

Abstract
Activation of hepatic stellate cells (HSCs) and subsequent uncontrolled accumulation of altered extracellular matrix (ECM) underpin liver fibrosis, a wound healing response to chronic injury, which can lead to organ failure and death. We sought to catalogue the components of fibrotic liver ECM to obtain insights into disease etiology and aid identification of new biomarkers. Cell-derived ECM was isolated from the HSC line LX-2, an in vitro model of liver fibrosis, and compared to ECM from human foreskin fibroblasts (HFFs) as a control. Mass spectrometry analyses of cell-derived ECMs identified, with ≥99% confidence, 61 structural ECM or secreted proteins (48 and 31 proteins for LX-2 and HFF, respectively). Gene ontology enrichment analysis confirmed the enrichment of ECM proteins, and hierarchical clustering coupled with protein–protein interaction network analysis revealed a subset of proteins enriched to fibrotic ECM, highlighting the existence of cell type-specific ECM niches. Thirty-six proteins were enriched to LX-2 ECM as compared to HFF ECM, of which Wnt-5a and CYR61 were validated by immunohistochemistry in human and murine fibrotic liver tissue. Future studies will determine if these and other components may play a role in the etiology of hepatic fibrosis, serve as novel disease biomarkers, or open up new avenues for drug discovery.
Keywords: CYR61, extracellular matrix, fibrosis, hepatic stellate cells, liver, LX-2, proteomics, Wnt-5a
Introduction
Extracellular matrix (ECM) is a critical component of the tissue microenvironment.1,2 Continuous ECM remodelling in the setting of chronic injury leads to an excessive accumulation of extracellular proteins, proteoglycans, and carbohydrates.3−5 The resultant pathological state, termed fibrosis, is responsible for the morbidity and mortality associated with organ failure in a variety of chronic diseases that affect a wide range of organs, including the liver, intestines, lungs, kidneys, eyes, heart, pancreas, and skin. To date, no clinically proven therapeutic strategy exists to reverse or prevent fibrosis. Elucidating the underlying mechanisms responsible for abnormal ECM deposition is therefore an increasingly pressing and important health challenge.
Central to this challenge is the need to characterize the qualitative and quantitative changes in ECM protein composition that occur during fibrotic progression and the effects these changes have on cell behavior. In liver fibrosis, changes in ECM composition are driven by hepatic stellate cells (HSCs), which upon chronic exposure to inflammatory cues become activated and trans-differentiate into proliferative myofibroblast cells.6 Once activated, HSCs up-regulate gene expression of ECM components, matrix-degrading enzymes, and their respective inhibitors, which in turn results in matrix remodelling and ECM accumulation at sites containing high densities of activated HSCs.6 The balance between ECM deposition and remodelling determines whether fibrosis progresses or regresses.7 In addition, there is increasing evidence that key facets of HSC biology are regulated by the pericellular ECM. For example, type I collagen, which is postulated to be the major component of fibrosis, has been reported to enhance proliferation of HSCs in vitro,7,8 whereas transfer of activated HSCs from a type I collagen substrate to a basement membrane-like matrix (Matrigel) can inhibit proliferation of HSCs as well as production of fibrogenic proteins.8,9 Furthermore, the regulation of HSC behavior by the extracellular microenvironment is believed to occur via integrin signaling.9,10 Thus, an understanding of the adhesion-dependent mechanisms that determine HSC behavior could help uncover novel therapeutic targets.
A large body of recent work has concentrated on elucidating mechanisms of fibrotic regression11 and identifying biomarkers of liver fibrosis.12 We hypothesized that a more detailed knowledge of the composition of fibrotic ECM would inform both of these aims. Therefore, we employed SDS-PAGE separation and mass spectrometry (GeLC–MS)-based proteomics to catalogue the constituents of fibrotic ECM. Though proteomics has been used to study liver fibrosis13 and ECM changes during transition from late-stage fibrosis to cancer,14 no previous studies have focused exclusively on an enriched liver ECM fraction, reflecting the fact that the ECM is a relatively unexplored proteome.15 A common approach taken to aid the successful analysis of a subcellular compartment using a mass spectrometry (MS)-based approach is the reduction of sample complexity. We therefore implemented a strategy that enriched for functionally relevant ECM16 (which has recently been employed upstream of MS analysis17), using the LX-2 cell line as a model of HSC-induced liver fibrosis in vitro(18) and dermal fibroblasts as a non-hepatic control. The resultant ECM catalogues were interrogated using a variety of bioinformatic tools, and immunohistochemical staining was used to validate potentially novel and interesting protein targets. Using this approach, we identified two previously unreported ECM proteins in fibrotic liver and provide evidence for the expression of many other ECM proteins within fibrotic tissue.
Experimental Section
Antibodies
Antibodies used for Western blotting and immunofluorescence were polyclonal rabbit immunoglobulin G (IgG) directed against human fibronectin (F3648; Sigma-Aldrich, Poole, U.K.) and fibrillin (provided by C. Kielty, University of Manchester, U.K.). Donkey anti-rabbit IgG conjugated to IRDye 800 (Rockland Immunochemicals, Gilbertsville, PA) was used for Western blotting detection, and donkey anti-rabbit IgG conjugated to an Alexa Fluor tag (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) was used for immunofluorescence. Antibodies used for immunohistochemistry were monoclonal rabbit IgG directed against human and mouse Wnt-5a (IMG-6075; Imgenex, San Deigo, CA), polyclonal rabbit IgG directed against human and mouse CYR61 (Abnova, Taipei City, Taiwan) and polyclonal rabbit IgG directed against human fibulin-2 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Goat anti-rabbit or anti-mouse IgG1 conjugated to biotin were used for immunohistochemical detection (ABC staining system, Santa Cruz Biotechnology, Inc.).
Cell Culture
The activated human HSC line LX-218 (provided by J. Iredale, Edinburgh University, U.K.) and human foreskin fibroblasts (HFFs; provided by K. Clark, University of Leicester, U.K.) were cultured in Dulbecco’s minimal essential medium (DMEM; Sigma-Aldrich) supplemented with 10% (v/v) fetal calf serum (FCS) and 2 mM l-glutamine. Cells were maintained at 37 °C in a humidified 5% (v/v) CO2 atmosphere. To isolate HFFs, human neonatal foreskins were sourced from the Cooperative Human Tissue Network. The foreskins were washed and minced into small chunks (<1 mm). The tissue chunks were dispersed in a cell culture dish containing DMEM supplemented with 50% (v/v) FCS, 2 mM l-glutamine, penicillin–streptomycin, Fungizone, and gentamicin. When cells migrated out of the tissue, FCS concentration was reduced to 10% (v/v), and cells were then passaged at confluence with trypsin/EDTA and cultured as described above.
Flow Cytometry
Details of flow cytometric analyses are provided in the Supporting Information.
ECM Purification
To generate cell-derived matrices (CDMs), 10 cm diameter cell culture dishes were coated with 0.2% (w/v) sterile gelatin (Sigma-Aldrich) for 60 min at 37 °C. After equilibration of gelatin-coated dishes with growth medium, primary fibroblasts (1 × 106 cells/mL) were plated onto dishes and cultured for a range of times (from 5 to 14 days), changing the growth medium every 3 days. To purify ECM, cells were removed using a modification of a previously published protocol.16 In brief, growth medium was aspirated and cells were washed with PBS. CDMs were denuded of cells by lysis with 20 mM NH4OH and 0.5% (v/v) Triton X-100 (Sigma-Aldrich) in PBS for 1 min at 37 °C, followed by digestion with 10 μg/mL DNase I (Roche Diagnostics, Burgess Hill, U.K.) for 60 min at 37 °C. CDMs were recovered in reducing sample buffer [50 mM Tris-HCl, pH 6.8, 10% (w/v) glycerol, 4% (w/v) sodium dodecylsulfate (SDS), 0.004% (w/v) bromophenol blue, 8% (v/v) β-mercaptoethanol] by scraping (Greiner Bio-One GmbH, Frickenhausen, Germany). Protein samples were resolved by SDS–polyacrylamide gel electrophoresis (PAGE) and subjected to Western blotting, immunofluorescence or MS as described below.
Western Blotting
Following SDS-PAGE, resolved proteins were transferred to nitrocellulose membrane (Whatman, Maidstone, U.K.). Membranes were blocked with casein blocking buffer (Sigma-Aldrich) and probed with primary antibodies diluted in blocking buffer containing 0.05% (v/v) Tween 20. Membranes were washed with Tris-buffered saline (10 mM Tris-HCl, pH 7.4, 150 mM NaCl) containing 0.05% (v/v) Tween 20 and incubated with species-specific fluorescent dye-conjugated secondary antibodies diluted in blocking buffer containing 0.05% (v/v) Tween 20. Membranes were washed in the dark and then scanned using an Odyssey infrared imaging system (LI-COR Biosciences, Cambridge, U.K.) to visualize bound antibodies.
Coomassie Blue Staining
Following SDS-PAGE, total protein was visualized by incubating gels in Coomassie staining solution [0.025% (w/v) Coomassie Brilliant Blue R-250, 10% (v/v) acetic acid, 25% (v/v) propanol] for 60 min at room temperature. Gels were then destained in 10% (v/v) acetic acid, washed with distilled H2O, and scanned using the Odyssey imaging system.
GeLC–MS and data analysis
In-Gel Proteolytic Digestion
In-gel digestion with trypsin was carried out as described by Shevchenko et al.19 with adaptations for processing in 96-well plates as described by Humphries et al.20 Briefly, following Coomassie Blue staining, gels were cut into 30 slices per lane and chopped into ∼1 mm3 pieces, washed with acetonitrile (ACN), reduced (10 mM dithiothreitol, 25 mM NH4HCO3), alkylated (55 mM iodoacetamide, 25 mM NH4HCO3), and digested with 12.5 ng/μL sequencing-grade modified trypsin (Promega, Southampton, U.K.). Peptides were extracted once with 20 mM NH4HCO3 and two times with 5% (v/v) formic acid in 50% (v/v) ACN, and then concentrated to 20 μL by vacuum centrifugation. Samples were stored at −20 °C until analysis by liquid chromatography–tandem mass spectrometry (LC–MS/MS).
LC–MS/MS Analysis
LC–MS/MS analysis was performed using a nanoACQUITY UltraPerformance LC system (Waters, Elstree, U.K.) coupled online to a 4000 Q TRAP triple-quadrupole linear ion trap analyzer (Applied Biosystems, Framingham, MA, USA), as described previously.20 Samples (5 μL) were concentrated and desalted on a Symmetry C18 preparative column (20 mm length, 180 μm inner diameter, 5 μm particle size, 100 Å pore size; Waters). Peptides were separated on an ACQUITY UltraPerformance LC bridged ethyl hybrid C18 analytical column (100 mm length, 75 μm inner diameter, 1.7 μm particle size, 130 Å pore size; Waters) using a 40-min linear gradient from 1% to 30% (v/v) ACN in 0.1% (v/v) formic acid at a flow rate of 300 nL/min at 50 °C. The mass spectrometer was instructed to acquire enhanced-resolution and product ion scans for peptides with ion counts greater than 250,000 counts/s, with a precursor ion mass-to-charge ratio (m/z) selection window of m/z 400–1600. Information-dependent acquisition (Analyst, version 1.4.1; Applied Biosystems) was used to acquire tandem mass spectra over the range m/z 140–1400 for the two most intense peaks, which were excluded for 12 s after two occurrences. Spectra were extracted, charge-state deconvoluted and deisotoped using the default setting of the Mascot Search script (mascot.dll, version 1.6b9; Matrix Science, London, U.K.) as a plug-in for Analyst.
Peak list files were searched against a modified version of the IPI human database (version 3.34, release date second October 2007, containing 67,756 sequences) containing 10 additional contaminant/reagent sequences of non-human origin. Searches were submitted to an in-house Mascot server (version 2.2.03; Matrix Science).21 Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionine was allowed as a variable modification. Only tryptic peptides were considered, with one missed cleavage permitted. Monoisotopic precursor mass values were used, and only doubly and triply charged precursor ions were considered. Mass tolerances for precursor and fragment ions were 1.5 and 0.5 Da, respectively.
To validate the proteomic data sets generated by GeLC–MS, multiple database search engines and rigorous statistical algorithms at both the peptide and protein level were employed.22,23 To achieve this, data validation was performed using Scaffold (versions Scaffold_2_06_00 and Scaffold_3.1.2; Proteome Software, Portland, OR). Database search files generated by Mascot were imported into Scaffold and further analyzed using the search engine X! Tandem (version 2007.01.01.1) implemented from within Scaffold. X! Tandem searches were conducted against the same protein sequence database and using the same search parameters as the associated Mascot search, except that X! Tandem allowed S-carbamoylmethylcysteine cyclization (pyro-carbamidomethylation of cysteine) or conversion of glutamine or glutamic acid to 2-pyrrolidone-5-carboxylic acid at N-termini as variable modifications by default. Peptide identifications were accepted if they could be established with at least 90% probability as determined by the PeptideProphet algorithm.22 Protein identifications were accepted if they were assigned at least two unique, validated peptides and could be established with at least 99% probability as determined by the ProteinProphet algorithm.23 These thresholds resulted in a protein false discovery rate (FDR) of 0.1% as calculated by Scaffold. Proteins that contained shared peptide matches were grouped in Scaffold to satisfy the principle of parsimony, such that the minimum set of proteins that adequately accounted for all identified peptide sequences was described. Further data analysis allowing oxidation of either lysine or proline as a variable modification in Mascot resulted in the identification of additional peptides, which matched almost exclusively to collagens. These peptides did not increase the number of proteins identified or significantly alter the abundance of any detected protein and, due to score suppression as a result of database searching with multiple variable modifications, these data were not used further.
MS data were converted using PRIDE Converter (version 2.5.3)24 and deposited in the PRIDE database (http://www.ebi.ac.uk/pride)25 under accession numbers 22483–22488.
Quantification Using Spectral Counting
Label-free quantification of relative protein abundance was performed by spectral counting26−28 using all spectra matched to a peptide sequence. Relative protein abundance was calculated on the basis of the unweighted spectral count assigned to each identified protein by Scaffold. The unweighted spectral count includes spectra matched to peptides shared between multiple proteins if there is independent evidence that these proteins are present. To normalize the data, spectral counts were expressed as a percentage of the total number of spectra observed in the entire sample. Mean normalized spectral counts were calculated using data from three independent LX-2 and HFF ECM isolations. Due to the method of protein normalization and to avoid overinterpretation of quantitative comparisons, normalized spectral counts were only compared between HFF and LX-2 for the same protein, and conclusions based on the stoichiometry of components of the ECM within an individual sample were not made.
Gene Ontology (GO) Enrichment Analysis
Official gene symbols were mapped to all protein identifications, and the LX-2 and HFF data sets were analyzed using the online bioinformatic tools available via the Database for Annotation, Visualization and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/home.jsp).29,30 For clarity, only top-level GO terms from the Cellular Component and Biological Process domains, second-level GO terms from the Molecular Function domain, and KEGG Pathway terms were considered. Furthermore, only terms with enrichment value ≥1.5, Bonferroni-corrected P-value <0.05, EASE score (modified Fisher Exact P-value) <0.05, and at least two genes per term were considered. The background data set for the analyses was the Homo sapiens genome, and the most relevant term relating to ECM or cell adhesion is shown for each category.
Hierarchical Clustering Analysis
Agglomerative hierarchical clustering using quantitative data (mean normalized spectral counts) was performed with Cluster 3.0 (C Clustering Library, version 1.37).31 Protein hits were hierarchically clustered on the basis of uncentered Pearson correlation, and distances between hits were computed using a complete-linkage matrix. Clustering results were visualized using Java TreeView (version 1.1.1)32 and MultiExperiment Viewer (version 4.1.01).33
Statistical Analysis of Relative Protein Abundance from MS Data Sets
Statistical analysis of differential spectral count data between samples was performed using QSpec (http://www.nesvilab.org/qspec.php/).34 QSpec uses Bayes statistics to test pairwise differences between spectral count data, which are modeled as observations from a Poisson distribution. Differential relative protein abundances with Bayes factors ≥10 and natural-logarithm-transformed fold changes ≥1.5 were selected. These parameters were chosen to provide a conservative FDR estimate of <5% in accordance with the modeled data of Choi et al.34 For this data set, positive fold changes represent proteins enriched to LX-2, negative fold changes represent proteins enriched to HFF, and values are represented as ln(fold change).
Interaction Network Analysis
Protein–protein interaction (PPI) network analysis was performed essentially as described by Humphries et al.20 The open-source platform Cytoscape (version 2.6.0)35 was used to visualize protein–protein interaction networks. Proteins annotated as part of the ECM or secreted in the UniProt Knowledgebase (http://www.uniprot.org/; 61 proteins in total) were selected and mapped onto the human Protein Interaction Network Analysis interactome (release date fourth March 2010; http://csbi.ltdk.helsinki.fi/pina/home.do),36 which consists of protein–protein interaction data integrated from six public curated databases. Interactions from the ECM-directed protein–protein interaction database MatrixDB37 (http://matrixdb.ibcp.fr) were added manually. It was possible to map 57 of the 61 ECM or secreted proteins onto this interactome. Proteins were assigned by hierarchical clustering as either LX-2-enriched, HFF-enriched, or shared LX-2 and HFF identifications. Clustering assignments were mapped as an attribute onto each protein (node) of the networks and represented visually by node color. Model layouts were constructed using the attribute circle layout implemented in Cytoscape.
Immunofluorescence
Cells were plated onto glass-bottom dishes (MatTek, Ashland, MA) coated with 0.2% (v/v) gelatin. LX-2 or HFF cells were grown for 11 days before preparing CDMs as described above. After DNase I treatment, CDMs were fixed with 3% (w/v) paraformaldehyde for 15 min and then incubated with the appropriate primary and secondary Alexa Fluor antibody conjugates for 60 and 45 min, respectively. Images were collected on a TCS SP5 AOBS inverted confocal (Leica Microsystems GmbH, Wetzlar, Germany) using a 60×/0.50 Plan Fluotar objective. The confocal settings were as follows: pinhole 1 airy unit, scan speed 1000 Hz unidirectional, format 1024 × 1024. Images were collected using the following detection mirror settings: FITC, 494–530 nm; Texas red, 602–665 nm; using the 488 nm (20%) and 594 nm (100%) laser lines for FITC and Texas red, respectively. When acquiring three-dimensional optical stacks, the confocal software was used to determine the optimal number of Z sections, and maximum intensity projections are shown in the results. Images were analyzed and processed using ImageJ (National Institutes of Health, Bethesda, MD) and Photoshop CS (Adobe Systems, Inc., San Jose, CA).
Immunohistochemistry
Archived fibrotic human liver tissue specimens obtained for the clinical staging of chronic hepatitis C infection were used, following approval from the Brent Research Ethics committee. All sections stained were graded as moderately fibrotic (Ishak score 3 to 4), and sections from a total of three patients for Wnt-5a and CYR61 were used. Archived murine fibrotic liver tissue was kindly supplied by Q. Anstee (Imperial College London, U.K.). C57BL/6J mice, aged 6–8 weeks old, were treated with carbon tetrachloride (CCl4) diluted in a corn-oil vehicle and administered by intraperitoneal injection on alternate week days for a period of 4 weeks, in accordance with local ethical approval and the Animal (Scientific Procedures) Act 1986. The dose of CCl4 administered was increased weekly in a stepwise fashion until a maximum dose of 1 mL CCl4 per kilogram body weight was reached after 3 weeks. At completion, animals were culled, and liver tissue was collected. Liver sections from three mice treated with CCl4 to induce liver fibrosis were stained for both CYR61 and Wnt-5a as described below.
All sections were cut at 5 μm from the corresponding formalin-fixed paraffin-embedded tissue blocks. Sections were dewaxed using xylene and then rehydrated in ethanol using a standard histological protocol. Sections were treated with 0.3% (v/v) hydrogen peroxide in methanol for 30 min to block endogenous peroxidase activity and then washed with PBS. For human liver tissue sections, antigen retrieval was performed by microwaving sections in 10 mM citrate buffer, pH 6.0, for 10 min and then washing with PBS. All sections were incubated with 1.5% (v/v) normal goat serum (ABC Staining System; Santa Cruz Biotechnology, Inc.) for 60 min. Sections were then incubated either overnight at 4 °C with anti-Wnt-5a (1:50 dilution for murine sections; 1:100 dilution for human sections), anti-CYR61 (1:100 dilution for murine sections), or anti-fibulin-2 (1:200 dilution for human sections), or for two hours at room temperature with anti-CYR61 (1:200 dilution for human sections). Sections were washed in PBS and incubated with a biotinylated goat anti-rabbit or goat anti-mouse secondary antibody (1:200 dilution; ABC Staining System) for 60 min and then washed again in PBS. Sections were incubated with an avidin-conjugated biotinylated horseradish peroxidase (1:50 dilution; ABC Staining System) for 30 min and then treated with 3,3′-diaminobenzidine tetrahydrochloride for 2 min, counterstained using hematoxylin, dehydrated, and mounted using a nonaqueous mounting medium, and coverslips were applied. Control slides were incubated as described above, but with the omission of the primary antibody. Slides were visualized using an Olympus BX50 microscope (Olympus, Southend-on-Sea, U.K.) under fixed lighting conditions, and images were captured using NIS-Elements (Nikon, Surrey, U.K.).
Results
Preparation and Isolation of HSC-Derived ECM
To gain insights into the etiology of liver fibrosis, we sought to catalogue the composition of fibrotic liver ECM using GeLC–MS. Remodelling of the ECM during liver fibrosis is driven by HSCs, and HSC–ECM interactions regulate HSC activation, proliferation, survival, and cell cycle.7 Isolation of HSC-derived ECM from the liver is, however, confounded by the variety and complexity of tissue structures and contributing cell types. We therefore isolated ECM produced by the HSC cell line LX-2 as an established in vitro model of liver fibrosis.7,18,38
As a further validation of the use of the LX-2 cell line, knowing that the integrin family of adhesion receptors plays a key role in mediating ECM engagement and organization,39 we determined the expression profile of integrins on LX-2 cells by flow cytometry. Using a panel of anti-integrin antibodies, we detected cell-surface expression of α1, α2, α5, α6, αV, β1, β3, and β5 integrin subunits, but not α3, α4, β6, or β8 (Supplementary Figure S1). This integrin expression profile was similar to the previously reported expression of α1, α2, α5, α6, αV, β1, β3, and β4 in primary HSCs,10,40,41 supporting the use of the LX-2 cell line as a model for investigating liver matrix biology.
We next performed experiments to optimize the isolation of CDM from LX-2 cells. We adapted a previously described ECM purification method to isolate CDMs from LX-2 cells by the rapid removal of cellular proteins after 5, 7, 11, or 14 days of culture.16 The synthesis and organization of ECM increased with time, and correspondingly higher levels of ECM were isolated at later time points (Figure 1A). Western blotting analysis of CDM for known components of fibrotic ECM revealed the presence of fibronectin and fibrillin. Enrichment of fibronectin was maximal at 11–14 days of culture, and enrichment of fibrillin was maximal at day 11 (Figure 1B). Thus, for all further analyses, we chose to isolate CDMs after 11 days of culture to coincide with maximal recovery of fibronectin and fibrillin and to reduce the potential impact of cell death that occurred as cells became highly confluent.
Figure 1.

Isolation and characterization of CDMs. CDMs were generated from LX-2 cells after 5, 7, 11, or 14 days. (A,B) Proteins were solubilized, separated by SDS-PAGE and stained with Coomassie Brilliant Blue (A) or Western blotted for fibronectin or fibrillin (B). Mkr denotes the position of molecular mass standards. (C) CDMs from LX-2 cells and HFFs were processed for immunofluorescence, stained with anti-fibronectin (green) and anti-fibrillin antibodies (red), and images were collected by confocal microscopy.
To characterize the isolated CDMs further, we performed immunofluorescence analyses of ECMs generated from LX-2 cells and compared them to HFFs as a non-hepatic myofibroblast control. ECM from both cell types stained positively for fibronectin (Figure 1C), revealing extensive networks of interconnecting fibronectin bundles. By contrast, immunofluorescence staining for fibrillin revealed cell type-specific differences between the CDMs, with increased incorporation of fibrillin into the ECM of LX-2 cells as compared to HFFs (Figure 1C). The staining of fibrillin in LX-2 CDM frequently co-aligned with that of fibronectin (Figure 1C), consistent with the presence of known binding sites for fibrillin within fibronectin.2 Moreover, the increased staining for fibrillin in LX-2-derived ECM is consistent with the previously reported increase in fibrillin within fibrotic liver.42,43 Together, these data support the use of purified LX-2 CDM as an in vitro model to study ECM changes during liver fibrosis.
Proteomic Analysis of LX-2 and HFF CDMs
CDMs isolated from LX-2 and HFF cells were analyzed by GeLC–MS. These experiments identified 277 proteins with ≥99% confidence in LX-2- and HFF-derived ECMs (Supplementary Tables S1 and S2). Cell type-specific proteins and proteins common to both cell types were identified (Figure 2A). Using the unbiased annotation provided by the UniProt Knowledgebase (http://www.uniprot.org/), a total of 61 proteins (22%) were defined as secreted or part of the ECM (Figure 2B). Thirty (49.2%) and 13 (21.3%) of these proteins were detected solely in LX-2 and HFF CDMs, respectively, and 18 (29.5%) proteins were detected in both CDMs (Figure 2B). Importantly, a large number of ECM components with well-defined roles in liver fibrosis were detected in LX-2-derived ECM. GO enrichment analysis was carried out to provide a global, unbiased assessment of the type and function of proteins identified by GeLC–MS analyses of cell-derived ECMs (Supplementary Table S3). Proteins detected in both LX-2- and HFF-derived ECM were enriched for identical GO terms relating to ECM and adhesion from multiple ontology categories (Figure 2C). This finding indicates that the method for isolating cell-derived ECM was effective and contrasts with the majority of MS-based analyses of liver tissue or whole cells, which do not readily detect ECM components.13 Together, these data indicate compositional differences in ECM and secreted proteins found within the ECM derived from the LX-2 cell model of liver fibrosis as compared to the non-fibrotic control.
Figure 2.
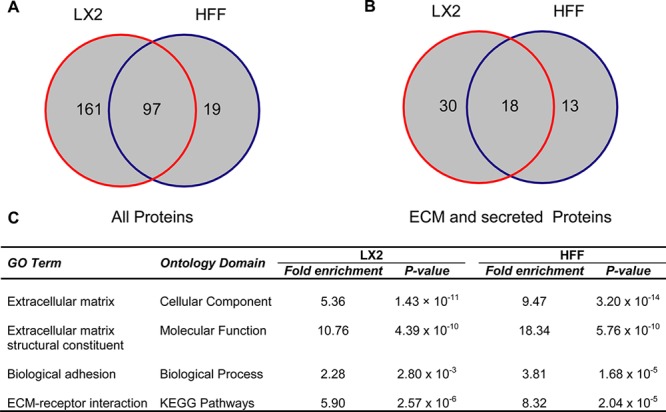
Proteomic analysis of LX-2 and HFF CDMs. (A,B) Venn diagrams display numbers of proteins identified by GeLC–MS analyses of LX-2- and HFF-derived ECM. In total, 277 proteins were identified (A), of which 61 were defined as ECM or secreted using the UniProt Knowledgebase (http://www.uniprot.org/) (B). Numbers indicate proteins identified with ≥99% confidence and ≥2 unique peptides. (C) GO enrichment analysis highlighting significantly enriched terms related to adhesion and ECM. Bonferroni-corrected P-values are displayed, and the background data set for analysis was the whole human genome.
To identify patterns of relative distribution of proteins in LX-2 and HFF CDMs, hierarchical clustering analysis was performed (Figure 3 and Supplementary Tables S4 and S5). On the basis of relative protein abundance, as measured by spectral counting,26−28 unbiased Pearson correlation identified three major groups of proteins, which corresponded to clusters of proteins enriched to HFF CDM (correlation = 0.89; Figure 3B), LX-2 CDM (correlation = 0.95; Figure 3C), or both HFF and LX-2 CDMs (correlation = 0.75; Figure 3D). On the basis of the assignment of proteins to clusters, the ECM and secreted components were distributed as follows: 36 proteins (59.0%) were enriched to LX-2 CDM, 16 proteins (26.2%) were enriched to HFF CDM, and 9 proteins (14.8%) were detected in both LX-2 and HFF CDMs. This quantitative analysis indicated a number of additional proteins that were either under-represented (Figure 3B) or over-represented (Figure 3C) in the ECM derived from LX-2 cells. Furthermore, statistical analysis using the QSpec method34 identified proteins that were significantly enriched in either LX-2 or HFF CDM (Figure 3B–D and Supplementary Table S6). Importantly, a number of proteins previously characterized as being up-regulated during liver fibrosis, such as fibrillin, nidogen, and laminin,44,45were found in the LX-2-enriched cluster and were significantly enriched (FDR ≤ 5%; Figure 3C). We therefore hypothesize that proteins with differential expression patterns identified via these analyses may reflect, or even direct, the mechanism of liver fibrosis.
Figure 3.

Hierarchical clustering of proteins identified by GeLC–MS analysis of LX-2 and HFF CDMs. (A) Complete output of unsupervised hierarchical clustering analysis of identified proteins. Quantitative heat maps display mean spectral counts as a percentage of the total number of spectra identified in each analysis. Associated dendrogram displays hierarchical clustering on the basis of uncentered Pearson correlation using complete linkage. Correlations at selected nodes are indicated. (B–D) ECM and secreted proteins are shown with gene names for HFF-enriched (blue vertical bar; B), LX-2-enriched (red vertical bar; C), and HFF and LX-2 shared (gray vertical bar; D) clusters. Asterisks (B–D) denote ECM and secreted proteins with statistically significant differential abundance between LX-2 and HFF CDMs at 5% FDR (Bayes factor ≥10, natural-log-transformed fold change ≥1.5).
LX-2- and HFF-Derived ECM Interaction Networks
To interrogate the molecular organization of the isolated cell-derived ECM, PPI network analysis was employed. To reduce the complexity of the networks, only the ECM and secreted proteins identified by GeLC–MS were mapped onto a human interactome. A total of 57 of the identified ECM and secreted proteins were mapped, resulting in PPI networks of similar size for both LX-2 CDM (28 proteins) and HFF CDM (21 proteins; Figure 4). Proteins (nodes) in the resulting PPI networks were colored according to the clusters they were assigned by hierarchical clustering (LX-2-enriched, red; HFF-enriched, blue; LX-2 and HFF shared, gray). Notably, the LX-2 network is largely formed from LX-2-enriched proteins (17 of the 28 nodes are red), and the converse is true for the HFF network (8 of the 21 nodes are blue), which suggests the existence of cell type- and/or disease-specific ECM niches. In addition, this analysis highlighted a larger number of components of the “basement membrane toolkit” (laminins, type IV collagens, perlecan, and nidogens46) incorporated into the LX-2 ECM network (10 proteins) in contrast to the HFF ECM network (2 proteins). We speculate that this observation may reflect a characteristic compositional and organizational change in the nature of fibrotic liver ECM. Furthermore, although some components were found in both networks, they exhibited different connectivities within each network. For example, fibulin-2 (FBLN2), a protein known to be up-regulated during liver fibrosis,47−49 was the third most connected node within the LX-2-derived ECM network, but only the 12th most connected in the HFF-derived ECM network. Thus, it seems likely that both protein abundance and connectivity within cell type-specific ECM niches may regulate the function of that tissue, such as the development or progression of fibrosis in the liver.
Figure 4.
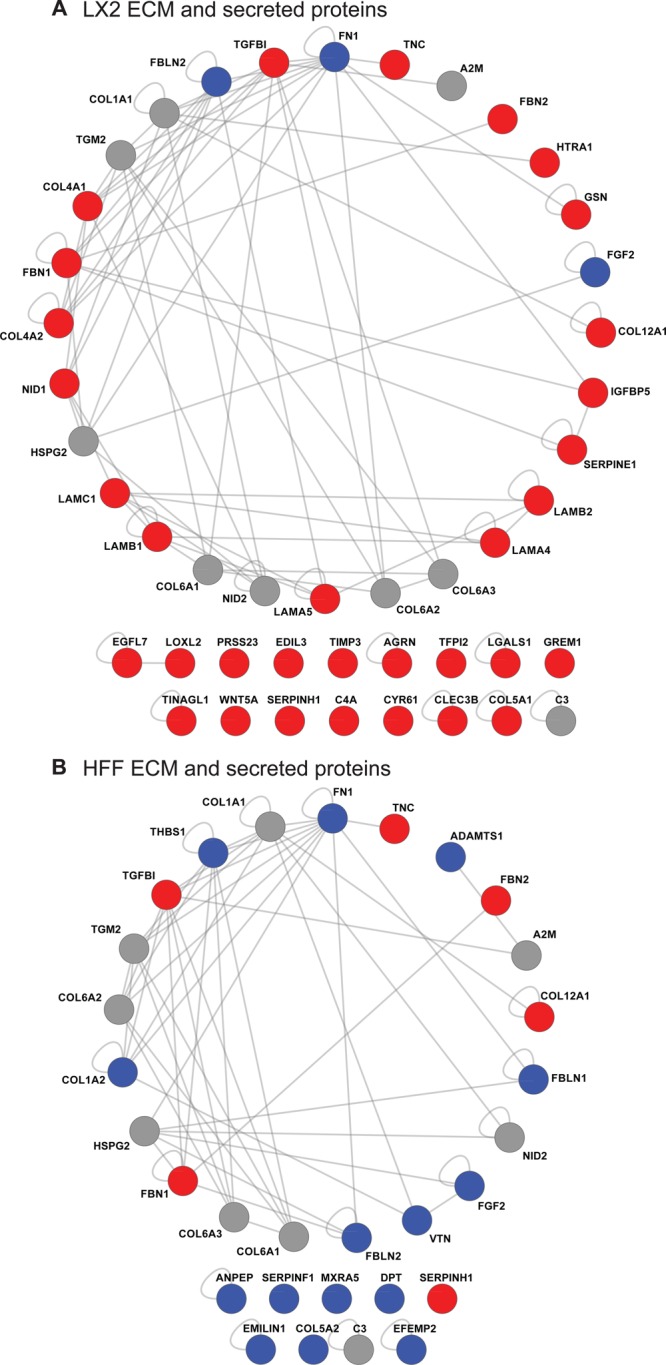
Protein–protein interaction network models for LX-2 and HFF CDMs. (A,B) ECM and secreted proteins identified from LX-2-derived (A) and HFF-derived (B) ECM were mapped onto a human interactome to generate interaction network models. Protein identifications (nodes labeled with gene names) are displayed as a degree-sorted circle (proteins are ordered anticlockwise by descending number of binding partners with the most connected protein at the top). Nodes are colored according to their hierarchical clustering assignment to LX-2-enriched (red), HFF-enriched (blue), or LX-2 and HFF shared (gray) clusters (vertical colored bars in Figure 3). Proteins not incorporated into the largest interconnected network are displayed below the respective network.
Expression of Wnt-5a and CYR61 in Liver Fibrosis
As a proof of principle, and to confirm the relevance of the proteins identified by proteomic analysis of LX-2-derived ECM, immunohistochemical staining of human and mouse fibrotic liver tissue was performed (Figures 5, 6 and Supplementary Figures S2, S3, and S4). A number of proteins identified in LX-2 or HFF CDMs were not incorporated into the PPI networks visualized in Figure 4 due to the lack of current knowledge of their interaction partners. Using spectral counts as a measure of abundance, some of these proteins are likely to represent abundant components of the ECM. In addition, a number of the identified proteins were statistically enriched to LX-2 CDM and have intriguing links with fibrotic processes. For example, WNT5A mRNA levels have been shown to be up-regulated in activated HSCs,50 but differential protein expression has not been reported. Also, CYR61 has been shown to trigger senescence of dermal fibroblasts,51 and senescence of activated HSCs has been shown to limit liver fibrosis.52 We therefore targeted Wnt-5a and CYR61 for validation by immunohistochemical analysis and used fibulin-2 as a positive control.
Figure 5.
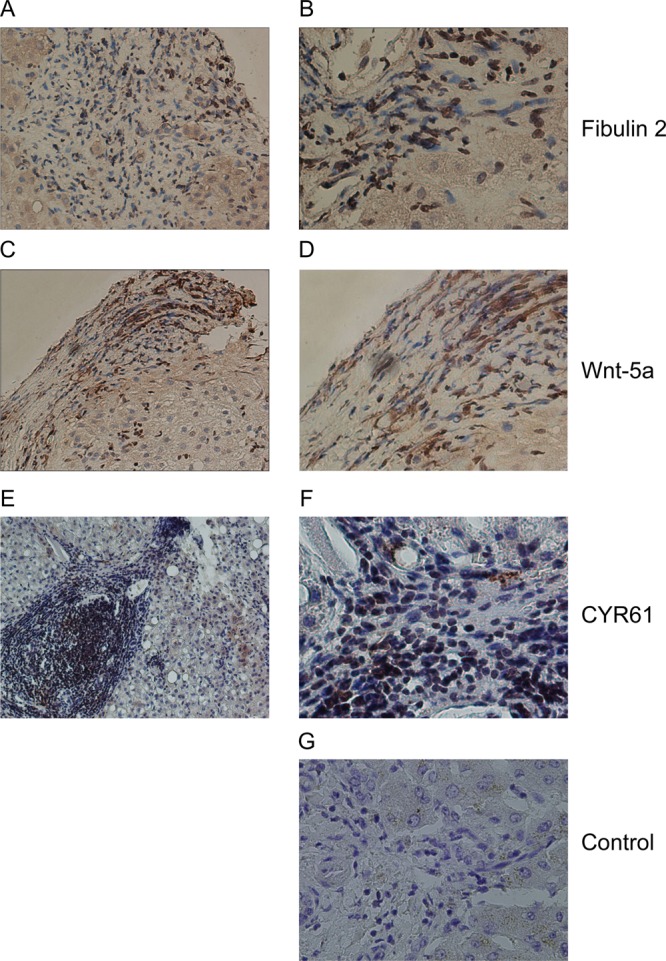
Expression of fibulin-2, Wnt-5a and CYR61 in human fibrotic liver sections. (A–F) Representative images of histological sections demonstrating immunohistochemical staining in human hepatitis C virus-induced liver fibrosis. Fibulin-2 staining (A and B) was observed within fibrotic areas. Wnt-5a (C and D) and CYR61 (E and F) staining was localized to fibrotic septa in human liver tissue. Sections displayed are from different individual patients. Control (G) indicates section stained with the omission of the primary antibody. Images were acquired using 10× (A, C, E) and 20× (B, D, F, G) objectives.
Figure 6.
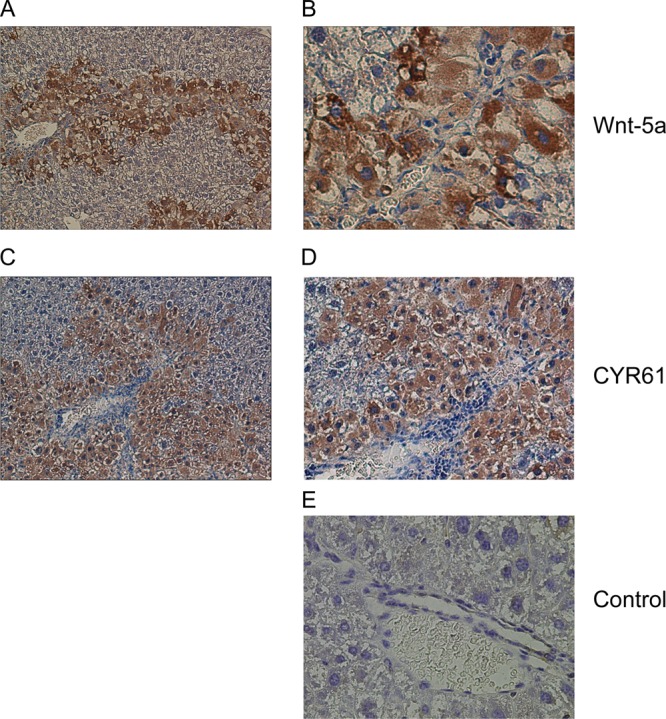
Expression of Wnt-5a and CYR61 in mouse fibrotic liver sections. (A–D) Representative images of histological sections demonstrating immunohistochemical staining in murine CCl4-induced liver fibrosis. Wnt-5a (A and B) and CYR61 (C and D) both heavily stained peri-fibrotic areas in murine liver tissue. Control (E) indicates section stained with the omission of the primary antibody. Images were acquired using 20× (A and C) and 40× (B,D, and E) objectives. Additional negative control tissue, in which the primary antibody was omitted and normal murine liver tissue did not stain positively for Wnt-5a or CYR61 is provided in Supplementary Figure S4.
As expected,49 fibulin-2 staining was localized to fibrotic areas in human hepatitis C virus-induced liver fibrosis tissue sections (Figure 5A and B). Positive staining for Wnt-5a (Figure 5C and D) and CYR61 (Figure 5E and F) was also observed in fibrotic septa from human hepatitis C virus-induced liver fibrosis tissue sections. In addition, both Wnt-5a (Figure 6A and B) and CYR61 (Figure 6,C and D) were found in peri-fibrotic areas in a murine model of liver fibrosis. Taken together, these analyses show that Wnt-5a is expressed in fibrotic areas of human tissue and perifibrotic areas of murine tissue, and CYR61 is found in peri-fibrotic areas of human and murine tissue. Collectively, these data confirm the identification of two novel components of fibrotic liver ECM.
Discussion
The extracellular environment is a key regulator of cell fate and function.53 Indeed, altering the composition of the ECM has previously been shown to induce deactivation of HSCs. In their activated forms, HSCs are postulated to drive fibrotic liver disease, and so remodelling of the fibrotic matrix represents a potential therapeutic strategy.6,54 Antagonism of αVβ3 integrin receptor, for example, has been shown to block binding of HSCs to ECM survival ligands, which induces HSC apoptosis and results in remodelling of the surrounding ECM.10 However, since relatively little is known about the constituents of the fibrotic liver ECM, disruption of binding of all αVβ3 ligands would likely cause catastrophic effects upon a wide range of cell-ECM interactions. Instead, more refined disease- and organ-specific targets of fibrosis need to be identified. A first step toward this goal is the holistic description of the constituents of the fibrotic liver ECM. Our study confirms that such targets can be identified using the GeLC–MS-based proteomics approach described here. Furthermore, the predictive nature of these analyses allows us to hypothesize that a number of the proteins we identified in this study will make excellent candidates for further investigation into the mechanisms, diagnosis, and treatment of fibrotic disease.
As this study of stellate cell ECM was a component of a program to identify suitable therapeutic targets to inhibit liver fibrosis, the control cell line was selected so that proteins or networks that are specific to liver fibrogenesis could be identified in order to avoid the potential adverse effects of systemic fibrogenesis inhibition. Dermal fibroblasts were chosen as their use with the ECM enrichment protocol has been established, resulting in the isolation of ECM that had both structurally and functionally relevant composition.55,56 We therefore anticipated a similarly functionally relevant LX-2 ECM to be isolated and characterized. Furthermore, the use of HFFs permitted an organ-specific comparison to be made as it is well established that liver fibrogenesis differs from general fibrosis (which is represented here by HFFs). A direct comparison between quiescent and activated HSCs was not possible due to technical limitations as freshly isolated quiescent HSCs activate spontaneously in culture. An alternative approach of inactivating HSCs by culturing cells on Matrigel8,9 was not used because Matrigel contamination of cell-derived ECM would confound the MS analysis.
It is important to note that a number of the proteins identified in this study have well reported links with fibrotic processes occurring in both liver and other organs. For example, fibrillin, previously implicated in the storage and activation of transforming growth factor-β (TGF-β), is a known profibrotic cytokine that drives HSC activation and secretion of ECM57,58 and is up-regulated in both liver fibrosis42,43 and sclerosis.59 Our analysis revealed significantly increased abundances of both fibrillins 1 and 2 in the LX-2-derived ECM as compared to the control. Similarly, plasminogen activator inhibitor 1 (PAI-1/SERPINE1), which was up-regulated in LX-2-derived ECM, has been shown to be a profibrotic matricellular protein with roles in cardiovascular and liver fibrotic disease,60 the knockout of which protects against experimental liver fibrosis.61 Other proteins found in our study to be specifically up-regulated in LX-2-derived ECM included LRRC17, a protein whose gene expression is increased in Dupuytren’s disease (a fibrotic disorder affecting the hand, characterized by abnormal proliferation of fibroblasts and their differentiation into myofibroblasts62), and agrin, a proteoglycan shown to stain specific regions of cirrhotic liver.63 Indeed, screens for alterations in gene expression during liver fibrosis have identified up-regulation of genes for gremlin, TGF-β-induced protein ig-h3 (BIGH3) and insulin-like growth factor-binding protein 5 (IGFBP-5),50,64 the protein products for all of which were overexpressed in LX-2-derived ECM. The significance of this validation is revealed by consideration of gremlin, which is a bone morphogenetic protein (Bmp) antagonist. Bmp family members are known to play a role in liver homeostasis by blocking TGF-β-related signaling pathways.65 Bmp antagonists, such as gremlin, could therefore play a profibrotic role by promoting TGF-β activity, and targeting their action has been postulated as a potential therapy for liver and kidney fibrosis.64 Thus, our GeLC–MS-based proteomics approach adds a new level of selective refinement to previous global analytical approaches, such as array-based gene expression studies, and identifies biologically significant protein targets for potential downstream investigations.
In contrast to candidates overexpressed in LX-2 CDM, proteins whose expression was down-regulated in LX-2-derived ECM may also be important for fibrotic progression. For example, elastin microfibril interfacer 1 (EMILIN-1) was absent from LX-2-derived ECM but robustly identified in the control dermal ECM. This may have important consequences for fibrosis as EMILIN-1 has been described as a negative regulator of TGF-β signaling that prevents maturation of pro-TGF-β by furin convertases, thereby regulating blood pressure homeostasis.66 EMILIN-1 may thus have a wider role in fibrosis, reinforcing the notion that proteins observed to be either up- or down-regulated in liver ECM could have important roles in the mechanism of fibrosis.
Two novel fibrotic protein constituents identified through this study were also validated in fibrotic liver, namely, CYR61 and Wnt-5a. CYR61 is a member of the CCN (cysteine-rich protein, connective tissue growth factor, and nephroblastoma overexpressed gene) family of matricellular proteins that typically signal through integrins to regulate a variety of disease processes, including cancer, inflammation, and for connective tissue growth factor (also known as CCN2) in particular, fibrosis.67,68 CYR61 (also known as CCN1) has not previously been reported as a constituent of fibrotic liver but is known to function as a ligand for a variety of integrins expressed by HSCs.68 Intriguingly, we detected by GeLC–MS increased abundance of CYR61 in LX-2-derived ECM as compared to the control, and we were able to validate this finding in both human and murine fibrotic liver samples. Activation of cellular senescence could be an attractive alternative antifibrotic therapy as senescence of activated HSCs has been shown to limit liver fibrosis.52 Indeed, senescence of dermal fibroblasts has been shown to be triggered by CYR61 signaling via integrin receptors.51 The relationship between increased levels of CYR61 in fibrotic tissue and its reported antifibrotic function requires further investigation but may reflect tissue-specific differences in CYR61 function, altered CYR61 signaling in fibrosis or a failed attempt by HSCs to activate senescence, and down-regulate fibrosis, in response to profibrotic insult.
In a similar manner to CYR61, Wnt-5a was found to be increased in LX-2-derived ECM and fibrotic liver in vivo. In a gene expression analysis of quiescent and activated rat HSCs, Wnt5a mRNA levels were up-regulated in activated HSCs,50 although up-regulation of Wnt-5a protein expression was not confirmed. We have therefore extended the findings of this previous study,50 and we describe here the expression of Wnt-5a protein in both a murine model of liver fibrosis and human fibrotic tissue. Wnt-5a is one member of a family of 19 secreted glycoproteins that play essential roles in many aspects of embryonic patterning, cell specification, cell growth, and differentiation.69,70 A role for Wnt-5a in fibrosis was suggested by its high expression level in fibroblasts taken from patients with interstitial lung fibrosis.71 Wnt-5a was postulated to act as a regulator of fibroblast proliferation and to induce resistance to apoptosis, as had previously been reported in human dermal fibroblasts.72 These findings are consistent with the hypothesis that Wnt-5a plays an analogous role in liver fibrosis by stimulating proliferation and inhibiting apoptosis of HSCs. Alternatively, given that the LX-2 cell line is an immortalized HSC line,38 detection of Wnt-5a may reflect residual neoplastic characteristics likely to remain present in such cells. The fact that expression of Wnt-5a has also been reported to alter during progression from cirrhosis to hepatocellular cancer73 suggests a possible molecular link between activated HSCs and hepatocellular carcinoma, reflecting the well established clinical association. In this regard, these data support the general notion that changes in hepatocellular carcinoma ECM composition could be used to aid diagnosis or treatment14 and suggest that altered expression of Wnt-5a may represent an urgently required biomarker for early detection of fibrosis and transition from late-stage fibrosis to cancer.73 In either case, validation of these possibilities is required using both primary derived HSCs and biopsy samples at different stages of disease pathogenesis ranging from quiescence through fibrosis to cancer. As CYR61 and Wnt-5a are both secreted matricellular proteins up-regulated in liver fibrosis, they represent ideal candidates as new type I, or direct, biomarkers for further evaluation.
Conclusions
In this study, the ECM proteins produced by HSCs have been catalogued using GeLC–MS-based proteomics coupled to an ECM-enrichment strategy and an in vitro model of liver fibrosis. Using this approach, the majority of all previously reported canonical ECM protein constituents of human fibrotic liver tissue were identified.74 Furthermore, two novel components of fibrotic liver tissue, CYR61 and Wnt-5a, have been identified and validated in vivo. These results confirm the biological and potential clinical relevance of this new strategy. Understanding the molecular changes underpinning the pathological process of fibrosis has been an unmet challenge to investigators for decades. We propose that the ECM protein cataloguing technique described here could present researchers with a valuable tool to advance the field. Application of this strategy to different in vitro disease models could therefore significantly advance the identification of tissue- and disease state-specific ECM proteins and provide the first step toward determining the mechanisms underlying fibrosis and the identification of novel therapeutic targets or biomarkers.
Acknowledgments
We thank Stacey Warwood (University of Manchester) for MS data acquisition, Emma Keevill (University of Manchester) for assistance with MS sample preparation, Jane Kott (University of Manchester) for assistance with microscopy, and Mike Jackson (University of Manchester) for flow cytometric data acquisition. We acknowledge Rebecca Aucott (University of Edinburgh), John Iredale (University of Edinburgh), and Scott Friedman (Mount Sinai Medical Center, New York, NY) for providing the LX-2 cell line; Kath Clark (University of Leicester) for providing the HFF cells; Quentin Anstee (Imperial College London) for providing the archived murine fibrotic liver tissue; and Cay Kielty (University of Manchester) for providing the fibrillin antibody. This work was supported by grants 045225 and 074941 from the Wellcome Trust (to M.J.H.). The Biomolecular Analysis Facility mass spectrometer and Bioimaging Facility microscopes used in this study were purchased with grants from the Biotechnology and Biological Sciences Research Council, Wellcome Trust. and the University of Manchester Strategic Fund. S.T.R. was funded by a National Institute for Health Research Academic Clinical Fellowship. M.T., R.D.G., and A.D. are grateful to the NIHR Biomedical Research Centre funding to Imperial College Healthcare for their infrastructure support.
Glossary
Abbreviations
- Bmp
bone morphogenetic protein
- CDM
cell-derived matrix
- DMEM
Dulbecco’s minimal essential medium
- ECM
extracellular matrix
- EMILIN-1
elastin microfibril interfacer 1
- FDR
false discovery rate
- GO
Gene Ontology
- HFF
human foreskin fibroblast
- HSC
hepatic stellate cell
- IgG
immunoglobulin G
- PPI
protein–protein interaction
- TGF-β
transforming growth factor-β
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interest.
Author Contributions
⊥ These authors contributed equally to this work.
Author Present Address
# Department of Medicine, University of Cambridge, Hills Road,Cambridge CB2 OQQ, U.K.
Supplementary Material
References
- Frantz C.; Stewart K. M.; Weaver V. M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R. O. The extracellular matrix: not just pretty fibrils. Science 2009, 326, 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gressner A. M.; Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J. Cell. Mol. Med. 2006, 10, 76–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller R.; Brenner D. A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox T. R.; Erler J. T. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman S. L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iredale J. P. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Invest. 2007, 117, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaso E.; Ikeda K.; Eng F. J.; Xu L. M.; Wang L. H.; Lin H. C.; Friedman S. L. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J. Clin. Invest. 2001, 108, 1369–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaca M. D. A.; Zhou X. Y.; Issa R.; Kiriella K.; Iredale J. P.; Benyon R. C. Basement membrane-like matrix inhibits proliferation and collagen synthesis by activated rat hepatic stellate cells: evidence for matrix-dependent deactivation of stellate cells. Matrix Biol. 2003, 22, 229–239. [DOI] [PubMed] [Google Scholar]
- Zhou X. Y.; Murphy F. R.; Gehdu N.; Zhang J. L.; Iredale J. P.; Benyon R. C. Engagement of alpha(v)beta(3) integrin regulates proliferation and apoptosis of hepatic stellate cells. J. Biol. Chem. 2004, 279, 23996–24006. [DOI] [PubMed] [Google Scholar]
- Snowdon V. K.; Fallowfield J. A. Models and mechanisms of fibrosis resolution. Alcohol: Clin. Exp. Res. 2011, 35, 794–799. [DOI] [PubMed] [Google Scholar]
- Cowan M. L.; Rahman T. M.; Krishna S. Proteomic approaches in the search for biomarkers of liver fibrosis. Trends Mol. Med. 2010, 16, 171–183. [DOI] [PubMed] [Google Scholar]
- Diamond D. L.; Proll S. C.; Jacobs J. M.; Chan E. Y.; Camp D. G.; Smith R. D.; Katze M. G. HepatoProteomics: applying proteomic technologies to the study of liver function and disease. Hepatology 2006, 44, 299–308. [DOI] [PubMed] [Google Scholar]
- Lai K. K. Y.; Shang S.; Lohia N.; Booth G. C.; Masse D. J.; Fausto N.; Campbell J. S.; Beretta L. Extracellular matrix dynamics in hepatocarcinogenesis: a comparative proteomics study of PDGFC transgenic and Pten null mouse models. PLoS Genet. 2011, 7, e1002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R. The extracellular matrix: an underexplored but important proteome. Expert Rev. Proteomics 2010, 7, 803–806. [DOI] [PubMed] [Google Scholar]
- Beacham D. A.; Amatangelo M. D.; Cukierman E.. Preparation of extracellular matrices produced by cultured and primary fibroblasts. Curr. Protoc. Cell Biol. 2001, Chapter 10, Unit 10.9 [DOI] [PubMed] [Google Scholar]
- Todorovic V.; Desai B. V.; Eigenheer R. A.; Yin T. F.; Amargo E. V.; Mrksich M.; Green K. J.; Patterson M. J. S. Detection of differentially expressed basal cell proteins by mass spectrometry. Mol. Cell. Proteomics 2010, 9, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.; Hui A. Y.; Albanis E.; Arthur M. J.; O’Byrne S. M.; Blaner W. S.; Mukherjee P.; Friedman S. L.; Eng F. J. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A.; Wilm M.; Vorm O.; Mann M. Mass spectrometric sequencing of proteins from silver stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Humphries J. D.; Byron A.; Bass M. D.; Craig S. E.; Pinney J. W.; Knight D.; Humphries M. J. Proteomic analysis of integrin-associated complexes identifies RCC2 as a dual regulator of Rac1 and Arf6. Sci. Signal. 2009, 2, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J. C.; Creasy D. M.; Cottrell J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [DOI] [PubMed] [Google Scholar]
- Keller A.; Nesvizhskii A. I.; Kolker E.; Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A. I.; Keller A.; Kolker E.; Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Barsnes H.; Vizcaíno J. A.; Eidhammer I.; Martens L. PRIDE Converter: Making proteomics data-sharing easy. Nat. Biotechnol. 2009, 27, 598–599. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Côté R.; Reisinger F.; Barsnes H.; Foster J. M.; Rameseder J.; hermjakob H.; Martens L. The Proteomics Identifications database: 2010 update. Nucleic Acids Res. 2010, 38, D736–D742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. B.; Sadygov R. G.; Yates J. R. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004, 76, 4193–4201. [DOI] [PubMed] [Google Scholar]
- Old W. M.; Meyer-Arendt K.; Aveline-Wolf L.; Pierce K. G.; Mendoza A.; Sevinsky J. R.; Resing K. A.; Ahn N. G. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 2005, 4, 1487–1502. [DOI] [PubMed] [Google Scholar]
- Zybailov B.; Coleman M. K.; Florens L.; Washburn M. P. Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal. Chem. 2005, 77, 6218–6224. [DOI] [PubMed] [Google Scholar]
- Dennis G.; Sherman B. T.; Hosack D. A.; Yang J.; Gao W.; Lane H. C.; Lempicki R. A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, R60. [PubMed] [Google Scholar]
- Huang D. W.; Sherman B. T.; Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [DOI] [PubMed] [Google Scholar]
- de Hoon M. J. L.; Imoto S.; Nolan J.; Miyano S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- Saldanha A. J. Java Treeview—extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Saeed A. I.; Sharov V.; White J.; Li J.; Liang W.; Bhagabati N.; Braisted J.; Klapa M.; Currier T.; Thiagarajan M.; Sturn A.; Snuffin M.; Rezantsev A.; Popov D.; Ryltsov A.; Kostukovich E.; Borisovsky I.; Liu Z.; Vinsavich A.; Trush V.; Quackenbush J. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [DOI] [PubMed] [Google Scholar]
- Choi H.; Fermin D.; Nesvizhskii A. I. Significance analysis of spectral count data in label-free shotgun proteomics. Mol. Cell Proteomics 2008, 7, 2373–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P.; Markiel A.; Ozier O.; Baliga N. S.; Wang J. T.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U.; Worm U.; Lalowski M.; Haenig C.; Brembeck F. H.; Goehler H.; Stroedicke M.; Zenkner M.; Schoenherr A.; Koeppen S.; Timm J.; Mintzlaff S.; Abraham C.; Bock N.; Kietzmann S.; Goedde A.; Toksoz E.; Droege A.; Krobitsch S.; Korn B.; Birchmeier W.; Lehrach H.; Wanker E. E. A human protein-protein interaction network: A resource for annotating the proteome. Cell 2005, 122, 957–968. [DOI] [PubMed] [Google Scholar]
- Chautard E.; Ballut L.; Thierry-Mieg N.; Ricard-Blum S. MatrixDB a database focused on extracellular protein-protein and protein-carbohydrate interactions. Bioinformatics 2009, 25, 690–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann J.; Gressner A. M.; Weiskirchen R. Immortal hepatic stellate cell lines: useful tools to study hepatic stellate cell biology and function?. J. Cell. Mol. Med. 2007, 11, 704–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries J. D.; Byron A.; Humphries M. J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carloni V.; Romanelli R. G.; Pinzani M.; Laffi G.; Gentilini P. Expression and function of integrin receptors for collagen and laminin in cultured human hepatic stellate cells. Gastroenterology 1996, 110, 1127–1136. [DOI] [PubMed] [Google Scholar]
- Milliano M. T.; Luxon B. A. Initial signaling of the fibronectin receptor (alpha(5)beta(1) integrin) in hepatic stellate cells is independent of tyrosine phosphorylation. J. Hepatol. 2003, 39, 32–37. [DOI] [PubMed] [Google Scholar]
- Dubuisson L.; Lepreux S.; Bioulac-Sage P.; Balabaud C.; Costa A. M. A.; Rosenbaum J.; Desmouliere A. Expression and cellular localization of fibrillin-1 in normal and pathological human liver. J. Hepatol. 2001, 34, 514–522. [DOI] [PubMed] [Google Scholar]
- Lorena D.; Darby I. A.; Reinhardt D. P.; Sapin V.; Rosenbaum J.; Desmouliere A. Fibrillin-1 expression in normal and fibrotic rat liver and in cultured hepatic fibroblastic cells: modulation by mechanical stress and role in cell adhesion. Lab. Invest. 2004, 84, 203–212. [DOI] [PubMed] [Google Scholar]
- Schuppan D.; Ruehl M.; Somasundaram R.; Hahn E. G. Matrix as a modulator of hepatic fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [DOI] [PubMed] [Google Scholar]
- Ramadori G.; Knittel T.; Saile B. Fibrosis and altered matrix synthesis. Digestion 1998, 59, 372–375. [DOI] [PubMed] [Google Scholar]
- Hynes R. O. The evolution of metazoan extracellular matrix. J. Cell Biol. 2012, 196, 671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knittel T.; Kobold D.; Piscaglia F.; Saile B.; Neubauer K.; Mehde M.; Timpl R.; Ramadori G. Localization of liver myofibroblasts and hepatic stellate cells in normal and diseased rat livers: distinct roles of (myo-)fibroblast subpopulations in hepatic tissue repair. Histochem. Cell Biol. 1999, 112, 387–401. [DOI] [PubMed] [Google Scholar]
- Knittel T.; Kobold D.; Saile B.; Grundmann A.; Neubauer K.; Piscaglia F.; Ramadori G. Rat liver myofibroblasts and hepatic stellate cells: Different cell populations of the fibroblast lineage with fibrogenic potential. Gastroenterology 1999, 117, 1205–1221. [DOI] [PubMed] [Google Scholar]
- Piscaglia F.; Dudas J.; Knittel T.; Di Rocco P.; Kobold D.; Saile B.; Zocco M. A.; Timpl R.; Ramadori G. Expression of ECM proteins fibulin-1 and −2 in acute and chronic liver disease and in cultured rat liver cells. Cell Tissue Res. 2009, 337, 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F.; Parsons C. J.; Stefanovic B. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J. Hepatol. 2006, 45, 401–409. [DOI] [PubMed] [Google Scholar]
- Jun J. I.; Lau L. F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V.; Yon M.; Dickins R. A.; Hearn S.; Simon J.; Miething C.; Yee H.; Zender L.; Lowe S. W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan M. R.; Humphries M. J.; Bass M. D. Synergistic control of cell adhesion by integrins and syndecans. Nat. Rev. Mol. Cell Biol. 2007, 8, 957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallowfield J. A.; Kendall T. J.; Iredale J. P. Reversal of fibrosis: no longer a pipe dream?. Clin. Liver Dis. 2006, 10, 481–497. [DOI] [PubMed] [Google Scholar]
- Cukierman E.; Pankov R.; Stevens D. R.; Yamada K. M. Taking cell-matrix adhesions to the third dimension. Science 2001, 294, 1708–12. [DOI] [PubMed] [Google Scholar]
- Bass M. D.; Roach K. A.; Morgan M. R.; Mostafavi-Pour Z.; Schoen T.; Muramatsu T.; Mayer U.; Ballestrem C.; Spatz J. P.; Humphries M. J. Syndecan-4-dependent Rac1 regulation determines directional migration in response to the extracellular matrix. J. Cell Biol. 2007, 177, 527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Border W. A.; Noble N. A. Transforming growth-factor-beta in tissue fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292. [DOI] [PubMed] [Google Scholar]
- Friedman S. L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000, 275, 2247–2250. [DOI] [PubMed] [Google Scholar]
- Brinckmann J.; Hunzelmann N.; Kahle B.; Rohwedel J.; Kramer J.; Gibson M. A.; Hubmacher D.; Reinhardt D. P. Enhanced fibrillin-2 expression is a general feature of wound healing and sclerosis: potential alteration of cell attachment and storage of TGF-beta. Lab. Invest. 2010, 90, 739–752. [DOI] [PubMed] [Google Scholar]
- Samarakoon R.; Goppelt-Struebe M.; Higgins P. J. Linking cell structure to gene regulation: Signaling events and expression controls on the model genes PAI-1 and CTGF. Cell. Signalling 2010, 22, 1413–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteel G. E. New role of plasminogen activator inhibitor-1 in alcohol-induced liver injury. J. Gastroenterol. Hepatol. 2008, 23, S54–S59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L. C.; Zhang A. Y.; Chong A. K.; Pham H.; Longaker M. T.; Chang J. Expression of a novel gene, MafB, in Dupuytren’s disease. J. Hand Surg. Am. 2006, 31, 211–218. [DOI] [PubMed] [Google Scholar]
- Tatrai P.; Dudas J.; Batmunkh E.; Mathe M.; Zalatnai A.; Schaff Z.; Ramadori G.; Kovalszky I. Agrin, a novel basement membrane component in human and rat liver, accumulates in cirrhosis and hepatocellular carcinoma. Lab. Invest. 2006, 86, 1149–1160. [DOI] [PubMed] [Google Scholar]
- Boers W.; Aarrass S.; Linthorst C.; Pinzani M.; Elferink R. O.; Bosma P. Transcriptional profiling reveals novel markers of liver fibrogenesis: Gremlin and insulin-like growth factor-binding proteins. J. Biol. Chem. 2006, 281, 16289–16295. [DOI] [PubMed] [Google Scholar]
- Weiskirchen R.; Meurer S. K.; Gressner O. A.; Herrmann J.; Borkham-Kamphorst E.; Gressner A. M. BMP-7 as antagonist of organ fibrosis. Front. Biosci. 2009, 14, 4992–5012. [DOI] [PubMed] [Google Scholar]
- Zacchigna L.; Vecchione C.; Notte A.; Cordenonsi M.; Dupont S.; Maretto S.; Cifelli G.; Ferrari A.; Maffei A.; Fabbro C.; Braghetta P.; Marino G.; Selvetella G.; Aretini A.; Colonnese C.; Bettarini U.; Russo G.; Soligo S.; Adorno M.; Bonaldo P.; Volpin D.; Piccolo S.; Lembo G.; Bressan G. M. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell 2006, 124, 929–942. [DOI] [PubMed] [Google Scholar]
- Kular L.; Pakradouni J.; Kitabgi P.; Laurent M.; Martinerie C. The CCN family: A new class of inflammation modulators?. Biochimie 2011, 93, 377–388. [DOI] [PubMed] [Google Scholar]
- Chen C. C.; Lau L. F. Functions and mechanisms of action of CCN matricellular proteins. Int. J. Biochem. Cell Biol. 2009, 41, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan C. Y.; Nusse R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [DOI] [PubMed] [Google Scholar]
- Kikuchi A.; Yamamoto H.; Sato A. Selective activation mechanisms of Wnt signaling pathways. Trends Cell Biol. 2009, 19, 119–129. [DOI] [PubMed] [Google Scholar]
- Vuga L. J.; Ben-Yehudah A.; Kovkarova-Naumovski E.; Oriss T.; Gibson K. F.; Feghali-Bostwick C.; Kaminski N. WNT5A is a regulator of fibroblast proliferation and resistance to apoptosis. Am. J. Respir. Cell Mol. Biol. 2009, 41, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii K.; Nishizawa K.; Kawasaki A.; Yamashita Y.; Katada M.; Ito M.; Nishimoto I.; Terashita K.; Aiso S.; Matsuoka M. Anti-apoptotic action of Wnt5a in dermal fibroblasts is mediated by the PKA signaling pathways. Cell. Signalling 2008, 20, 1256–1266. [DOI] [PubMed] [Google Scholar]
- Liu X. H.; Pan M. H.; Lu Z. F.; Wu B.; Rao Q.; Zhou Z. Y.; Zhou X. J. Expression of Wnt-5a and its clinicopathological significance in hepatocellular carcinoma. Dig. Liver Dis. 2008, 40, 560–567. [DOI] [PubMed] [Google Scholar]
- Gressner O. A.; Rizk M. S.; Kovalenko E.; Weiskirchen R.; Gressner A. M. Changing the pathogenetic roadmap of liver fibrosis? Where did it start; where will it go?. J. Gastroenterol. Hepatol. 2008, 23, 1024–1035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.