically similar in terms of surface area burial to those adopted by globular proteins and that the unfolded states are similar. However, although seemingly unlikely, we cannot exclude the possibility that both the folded and unfolded states of ID and globular proteins are different from each other and respond differently to TMAO but that the differences are quantitatively canceled out in the overall m-value. Regardless, the observed similarity in the size dependence of the m-value between ID and globular proteins even when negative cooperativity causes the transition to deviate from two-state (see Fig. 6 and accompanying discussion) is indicative of a significant burial of the surface upon unfolding.
FIGURE 4.

A, dependence of m-values on protein chain length (L) for TMAO-induced folding transitions. Shown are the m-values fitted from TMAO-induced two-state folding transitions for Barstar (closed star 1; Ref. 36), RCAM-T1 (closed star 2; Ref. 22), P protein (closed star 3; Ref. 37), Nank 1–7 (closed star 4; Ref. 38), GR A-NTD (open square), GR C2-NTD (open circle), and GR C3-NTD (open triangle). All seven data points can be fitted to a linear line; i.e. m-value = 0.0215 × L with a correlation coefficient of R2 = 0.97. This indicates that the TMAO-induced folding of GR A-NTD, GR C2-NTD, and GR C3-NTD followed the cooperatively two-state transition processes. In addition, the folded states of GR NTDs induced by TMAO are thermodynamically similar to globular proteins of similar length. B, dependence of calculated m-values on protein length (L) and sequence composition. WT sequences are depicted in black circles and can be fitted to the line m-value = 0.0247 × L (shown in gray) with a correlation coefficient of R2 = 0.98. BLOSUM62 (32)-mutated sequences are shown as dark gray stars and can be fitted to the line m-value = 0.0249 × L (shown in black short dash) with a correlation coefficient of R2 = 0.99. DISORDER-mutated sequences are shown as light gray boxes and can be fitted to the line m-value = 0.0248 × L (shown in black long dash) with a correlation coefficient of R2 = 0.99. All three fits have similar dependence on L. The absolute changes in slope are only minutely dependent on variations in sequence composition because although ∼25% of the m-value contribution comes from side chain contributions (with the remaining 75% coming from backbone contributions; supplemental Table S1) the relatively minor differences in amino acid usage between ID and globular proteins amount to only a small fraction of the computed m-value. For comparison, GR A-NTD, GR C2-NTD, and GR C3-NTD are depicted as the unfilled triangle, circle, and square, respectively.
FIGURE 6.

A, schematic representation of the ensemble model of allosteric regulation in full-length GR NTD containing the R region and F region. Each region can be folded or unfolded, resulting in four possible states (i.e. N, I1, I2, and U) (42). S.W. is the statistical weight of each state. Q is the partition function, which is the sum of the statistical weights of all the states in the ensemble. KF and KR are equilibrium constants between the folded and unfolded state for F and R region, respectively, i.e., KF = Exp(−ΔGF/RT), KR = Exp(−ΔGR/RT). Φint is the statistical weight of the interaction between R and F region, Φint = Exp(−Δgint/RT). B, dependence of the population of the states with F region folded (i.e. N and I2) on TMAO concentration. Raw data for GR A-NTD (open squares) and GR C3-NTD (open triangles) are calculated from the TMAO folding curve as shown in Fig. 2. The underlying assumption made is that the fully unfolded state U and the intermediate state I1 (where the F region is unfolded) have the same fluorescence emission intensity (set to 0) and that the fully folded state N and the intermediate state I2 (wherein the F region is folded) have the same fluorescence emission intensity (set to 1) as both of the two NTD tryptophans are in the F region. The fitted curve is based on the linear extrapolation method, fitting to the equation Observable = PN + PI1. The fitted parameters are as follows: ΔG0F→U (F) = −8.4 ± 0.5 kcal/mol, m (F) = 7.4 ± 0.4 kcal/(mol × m); ΔG0F→U (R) = 0 kcal/mol, m (R) = 2.1 ± 0.4 kcal/(mol × m); Δg0int = −3.0 ± 1.0 kcal/mol, and mint = 2.0 ± 0.9 kcal/(mol × m). C, histogram plot of the extrapolated values for stabilities of the F region (ΔGF), R region (ΔGR), the interaction energy (Δgint), and the calculated free energy value for the fully unfolded state U (ΔGU = ΔGF + ΔGR + Δgint). D, dependence of the free energy of the F region, R region, interaction interface, and fully unfolded state on TMAO concentration, based on the fitted parameters from Fig. 6B.
To explore this dependence, we note that TMAO-induced m-values appear to be primarily determined by energetically unfavorable interactions between TMAO and the peptide backbone and are expected to be affected to a lesser degree by side chain contributions (12). This expectation is especially important given that ID proteins have more charged residues, fewer hydrophobic residues, and lower sequence complexity compared with globular proteins (3, 4), differences that could potentially undermine the interpretation that the similarity in m-value reflects similar backbone burial. To test whether the sequence differences between ID and globular proteins would dramatically impact the measured m-value, simulated m-values (based on two-state native to unfolded state transitions) for a database of globular proteins were computed using artificial sequences generated in two ways. First, sequences were generated using property-based similarity (using BLOSUM62 (32)). This produced sequences with properties similar to the parent structure but with the amino acid usage characteristic of globular proteins. Second, sequences were generated by randomly assigning amino acids to positions keeping the distributions consistent with ID proteins (33). As Fig. 4B reveals, regardless of the model chosen, the amino acid composition does not dramatically impact the expected m-value, confirming that m-values arise primarily from the backbone and that backbone burial must occur to obtain a large m-value. Specifically, ∼75% of the m-value contributions from an average globular protein arises from the backbone, whereas the remaining 25% stems from the specific side chains (supplemental Table S1).
The importance of the results presented in Fig. 4B is 2-fold. First, they reveal that sequence differences between globular and ID proteins are not a strong determinant of the measured m-values and thus validate the apparent similarity between the folding reactions of globular and ID proteins inferred from m-values. Second and perhaps most important, they provide an important new tool to quantitatively compare the folding reaction(s) of both ID and globular proteins. To date, attempts to study differences between globular and ID proteins have focused on the unfolded states and the interconversion between disordered species (26), a bias that stems from the uncertainty as to whether all ID proteins must fold to function and the difficulty in identifying conditions that can facilitate folding for those ID proteins. Until now, such factors have undermined the notion that ID proteins have the capacity to adopt folded structures similar to their globular counterparts or that the thermodynamics of globular and ID proteins could even be quantitatively compared.
The uniqueness of the results for the full-length NTD and the two truncation isoforms can be further demonstrated by noting that not all ID regions undergo osmolyte-induced cooperative two-state transitions that produce globular protein-like m-values. For example, TMAO-induced folding transition of the sequence corresponding to only the AF1 region of GR (i.e. GR 77–262) results in an m-value that deviates significantly from the value expected for a two-state transition. The m-value for the AF1 construct we report here, m = 1.4 ± 0.2 kcal/(mol × m), which was obtained in a manner similar to the GR A-NTD, GR C2-NTD, and GR C3-NTD (supplemental Fig. 1), is in quantitative agreement with the previously reported m-value of 1.6 ± 0.3 kcal/(mol × m) (39). As can be inferred from the line in Fig. 4A, however, the expected m-value for a protein with 186 amino acids (i.e. 77–262) undergoing a cooperative two-state transition in TMAO is ∼4.0 kcal/(mol × m), a value that is significantly greater (>2-fold) than the experimental value. The dramatically lower than expected m-value indicates that either 1) the folded state is not completely folded, 2) the unfolded state has structure (40), or 3) an equilibrium intermediate (I) state (or states) is being populated during the folding transition (41). A consequence of the presence of an intermediate(s) (which makes the transition (at least) three-state) is that the fitted ΔG0U→F for the construct corresponding to the AF1 region cannot be interpreted (because the two-state assumption is invalid) as a true measure of the stability difference between the folded and ID states nor can the m-value be interpreted in terms of surface area burial (as determined by Fig. 4). Only in the case where the transition is well approximated as two-state and the m-value quantitatively agrees with expectation (based on size), can the m-value be unambiguously interpreted in terms of surface area.
Thus, the facts that (i) the fitted parameters for each NTD construct studied here are independent of fluorescence emission wavelength and (ii) the m-values correspond (within error) to what would be expected for the folding of proteins of the respective sizes strongly suggest that each translational isoform is undergoing a transition into a globular protein-like fold that buries the majority of backbone surface area in a cooperative two-state manner (12) and that the free energies reported in Table 1 provide a reasonable representation of the stabilities of those folds. What role, if any, such fully folded states play in function is not known.
Full-length GR NTD Contains at Least Two Distinct Thermodynamically Coupled Regions
The stability data in Table 1 are also suggestive of a unique thermodynamic architecture and regulation strategy for GR activity. As noted, the functionally defined AF1 region is mapped to residues 77–262 within the NTD of human GR and is indispensable for its maximal transcriptional activity (16). However, the role of the remaining ID sequence of the NTD is not known. Previous studies, which show that binding of GR AF1 to TATA box-binding protein induces structure in the AF1 (10), suggest that the folded conformation of GR AF1 is a functionally important state. Indeed, consistent with this hypothesis is the observation that the stabilities of the various translation isoforms of GR from Table 1 are correlated with activity of the corresponding full-length constructs (19) (Fig. 5). The truncated isoform with the highest stability (i.e. the C3 isoform) also has the highest activity. These results are consistent with a coupled folding/binding mechanism for activity (supplemental Appendix), although the precise role this coupling plays in the activity of the full-length construct remains an open question.
FIGURE 5.

Relative activities of GR translational isoforms as reported in the literature (19) versus the extrapolated stabilities (ΔG0U→F) of their corresponding ID NTDs. The relationship indicates that GR translational isoforms with higher activity had more stable N-terminal domains. Errors for the extrapolated stabilities are from nonlinear least squares fitting of the TMAO folding data. Errors for the relative activities are calculated from the absolute errors reported in the literature (19) based on an error propagation method.
Nonetheless, due to the fact that the activity of GR can be linked to at least a subset of the residues between 98 and 420 (i.e. within the GR C3 N-terminal domain), the GR NTD can be viewed as having at least two functionally important regions, which we operationally define as the F and R regions. The F region is so defined because it contains the cofactor binding or AF1 sites (as noted above). As a consequence of the observation that truncating different size segments of the first 97 amino acids can modulate the stability of the remaining ID NTD, GR 1–97 can be viewed as a variable sized R region (Fig. 6). Removing a portion of the R region (i.e. GR 1–89) results in an increase in stability (and activity) for the functional domain, whereas removing the entire R region (i.e. GR 1–97) results in maximal stability and activity. This behavior (i.e. increasing stability by removing sequence segments) appears to end once truncation proceeds into the F region (presumably because the specific functionally important sites are removed). Indeed, sequence alignment of GR from different species (supplemental Fig. 2) reveals multiple conserved translation start sites within the first 97 amino acids (but none between 97 and 316), suggesting that the proposed functional division within the GR NTDs is conserved across different species and not limited to humans.
Importantly, the folding data for the various isoforms (Fig. 2) can be used to show that the F and R regions are thermodynamically negatively coupled. Using a simple cooperative ensemble allosteric mode described previously (42), the experimental folding free energies for the different constructs can be used to deduce the stability of the component parts. Shown in Fig. 6A is a schematic representation of the NTD ensemble that would result if the NTD was indeed composed of two domains. We note that for a two-domain protein there are four possible states representing folding and unfolding of each domain both with and without the other domain. Shown also in the figure are the relative stabilities, statistical weights, and probabilities for each state. The quintessential feature of this model, which is the reason that each domain can “sense” the other, is that the free energy of each state relative to the reference state is the free energy of unfolding each domain (i.e. ΔGR or ΔGF) plus the energy involved in breaking the interactions between the domains, Δgint.
In the case where Δgint = 0, there is no coupling, and the domains act simply as if they are tethered. However, if Δgint ≠ 0, the unfolding of one domain will necessarily affect the stability of the state with the remaining domain folded (43). In the case where Δgint > 0, the two domains are positively coupled because the unfolding of just one domain results in an energetic penalty that makes the state with the one remaining domain less stable. If this were the case, removing one domain (such as was done in the C3 construct) would result in an apparent decrease in the stability of the second domain. In the case where Δgint < 0 on the other hand, the two domains would be negatively coupled, meaning that the presence of each domain would act to destabilize the other domain. If this were the case, removing one domain would result in an apparent stabilization of the remaining domain. As both the stability (Fig. 2 and Table 1) and the protease sensitivity (Fig. 3) results indicate, the latter scenario appears to be valid. Removal of the R domain indeed stabilizes the F domain, indicating that the F and R domains are negatively coupled. More quantitative and mechanistic insight can be gained, however, by reanalyzing the data in Fig. 2 to the model shown in Fig. 6A to obtain estimates of the intrinsic stabilities and interaction energies (i.e. ΔGR, ΔGF, and Δgint).
To facilitate this end, we note that the stability of the F region can be directly obtained from the TMAO-induced folding of the C3 construct. The value of ΔGF = −ΔG0U→F (GR C3-NTD) = −7.6 kcal/mol indicates that the F domain is significantly unstable in isolation, which is the expected result given the ID nature of this protein. Determination of the stability of the R domain is less straightforward. The small size (97 amino acids) and the lack of Trp residues in this region make it impossible to observe and thus thermodynamically characterize a cooperative transition to a fully folded state with TMAO (as done with the F region). Nonetheless, the CD signal of the GR 1–97 construct (in the absence of TMAO) provides an important clue about the range of possible stabilities of the R domain. As shown in Fig. 7, the relative CD signals at 200 and 222 nm for the GR 1–97 construct place it somewhere between the average signal for folded (27) and ID proteins (26). This point is important because it reveals the presence of residual structure even in the absence of TMAO. In thermodynamic terms, this means that the stability of the R domain is such that both folded and disordered states must be populated to a significant degree. As only a narrow window of stability values allows for both folded and unfolded species to co-exist in substantial quantities simultaneously (i.e. ∼−1.5 < ΔGR < ∼1.5 kcal/mol), to a first approximation, the folded and unfolded states can be estimated to be equally probable (i.e. ΔGR ∼ 0 kcal/mol).
FIGURE 7.

Analysis of the circular dichroism data for the R region (GR 1–97) in terms of Δϵ222 nmversus Δϵ200 nm supports the conclusion that the R region in isolation is marginally stable even in the absence of TMAO. Open triangles represent the data from the Protein Circular Dichroism Data Bank (27), and open circles represent the data from Ref. 26. The filled star, triangle, and circle represent the data for GR A-NTD, C2-NTD, and C3-NTD, respectively, measured in this study. The filled square represents the data for GR 1–97, which is midway between the clusters for ID and globular folded proteins. deg, degrees.
Using these estimates for the stabilities of the F and R domains (i.e. ΔGF = −8.4 kcal/mol and ΔGR = 0 kcal/mol are fixed), the data for the TMAO dependence of the fluorescence of GR A-NTD can be fitted to obtain an estimate of the interaction energy. The fitted value of Δgint in the absence of TMAO (Δg0int = −3.0 ± 1.0 kcal/mol) indicates that the F and R domains are indeed negatively coupled and that the parameters are able to accurately reproduce both the GR A-NTD and GR C3-NTD TMAO refolding curves (Fig. 6B), which differ dramatically in their apparent steepness (i.e. the m-value for the GR A-NTD is considerably higher than that of the GR C3-NTD (Table 1)). We also note that the results are not qualitatively sensitive to the precise value of ΔGR; systematically varying the magnitude between −1.0 and 1.5 kcal/mol nonetheless results in a negative Δgint.
As expected, the free energy profiles of the different species from Fig. 6A reveal that the lowest energy state is the one in which the F region is disordered (Fig. 6, C and D). Inspection of the osmolyte dependences of the stabilities of the various states in the two-domain model (Fig. 6D) reveals the underlying basis for the stability increase upon truncation. In the case of the full-length GR A-NTD, the F domain is unstable by 7.6 kcal/mol, and there is approximately no difference in energy between the folded and unfolded states of the R domain (Fig. 6C). However, because the stability of the fully disordered state in the GR A-NTD is the sum of the stabilities of the F and R domains and their interaction energies (i.e. ΔG0F→U (GR A-NTD) = ΔGU = ΔGF + ΔGR + Δgint = −10.1 kcal/mol), the apparent stability of the F region will depend on whether it is coupled to the stability of the R region. This is shown in Fig. 6, C and D. As noted, the stability of the F region, ΔGF, is −7.6 kcal/mol in isolation. For the apparent stability to increase in the presence of the R domain (i.e. for ΔG0F→U to be less negative), the sum of the contributions of the stability of the R domain and the interaction energy must be positive (i.e. ΔGR + Δgint > 0). For the stability to decrease on the other hand, the sum must be negative (i.e. ΔGR + Δgint < 0). As the results presented in Figs. 2, 3, and 6 clearly indicate, the full-length construct is considerably less stable than the truncated form. In short, the results reveal that by coupling the stabilities of domains that are unstable in isolation, ID molecules can effectively tune the stabilities of folded (and presumably active) forms of the molecule.
DISCUSSION
The results presented here show that thermodynamic coupling does exist between different regions of the disordered NTD of GR, allowing truncation of the extreme N terminus to modulate the stability of the functionally important AF1 region when the NTD is studied in isolation. Of course, how this stability tuning is manifested at the level of the full-length GR (which also contains the DNA binding and ligand binding domains) remains an open question. Nonetheless, the results presented here provide significant insight into the organizing principles describing how changes in one domain can affect structure, stability, and activity in other domains, principles that in many ways are clearer when presented in the context of allosteric communication in ID proteins.
For more than 40 years, allostery has been described in terms of two classic models: Monod, Wyman, and Changeaux (44) and Koshland, Némethy, and Filmer (45). Both models describe the quantitative relationship between ligand binding at two different coupled sites. A fundamental limitation of both models, however, is that they provide little insight into the energetic determinants for “how” the coupling is facilitated. Are there ground rules that determine whether binding at one site can facilitate an affinity change at the second site? In other words, are there quantitative energetic relationships that must exist to propagate signal from one site to another?
Recently, this question was addressed by recasting allostery in terms of the ensemble of the high and low affinity states for each domain (42, 55). The results revealed that allosteric phenomena (at least those under thermodynamic control) are a manifestation of a set of energetic ground rules that govern whether conformational changes can be propagated to distal sites (42, 55). In the context of these ground rules, allostery is determined by (i) the local conformational equilibrium in each region, (ii) the intrinsic ligand affinity at each site, (iii) the ligand concentrations, and (iv) the coupling energies between those local equilibria (42, 55). In essence, allostery can be described in terms of the same types of energetic parameters determined in this study.
An important implication of this realization is the notion that allostery can evolve in systems that lack unique structure just as easily as it can evolve in ordered, folded structures. This observation stands in stark contrast to the classic structural view of allostery that has emerged over the past 50 years of structural biology research (46). According to the structural view, site-to-site coupling results from ligand-induced structural changes that propagate from one site to the other presumably as a series (or pathway) of structural distortions that can be more or less ascertained through an inspection of the high resolution structure. The theoretical as well as experimental realization that simply changing the breadth of conformational distributions without significant changes in average position of atoms can nonetheless affect allostery (47–51) reveals an entirely new spectrum of possible regulatory strategies available to proteins. Indeed, the regulatory strategy apparently at play in the GR system reveals an efficient mechanism of “overdesign.” As the stability and activity results show, the full-length GR is designed to operate at less than half of the potential maximum activity (Fig. 5) with activity being increased by the removal of sequence segments.
Much in the same way that the activity of a folded allosteric protein may be tuned by stabilization of its active conformation (through phosphorylation, binding of ligands, pH changes, etc.), disordered proteins can achieve the same ends by merely removing sequence segments. Interestingly, although seemingly much different from allosteric mechanisms utilized by folded proteins (i.e. a ligand-induced change in the structure of the active site), the ID domain-mediated strategy outlined here reveals the universality of the principles at play in all (thermodynamically controlled) allosteric systems. In the case of allostery within folded proteins, the allosteric ligand binds to the active state of the molecule, thus stabilizing it. For the mechanism described here, the active, folded state of the F region is affected not by mutational changes that directly stabilize the folded state but by removing residues that stabilize the unfolded state (i.e. that destabilize the folded state). In this respect, although the underlying principles governing signaling in folded and ID allosteric systems are the same (i.e. the relative stability of the active state must be increased), ID proteins appear to have capitalized on an alternative approach to achieve those ends. It should be noted, however, that traditional modes of inducing allosteric effects (i.e. binding ligands, post-translational modification, etc.) may also be at play in GR. In fact, recent work has shown that phosphorylation of the AF1 region of the NTD can also be used to tune the activity of GR (52), thus demonstrating that many of the same thermodynamic strategies utilized for signal propagation in folded proteins can also be applied to ID proteins. How classical and ID domain-mediated allosteric strategies combine to explain the complex regulation of GR controlled genes is an intriguing and as yet unanswered question.
We also note once again that the analysis described here was performed on the isolated NTDs. The DNA binding and ligand binding domains may also be coupled to the R and F regions of the NTD as well as to each other. As described elsewhere, couplings between three or more domains can potentiate a wide range of regulatory strategies that can even involve a conversion from activation to repression and vice versa (55). Thus, an understanding of the energetics of the DNA binding and ligand binding domains and their coupling to the NTD is essential to an understanding of how the functional division within the NTD described here will be manifested in the overall functional response of the protein. These studies are currently underway.
In conclusion, based on well established criteria for determining cooperativity of folding-unfolding transitions (i.e. insensitivity of the extrapolated thermodynamic parameters to the observables being monitored and the comparability of the m-value with that of a globular protein of similar size), the ID NTDs of three GR translational isoforms were found to cooperatively fold into stable folded structures when induced by TMAO. The folded conformations are thermodynamically similar to folded native states of globular proteins based on m-value comparison (although negative cooperativity within the NTD slightly affects this interpretation). The correlation between the activity of each isoform and the stability of their ID NTDs supports previous conclusions (10) that the folded state is needed for transcriptional activation.
Most importantly, our thermodynamic analysis reveals that within GR not all of the ID NTD serves the same functional role. There are at least two evolutionarily conserved, functionally distinct regions, which can be operationally defined as the F and R regions that may vary in their boundaries. For GR, the F and R regions are thermodynamically negatively coupled, a situation that provides a regulatory strategy possibly unique to ID proteins. Truncation of the R region results in a corresponding increase in the stability of the remaining ID NTD and an increase in its transcriptional activity. These results are suggestive of a general paradigm for allosteric control whereby the folding of disordered protein regions can control and be controlled by the folding of other ID regions (53, 54). This strategy provides proteins with a general mechanism that can be utilized within all disordered segments, perhaps answering why ID sequences are found in such abundance in transcription factors and other cell signaling proteins (2).
This work was supported by National Science Foundation Grants MCB-0446050 (to V. J. H.) and DBI-1005027 (to C. C.).

This article contains supplemental Figs. 1 and 2, Table S1, and Appendix.
- ID
- intrinsically disordered
- GR
- glucocorticoid receptor
- NTD
- N-terminal domain
- TMAO
- trimethylamine N-oxide
- AF1
- activation function-1 domain
- R
- regulatory
- F
- functional.
REFERENCES
- 1. Tantos A., Han K. H., Tompa P. (2012) Intrinsic disorder in cell signaling and gene transcription. Mol. Cell. Endocrinol. 348, 457–465 [DOI] [PubMed] [Google Scholar]
- 2. Liu J., Perumal N. B., Oldfield C. J., Su E. W., Uversky V. N., Dunker A. K. (2006) Intrinsic disorder in transcription factors. Biochemistry 45, 6873–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tompa P. (2002) Intrinsically unstructured proteins. Trends Biochem. Sci. 27, 527–533 [DOI] [PubMed] [Google Scholar]
- 4. Uversky V. N. (2011) Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 43, 1090–1103 [DOI] [PubMed] [Google Scholar]
- 5. Dyson H. J., Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 6. Fink A. L. (2005) Natively unfolded proteins. Curr. Opin. Struct. Biol. 15, 35–41 [DOI] [PubMed] [Google Scholar]
- 7. Dyson H. J., Wright P. E. (2002) Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 12, 54–60 [DOI] [PubMed] [Google Scholar]
- 8. Babu M. M., van der Lee R., de Groot N. S., Gsponer J. (2011) Intrinsically disordered proteins: regulation and disease. Curr. Opin. Struct. Biol. 21, 432–440 [DOI] [PubMed] [Google Scholar]
- 9. Midic U., Oldfield C. J., Dunker A. K., Obradovic Z., Uversky V. N. (2009) Unfoldomics of human genetic diseases: illustrative examples of ordered and intrinsically disordered members of the human diseasome. Protein Pept. Lett. 16, 1533–1547 [DOI] [PubMed] [Google Scholar]
- 10. Kumar R., Lee J. C., Bolen D. W., Thompson E. B. (2001) The conformation of the glucocorticoid receptor af1/tau1 domain induced by osmolyte binds co-regulatory proteins. J. Biol. Chem. 276, 18146–18152 [DOI] [PubMed] [Google Scholar]
- 11. Baskakov I., Bolen D. W. (1998) Forcing thermodynamically unfolded proteins to fold. J. Biol. Chem. 273, 4831–4834 [DOI] [PubMed] [Google Scholar]
- 12. Auton M., Bolen D. W. (2005) Predicting the energetics of osmolyte-induced protein folding/unfolding. Proc. Natl. Acad. Sci. U.S.A. 102, 15065–15068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Griekspoor A., Zwart W., Neefjes J., Michalides R. (2007) Visualizing the action of steroid hormone receptors in living cells. Nucl. Recept. Signal. 5, e003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McEwan I. J., Lavery D., Fischer K., Watt K. (2007) Natural disordered sequences in the amino terminal domain of nuclear receptors: lessons from the androgen and glucocorticoid receptors. Nucl. Recept. Signal. 5, e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lavery D. N., McEwan I. J. (2005) Structure and function of steroid receptor AF1 transactivation domains: induction of active conformations. Biochem. J. 391, 449–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hollenberg S. M., Evans R. M. (1988) Multiple and cooperative trans-activation domains of the human glucocorticoid receptor. Cell 55, 899–906 [DOI] [PubMed] [Google Scholar]
- 17. Dahlman-Wright K., Almlöf T., McEwan I. J., Gustafsson J. A., Wright A. P. (1994) Delineation of a small region within the major transactivation domain of the human glucocorticoid receptor that mediates transactivation of gene expression. Proc. Natl. Acad. Sci. U.S.A. 91, 1619–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bray P. J., Cotton R. G. (2003) Variations of the human glucocorticoid receptor gene (NR3C1): pathological and in vitro mutations and polymorphisms. Hum. Mutat. 21, 557–568 [DOI] [PubMed] [Google Scholar]
- 19. Lu N. Z., Cidlowski J. A. (2005) Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol. Cell 18, 331–342 [DOI] [PubMed] [Google Scholar]
- 20. Bolen D. W., Baskakov I. V. (2001) The osmophobic effect: natural selection of a thermodynamic force in protein folding. J. Mol. Biol. 310, 955–963 [DOI] [PubMed] [Google Scholar]
- 21. Bolen D. W. (2001) Protein stabilization by naturally occurring osmolytes. Methods Mol. Biol. 168, 17–36 [DOI] [PubMed] [Google Scholar]
- 22. Baskakov I. V., Kumar R., Srinivasan G., Ji Y. S., Bolen D. W., Thompson E. B. (1999) Trimethylamine N-oxide-induced cooperative folding of an intrinsically unfolded transcription-activating fragment of human glucocorticoid receptor. J. Biol. Chem. 274, 10693–10696 [DOI] [PubMed] [Google Scholar]
- 23. Kumar R., Baskakov I. V., Srinivasan G., Bolen D. W., Lee J. C., Thompson E. B. (1999) Interdomain signaling in a two-domain fragment of the human glucocorticoid receptor. J. Biol. Chem. 274, 24737–24741 [DOI] [PubMed] [Google Scholar]
- 24. Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsumoto K., Umetsu M., Kumagai I., Ejima D., Philo J. S., Arakawa T. (2004) Role of arginine in protein refolding, solubilization, and purification. Biotechnol. Prog. 20, 1301–1308 [DOI] [PubMed] [Google Scholar]
- 26. Uversky V. N. (2002) Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11, 739–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Whitmore L., Woollett B., Miles A. J., Klose D. P., Janes R. W., Wallace B. A. (2011) PCDDB: the Protein Circular Dichroism Data Bank, a repository for circular dichroism spectral and metadata. Nucleic Acids Res. 39, D480–D486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Santoro M. M., Bolen D. W. (1988) Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27, 8063–8068 [DOI] [PubMed] [Google Scholar]
- 29. Wu P., Bolen D. W. (2006) Osmolyte-induced protein folding free energy changes. Proteins 63, 290–296 [DOI] [PubMed] [Google Scholar]
- 30. Tanford C. (1964) Isothermal unfolding of globular proteins in Aqueous Urea Solutions. J. Am. Chem. Soc. 86, 2050–2059 [Google Scholar]
- 31. Auton M., Bolen D. W. (2007) Application of the transfer model to understand how naturally occurring osmolytes affect protein stability. Methods Enzymol. 428, 397–418 [DOI] [PubMed] [Google Scholar]
- 32. Henikoff S., Henikoff J. G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 89, 10915–10919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Midic U., Dunker A. K., Obradovic Z. (2009) Protein sequence alignment and structural disorder: a substitution matrix for an extended alphabet. StReBio 9, 27–31 [Google Scholar]
- 34. Wright P. E., Dyson H. J. (1999) Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 293, 321–331 [DOI] [PubMed] [Google Scholar]
- 35. Ghosh K., Dill K. A. (2009) Computing protein stabilities from their chain lengths. Proc. Natl. Acad. Sci. U.S.A. 106, 10649–10654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pradeep L., Udgaonkar J. B. (2004) Osmolytes induce structure in an early intermediate on the folding pathway of barstar. J. Biol. Chem. 279, 40303–40313 [DOI] [PubMed] [Google Scholar]
- 37. Henkels C. H., Kurz J. C., Fierke C. A., Oas T. G. (2001) Linked folding and anion binding of the Bacillus subtilis ribonuclease P protein. Biochemistry 40, 2777–2789 [DOI] [PubMed] [Google Scholar]
- 38. Mello C. C., Barrick D. (2003) Measuring the stability of partly folded proteins using TMAO. Protein Sci. 12, 1522–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kumar R., Serrette J. M., Khan S. H., Miller A. L., Thompson E. B. (2007) Effects of different osmolytes on the induced folding of the N-terminal activation domain (AF1) of the glucocorticoid receptor. Arch. Biochem. Biophys. 465, 452–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shortle D. (1995) Staphylococcal nuclease: a showcase of m-value effects. Adv. Protein Chem. 46, 217–247 [DOI] [PubMed] [Google Scholar]
- 41. Whitten S. T., Wooll J. O., Razeghifard R., García-Moreno E. B., Hilser V. J. (2001) The origin of pH-dependent changes in m-values for the denaturant-induced unfolding of proteins. J. Mol. Biol. 309, 1165–1175 [DOI] [PubMed] [Google Scholar]
- 42. Hilser V. J., Thompson E. B. (2007) Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 8311–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luque I., Leavitt S. A., Freire E. (2002) The linkage between protein folding and functional cooperativity: two sides of the same coin? Annu. Rev. Biophys. Biomol. Struct. 31, 235–256 [DOI] [PubMed] [Google Scholar]
- 44. Monod J., Wyman J., Changeux J. P. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118 [DOI] [PubMed] [Google Scholar]
- 45. Koshland D. E., Jr., Némethy G., Filmer D. (1966) Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5, 365–385 [DOI] [PubMed] [Google Scholar]
- 46. Hilser V. J. (2010) Biochemistry. An ensemble view of allostery. Science 327, 653–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cooper A., Dryden D. T. (1984) Allostery without conformational change. A plausible model. Eur. Biophys. J. 11, 103–109 [DOI] [PubMed] [Google Scholar]
- 48. Bai F., Branch R. W., Nicolau D. V., Jr., Pilizota T., Steel B. C., Maini P. K., Berry R. M. (2010) Conformational spread as a mechanism for cooperativity in the bacterial flagellar switch. Science 327, 685–689 [DOI] [PubMed] [Google Scholar]
- 49. Pan H., Lee J. C., Hilser V. J. (2000) Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proc. Natl. Acad. Sci. U.S.A. 97, 12020–12025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Popovych N., Sun S., Ebright R. H., Kalodimos C. G. (2006) Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 13, 831–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reichheld S. E., Yu Z., Davidson A. R. (2009) The induction of folding cooperativity by ligand binding drives the allosteric response of tetracycline repressor. Proc. Natl. Acad. Sci. U.S.A. 106, 22263–22268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kobayashi Y., Mercado N., Barnes P. J., Ito K. (2011) Defects of protein phosphatase 2A causes corticosteroid insensitivity in severe asthma. PLoS One 6, e27627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Freiburger L. A., Baettig O. M., Sprules T., Berghuis A. M., Auclair K., Mittermaier A. K. (2011) Competing allosteric mechanisms modulate substrate binding in a dimeric enzyme. Nat. Struct. Mol. Biol. 18, 288–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Garcia-Pino A., Balasubramanian S., Wyns L., Gazit E., De Greve H., Magnuson R. D., Charlier D., van Nuland N. A., Loris R. (2010) Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 142, 101–111 [DOI] [PubMed] [Google Scholar]
- 55. Motlagh H. N., Hilser V. J. (2012) Agonism/antagonism switching in allosteric ensembles. Proc. Natl. Acad. Sci. U.S.A. 109, 4134–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]