

Abstract
Significance: S-nitrosothiol formation and protein S-nitrosation is an important nitric oxide (NO)-dependent signaling paradigm that is relevant to almost all aspects of cell biology, from proliferation, to homeostasis, to programmed cell death. However, the mechanisms by which S-nitrosothiols are formed are still largely unknown, and there are gaps of understanding between the known chemical biology of S-nitrosothiols and their reported functions. Recent Advances: This review attempts to describe the biological chemistry of S-nitrosation and to point out where the challenges lie in matching the known chemical biology of these compounds with their reported functions. The review will detail new discoveries concerning the mechanisms of the formation of S-nitrosothiols in biological systems. Critical Issues: Although S-nitrosothiols may be formed with some degree of specificity on particular protein thiols, through un-catalyzed chemistry, and mechanisms for their degradation and redistribution are present, these processes are not sufficient to explain the vast array of specific and targeted responses of NO that have been attributed to S-nitrosation. Elements of catalysis have been discovered in the formation, distribution, and metabolism of S-nitrosothiols, but it is less clear whether these represent a specific network for targeted NO-dependent signaling. Future Directions: Much recent work has uncovered new targets for S-nitrosation through either targeted or proteome-wide approaches There is a need to understand which of these modifications represent concerted and targeted signaling processes and which is an inevitable consequence of living with NO. There is still much to be learned about how NO transduces signals in cells and the role played by protein S-nitrosation. Antioxid. Redox Signal. 17, 969–980.
Introduction
S-nitrosation, the modification of a thiol group that forms an S-nitrosothiol, has been a recognized reaction for more than 100 years. Originally termed thionitrites (as nitrous acid thioesters), S-nitrosothiols were found to be much less stable than corresponding esters of alcohols (alkyl nitrites). As early as 1840 (75), they were originally observed as transient color changes on the treatment of thiols with nitrous acid, and can be chemically synthesized by reacting thiols with nitrosyl chloride, dinitrogen trioxide, dinitrogen tetraoxide, and nitrous acid. Alkyl S-nitrosothiols are generally pink/red in color and have been difficult to isolate as powders. A fortunate exception to this is S-nitrosoglutathione (GSNO), which can be easily synthesized and isolated as a pure compound. Similar to tertiary S-nitrosothiols, such as (Ph)3CSNO and t-BuSNO, S-nitroso-N-acetyl penicillamine (SNAP) is green in color and is one of the most commonly used S-nitrosothiols in biological applications (See Fig. 1 for the structures of S-nitrosothiols that are commonly used in biological experiments).
FIG. 1.

Chemical structures of common S-nitrosothiols. D-CysNO, S-nitroso-D-cysteine; GSNO, S-nitrosoglutathione; L-CysNO, S-nitroso-l-cysteine; SNAP, S-nitroso-N-acetyl penicillamine.
The formation of S-nitrosothiols on the cysteine residues of proteins had been speculated on (75), and low-molecular-mass S-nitrosothiols had been used as chemical agents in biological systems [mainly for studying vessel relaxation (48)]. However, the idea that post-translational modification of proteins by the formation of S-nitrosothiols is a major biological consequence of nitric oxide (NO) formation was first advanced by Stamler and co-workers from their seminal discovery that S-nitrosothiols were present in the human circulation (89). This group has advanced the study and understanding of S-nitrosothiol formation in biology to what is now recognized as a major NO-dependent post-translational modification with implications in multiple pathologies (43). This review will not attempt to examine the biological and pathological implications of S-nitrosation, but will rather focus on the relationship between biological observations and the physical and chemical nature of NO and will highlight where these appear somewhat at odds.
The major pathways for the chemical reaction of NO with proteins are illustrated in Figure 2. By far, the fastest reactions of NO with proteins occur at inorganic centers, and, in particular, in iron-containing prosthetic groups. The rate constants of NO with ferrous heme groups are in the order of 107 M−1s−1. Only if an amino-acid residue has been oxidized to a radical can reactivity compete with the organic moiety of the protein. Studies have demonstrated that such reactions can give rise to nitrated and nitrosated products. NO is chemically unable to directly oxidize amino-acid side chains at any meaningful biological rate, and, therefore, NO-dependent amino-acid oxidation occurs via secondary reactions after the oxidation of NO to nitrogen dioxide, dinitrogen trioxide, or peroxynitrite. The oxidation of NO may occur through a reaction with oxygen (as will be extensively discussed next), through a reaction with superoxide, and through a reaction with metal centers (such as peroxidases). S-nitrosation is, therefore, an indirect reaction of NO that results in a chemical modification of a thiol group. It is not a reversible association of NO with a thiol, and it is essential to comprehend this difference in order to understand the biological chemistry of S-nitrosation. Despite recent advances, there is still much to be learned about how NO transduces signals in biological systems and what role S-nitrosation plays in such processes.
FIG. 2.

Protein-based targets of NO and its oxidation products in biological systems. NO, nitric oxide.
The Chemistry of the NO, Oxygen, and Thiol System
There is little doubt that the concept of NO-dependent signal transduction through the formation of S-nitrosothiols derives, at least in part, from the observation that a mixture of NO, oxygen, and a thiol generates an S-nitrosothiol (36, 99). Consequently, this review will examine this complex reaction system in some detail. The importance of these reactions to biological processes is under some debate; however, an understanding of the underlying chemistry presented here is essential in order to understand how NO can and cannot act in biologically relevant conditions.
A schematic of the possible reactions that can occur when NO is released in the presence of oxygen and a thiol is shown in Figure 3 and, as can be seen, is somewhat complex. The overall mechanism can be divided into three main pathways, as illustrated in the figure. A critical distinction between pathways 1 and 2, and pathway 3 (which will be discussed later), is that these two pathways rely on the initial oxidation of NO by oxygen. This reaction has been studied in some detail (17, 35, 98) and can be subdivided into three fundamental reactions given by equations 1 and 2. NO will reversibly associate
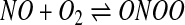 |
[1] |
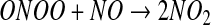 |
[2] |
FIG. 3.

Pathways of S-nitrosothiol formation from NO, oxygen, and GSH. Reprinted with permission from Keszler et al. (58). GSH, glutathione. (To see this illustration in color the reader is referred to the web version of this article at www.liebertpub.com/ars).
with oxygen to generate a peroxynitrite radical, which may then react with a second NO to generate two molecules of nitrogen dioxide. Kinetically, this reaction is limited by an apparent third-order rate law, as depicted in equation 3. The value of k has been determined to be 2.5×106 M−2s−1 (36). The
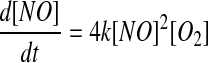 |
[3] |
dependence of this rate on the squared concentration of NO underpins many of the problems and misconceptions concerning NO reactivity. At high concentrations of NO (μM to mM), this reaction takes place in seconds or less, whereas at concentrations in the physiological range (≤100 nM), the reaction is much slower. For example, the initial rate for the reaction between 1 mM NO and 250 μM oxygen is about 2.5 mM/s, whereas at more physiological levels of 10 nM NO with 50 μM oxygen, it is 50 fM/s. The first half life in the former case is about 0.5 s, and in the latter, it is about 50 h. The administration of NO at concentrations above physiological levels can, therefore, promote a chemistry that is too slow to occur under physiological conditions. In addition, when a solution of NO is added to an experiment in a small volume, but at a high concentration, it may react before it has time to mix (so-called bolus addition effects), giving rise to perceived unexpected chemical reactivity (40, 55, 104).
The next step of the nitrosation process is where pathways 1 and 2 (Fig. 3) diverge. In pathway 1,
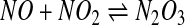 |
[4] |
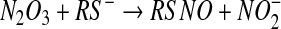 |
[5] |
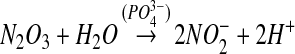 |
[6] |
nitrogen dioxide reacts with a second molecule of NO to form dinitrogen trioxide (equation 4). This is a reversible reaction with well-defined kinetics [see (17) and references therein]. Dinitrogen trioxide is a good nitrosating agent that is able to directly form S-nitrosate thiols according to equation 5. An alternative fate of dinitrogen trioxide is the hydrolysis to form nitrite (equation 6), a process that is accelerated by the presence of phosphate. In the absence of a nucleophile other than water, nitrite is by far the major product of NO oxidation (4).
The major objection to this pathway as a mechanism of S-nitrosothiol formation is the fact that the nitrogen dioxide radical may have many competing reactions, including a direct reaction with the thiol (equation 7). Although the rate constant for this reaction is slower than that for reaction 4, it is irreversible, and there
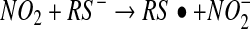 |
[7] |
is usually much more thiol (RSH) than NO to react with. Consequently, the reaction shown in equation 7 will effectively compete with that in equation 4. Another major objection to pathway 1 is the fact that S-nitrosothiols are actually a minor product (∼20%) of the reaction between NO, oxygen, and thiols (56). The major product formed is thiol disulfide, and pathway 1 gives no clear mechanism for how this can be formed. Equation 7, however, can lead to S-nitrosothiol formation via pathway 2. The formation of the thiyl radical from equation 7 gives a clear path to the formation of both S-nitrosothiol and
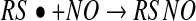 |
[8] |
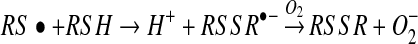 |
[9] |
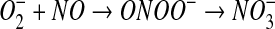 |
[10] |
thiol disulfide, as shown in equations 8 and 9. The latter reaction also generates superoxide, which, after a reaction with NO to form peroxynitrite, may also be the source of the small amounts of nitrate detected during the course of the reaction (equation 10) (56).
The third pathway depicted in Figure 3 involves the direct addition of NO to a thiol, to form an aminoxyl radical followed by a one-electron oxidization of this complex by oxygen to form an S-nitrosothiol (equations 11 and 12) (37).
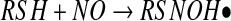 |
[11] |
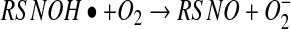 |
[12] |
It is clear that this reaction does not occur in studies that have examined the kinetics of this process, as S-nitrosation has always been shown to have a second-order dependence on NO, and not a first-order dependence, as predicted by this mechanism. However, the possibility that this reaction occurs at low NO levels (where reaction order is difficult to ascertain) is intriguing, but remains speculative. The aminoxyl radical was first reported to be an intermediate in the slow, anerobic mechanism by which NO oxidizes thiols to disulfides (45, 78). The formation of this radical, and its subsequent rearrangement to the more stable thionitroxide radical, was reported to be the best explanation of the crystal structure obtained after crystallized hemoglobin is exposed to NO gas (16, 108). This suggests that at least the transient formation of a direct thiol/NO adducts is possible, but there is no direct evidence for their generation in solution. It is possible that the transient association of NO with thiols to form quasi-stable radical adducts is a mechanism of direct and reversible NO-dependent signaling through protein thiol groups, and this has been suggested in the case of the NO-dependent activation of soluble guanylyl cyclase activity (sGC) (26). The possibility that another electron acceptor besides oxygen could participate in S-nitrosothiol formation in a similar way to reaction 12 will be discussed later.
Transition Metal Ion/Protein Catalyzed Formation of S-Nitrosothiols
The reaction shown in equation 8 indicates that the reaction between a thiyl radical and NO can give rise to an S-nitrosothiol. It follows that any reaction or process that can generate a thiyl radical has the potential to generate S-nitrosothiols. Transition metal ions with one-electron redox reactions within the reach of physiological redox potentials are good candidates for such chemistry. There is evidence that copper and iron ions are able to generate S-nitrosothiols presumably via one-electron oxidation of thiols to thiyl radicals (equation 13) or through the formation of NO/metal complexes (91, 96), but these observations are always confounded by the
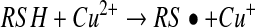 |
[13] |
fact that such metal ions are extremely good catalysts for S-nitrosothiol decomposition. However, the possibility remains that metal ions and metal centers are involved in nitrosation chemistry. The clearest enunciation of this mechanism is the observation that ceruloplasmin (CP) (49), a copper-containing plasma protein, can increase the generation of GSNO from a mixture of NO and glutathione (GSH). It was suggested that the mechanism of CP-dependent S-nitrosothiol production involves the oxidation of NO, rather than the thiol, to generate nitrosonium (NO+) by the type I copper of the protein. The NO+ then reacts with thiol to form S-nitrosothiol (equation 14 and 15). One issue with this mechanism, and all similar mechanisms that involve NO+ as an intermediate, is that the reaction should be concerted, and NO+ can never be released into a solution as a free entity due to the fact that it is instantly hydrolyzed in water to form nitrite (5). Additional evidence of an NO oxidase activity of CP comes from the observation that nitrite can be generated from NO in plasma and in whole blood by a CP-dependent mechanism (87), implying that plasma CP is at least partially competitive with erythrocyte hemoglobin in its ability to oxidize NO. Despite these findings, the ability of CP to catalyze the formation of S-nitrosothiols in whole blood has not been assessed, and no intracellular copper proteins have been determined to possess such activity.
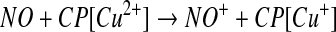 |
[14] |
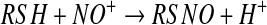 |
[15] |
Similarly, peroxidases represent potential S-nitrosothiol generating enzymes, but there is little experimental evidence for such activity. Several possible mechanisms of S-nitrosothiol formation present themselves. (i) The direct oxidation of NO by peroxidase compounds I and II, to form NO+ (15, 34); (ii) the direct or indirect oxidation of thiol to the thiyl radical (15) followed by the addition of NO; and (iii) the oxidation of nitrite to nitrogen dioxide and the subsequent formation of the nitrosating agent dinitrogen trioxide after a reaction with NO (93). There is evidence that myeloperoxidase can promote N-nitrosation of morpholine and derivatives (62), and also the horseradish peroxidase/H2O2/nitrite system-generated peptides with a 29 Da mass-shift difference, though these were most likely linked to tyrosine modification (84). Despite these possibilities, there is no evidence that peroxidases are able to generate S-nitrosothiols, either in vivo or in vitro.
A novel and interesting idea related to the ability of peroxidases to enhance S-nitrosation relates tyrosine oxidation by peroxidases to thiyl radical formation (and subsequent potential S-nitrosation) via electron transfer processes within proteins (103). By this process, a peroxidase may generate a tyrosyl radical on a protein, which may then transfer one oxidizing equivalent to a thiol residue to generate a thiyl radical. This type of process has been shown to inhibit the nitration of oxidized tyrosine and the formation of disulfides. While this idea is speculative as a mechanism of S-nitrosothiol formation, it highlights the fact that radicals are not necessarily fixed in proteins at the site of oxidation and that the S-nitrosation of a protein could occur at some distance from the original point of oxidation.
The inter-conversion of dinitrosyl iron complexes (DNIC) that form S-nitrosothiols was first proposed by Boese et al. (7). According to this report, the formation of S-nitrosthiols on bovine serum albumin requires both iron and free cysteine and occurs through the intermediacy of DNIC formation. Non-heme iron nitrosyl complexes have been observed in cells by electron paramagnetic resonance, but remain poorly understood (63). A substantial literature has been developed around the concept of DNICs as mediators of NO activity (95). A mixture of iron, thiol, and NO will form a DNIC spontaneously in solution, and it has been suggested that these more-stable metabolites of NO actually are the molecular identity of endothelium-derived relaxing factor (EDRF) (94). Recent studies by Bosworth et al. (8) suggest that DNICs may well be precursors of cellular S-nitrosothiols, which, if true, would incorporate an essential role for the so-called “labile iron” pool of cells in S-nitrosothiol synthesis. This would also link the formation and potential targeting of S-nitrosothiols to iron uptake, distribution, and metabolism. Significantly, these authors indicate that S-nitrosothiol formation is zero order with regard to NO and inhibited by oxygen, suggesting that this mechanism may be more important in hypoxia.
We just discussed a potential mechanism for S-nitrosothiol formation involving the direct addition of NO to a thiol followed by a reaction of the putative aminoxyl radical with an electron acceptor (e.g., oxygen or NAD+), as first proposed by Gow et al. (37). Our own studies examining this mechanism have not found evidence that either oxygen or NAD+ is able to act in such a way; however, we observed robust and efficient S-nitrosation under anerobic conditions in the presence of ferric cytochrome c (3). It has long been known that GSH has a weak binding site on cytochrome c and can slowly reduce it. We observed that NO, at low concentrations, could greatly enhance the rate of cytochrome c reduction by GSH with the concomitant formation of GSNO (3). In contrast, higher concentrations of NO inhibited this process. We invoked a mechanism whereby NO reacts with GSH-bound ferric cytochrome c to form a ternary complex that can either react with excess NO in an unproductive reaction to form glutathione disulfide or can undergo an electron transfer reaction to reduce the heme iron and release GSNO. While evidence is still lacking for this mechanism, it is consistent with the observed reaction profile. The most striking feature of this mechanism is that it becomes more efficient at lower NO concentrations, as side reactions are minimized, approaching 100% conversion of NO to GSNO. We know of no other mechanism of S-nitrosothiol formation that gets close to this level of efficiency. Although most of our studies were done anerobically to preclude GSNO formation from the NO/O2 mechanism, we have recently shown that even under atmospheric oxygen levels, the presence of cytochrome c is still able to enhance GSNO formation. The reaction between GSH, cytochrome c, and NO is illustrated in Figure 4. In addition, we have demonstrated that cells deficient in cytochrome c generate much lower levels of S-nitrosothiols after exposure to NO (10). While not intrinsically catalytic, the reoxidation of ferrous cytochrome c by other cellular processes would provide for the recycling of cytochrome c.
FIG. 4.

Cytochrome c-dependent S-nitrosothiol formation. Ferric cytochrome c weakly binds glutathione, and this complex reacts with NO to form GSNO and ferrous cytochrome c. The process becomes catalytic if the cytochrome c is further oxidized by, for example, mitochondrial complex IV. (To see this illustration in color the reader is referred to the web version of this article at www.liebertpub.com/ars).
Transnitrosation
Probably the most important reaction of an S-nitrosothiol inside a cell or in a biological fluid is transnitrosation (1, 85). The S-nitroso functional group can be transferred to a thiolate in a reversible reaction, as shown in equation 16.
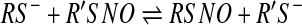 |
[16] |
This reaction involves the nucleophilic attack of a thiolate anion on the nitroso nitrogen. Since the products of this reaction are an S-nitrosothiol and a thiol, a similar reaction in reverse will yield the original reactants. If one of the species in equation 16 is a protein, then this reaction represents a mechanism of S-nitrosation or S-denitrosation of a protein. The equilibrium position of equation 16 will depend on the ratio of the forward- and reverse-rate constants (44). Transnitrosation reactions are not particularly fast and can vary between about 0.1 and 500 M−1s−1 (19, 44, 70, 77, 83), depending on the nature of the reactants. Measured equilibrium constants often approach the value of one for a given combination of thiol and nitrosothiol (44). In the absence of all other factors, the distribution of S-nitrosothiols within a cell will depend on the kinetics and equilibria of such reactions, and while the catalysis of transnitrosation by enzymes such as thioredoxin (71) may change the kinetics of this reaction, it will not change the equilibrium position. It is clear that not all protein thiols are equal with regard to transnitrosation, and some thiols in an individual protein are preferentially S-nitrosated. There are many examples of studies of isolated proteins that show preferential S-nitrosation of a single site on the protein after exposure to a low-molecular-mass S-nitrosothiol. A classic example is human hemoglobin that is relatively selectively S-nitrosated on the cysteine residue at position 93 of the beta chain after exposure to S-nitrosocysteine (100) or GSNO (54). The factors influencing this selectivity are complex, but both kinetic and thermodynamic factors may come into play. Steric hindrance is a major contributor, and solvent-exposed thiols are much more susceptible to modification by transnitrosation than thiols buried within the protein. Other factors such as thiol pKa and neighboring amino-acid effects may also contribute toward thermodynamically stabilizing the protein S-nitrosothiol (18, 90). While a “consensus motif” for S-nitrosation has been discussed (90), such motifs are more often associated with protein-protein interactions that dictate the site of action of an enzyme (such as a kinase), whereas protein thiol modification by transnitrosation more likely depends on the three-dimensional environment of the thiolate than a specific motif in the primary structure (2).
Transnitrosation kinetics/thermodynamics are clearly important when examining the pattern of S-nitrosation that occurs when exposing cells to low-molecular-mass S-nitrosothiols, which may largely modify proteins through transnitrosation without the involvement of NO (106). When NO itself is examined, the situation is more complex, as S-nitrosothiols may be formed outside the cell and be subsequently transported inside, may be formed on low-molecular-mass thiols within the cell with subsequent transnitrosation to protein, or may be formed directly on protein thiols. Consequently, the factors influencing the S-nitrosation of proteins by NO may be different from those for S-nitrosothiols, and the proteome of S-nitrosated proteins may be different depending on the nature of the terminal protein nitrosating agent. Evidence so far collected suggests that this may be the case (39).
Mechanisms in Cells, Tissues, and Organs
It is clear that the formation of S-nitrosothiols occurs as a product of the formation of NO and that a certain amount of S-nitrosation accompanies NO formation. These levels of S-nitrosothiols have been prescribed functions and activities that are crucial to the regulation of many important physiological and pathophysiological events. If S-nitrosation does indeed play such a role, then it would seem essential that the formation and decay of these species should be regulated and controlled, and not left to the random NO chemistry just described. There is currently very little evidence for such controlling mechanisms. CP has been reported to be a nitrosothiol synthase in plasma as just discussed. The idea that superoxide dismutase represents a catalyst for the formation of S-nitrosohemoglobin (38, 43), and that hemoglobin undergoes self-nitrosation during oxygenation deoxygenation cycles, is controversial (47, 54, 68, 101). The lack of a specific and dedicated S-nitrosothiol synthetic machinery is a major weakness of the concept that S-nitrosothiol synthesis is a targeted and directed signaling process (64). In the absence of specific “kinase” analogs, the targeting of S-nitrosation would be determined by mechanisms based on thiol pKa, hydrophobicity and localization of source and target. Each of these concepts will be discussed in detail.
Thiol pKa is the primary determinant of thiol reactivity. Although most protein thiols possess a pKa within the range of 8–9 (14), some thiols exhibit proton dissociation constants that are several orders of magnitude outside this range, from pKa values of ∼4–11. Thiolate is one of the most reactive protein functional groups and is a much stronger nucleophile than a thiol. Thus, it will generally react many times faster with biologically relevant oxidants and electrophiles. Proteins that contain thiols with particularly low pKa values are more susceptible to oxidative modification due to the higher proportion of ionization at physiological pH. Thiols are positioned in environments that engender a low pKa for a multiplicity of apparent reasons, including catalytic activity [e.g., thiol disulfide exchange in thioredoxin (22, 32)], facilitation of substrate binding (30), or for purposes of allosteric regulation (41). It may well be the case that cysteines with low pKa are good candidates for protein S-nitrosation, but this should not be invoked as a specific targeting mechanism in the same way, for example, as a consensus motif for kinase-dependent phosphorylation (2, 90). The ability of low pKa thiols to respond to oxidants and nitrosating agents is more akin to a stress response in which a certain threshold of oxidant concentration results in enough modification of the thiol target to elicit a biological response. A classic example of this kind of mechanism is the Keap1 protein that dissociates from Nrf2 in response to the electrophilic and oxidative modification of key cysteine residues which allows Nrf2 to translocate to the nucleus and act as a regulator of transcription (61). The difference in pKa of a cysteine residue is possibly one of the major reasons, in combination with steric constraints and solvent accessibility, why a purified protein will exhibit selective S-nitrosation of one or more thiols, in the presence of nitrosating agents. It is likely that thiols with low pKa values are critical to some aspect of protein function (e.g., active site cysteines); thus, S-nitrosation of these thiols can potentially have a functional consequence.
It has been postulated that thiols in hydrophobic regions of membranes or proteins are more prone to S-nitrosation (43, 74, 79). Hydrophobicity enhances the rate of reaction between NO and oxygen by a significant factor (30–300-fold) due to the fact that both NO and oxygen are hydrophobic gasses and preferentially partition into hydrophobic phases (64, 72). As shown by Moller et al. (72), the presence of low-density lipoprotein significantly increased the yield of GSNO formation from the NO donor PROLI/NO. However, these studies do not say, and in some cases explicitly warn against the idea (72), that this would facilitate the S-nitrosation of thiols in hydrophobic pockets. The major reason that hydrophobic thiol nitrosation is unlikely is that thiol groups in aprotic regions will be protonated and, thus, will be poorly reactive with S-nitrosating agents. Recent studies looking at the S-nitrosation of transmembrane spanning model peptides clearly show dramatically decreased S-nitrosation the deeper the thiol is placed within a model membrane (102). It may be that low pKa thiols in the vicinity of membranes will experience a higher steady state of S-nitrosating agents than cytosolic proteins at a distance from the membrane, but it is not currently clear, considering the kinetics and dynamics of S-nitrosation chemistry and the diffusivity of NO and derivative nitrosating species, how much of a localization effect this will have. Moller et al. have estimated that if nitrogen dioxide is formed in a membrane, it could travel 40-membrane thicknesses in the presence of 5 mM GSH (72). If GSH is the major target for nitrogen dioxide resulting in GSNO formation, the nitrosation envelope is then defined by the diffusivity and reactivity of GSNO with protein thiols. The work of Nudler and co-workers has been especially influential in suggesting that hydrophobic motifs of proteins may promote S-nitrosation (74, 79). These authors suggested that bovine serum albumin could catalyze thiol S-nitrosation by providing a hydrophobic sub-domain. These studies did not directly measure S-nitrosothiols but electrochemically measured NO release after the addition of large amounts of Cu2+ and assumed that this was quantitatively related to the level of S-nitrosothiol formed. Our studies using a more direct detection of S-nitrosothiols (high performance liquid chromatography [HPLC] and mercury-inhibitable tri-iodide-based chemiluminescence) have shown both bovine and human serum albumin as causing a small but significant decrease in S-nitrosothiol yield (58). The concept of hydrophobicity-targeted S-nitrosation can, therefore, be regarded as unproven and chemically unlikely.
It is possible that specificity is engendered by rates of degradation rather than by rates of formation, such that rapidly degraded S-nitrosothiols would have less influence than more resistant ones. Although limited evidence suggests that different S-nitrosothiols can have different biological lifetimes, there is little evidence for specific enzymatic denitrosating activities. Although GSH-dependent formaldehyde (dehydrogenase) catalyzes the decomposition of GSNO (53, 67) and metabolizes cellular S-nitrosothiols, it lacks activity toward protein targets. Thioredoxin and protein disulfide isomerase have been shown to catalyze protein denitrosation (6, 88), but it is not clear whether these enzymes have the specificity to influence selective pathways and, thus, modulate the direction of NO signaling. At minimum, these enzymes likely play a role in the modulation of the overall S-nitrosothiol level. Although more is known about S-nitrosothiol decomposition than about biosynthesis, much still needs to be understood about how these pathways influence NO-dependent signaling.
There has been significant recent discussion about the issue of targeting S-nitrosation by the co-localization of NO synthase (NOS) with the target thiols (23, 60). In addition, it has been shown that altering endothelial NOS (eNOS) localization can alter the distribution of fluorescent dye after the labeling of S-nitrosoproteins (50). These studies used “switch” methods and so can be regarded as somewhat indirect; however, the fact that the localization of eNOS alters patterns of labeling gives rise to some intriguing possibilities concerning mechanism. Biological localization is easy to understand, as it relies on the recognition of specific protein structural motifs to bring, for example, a kinase in close proximity to its protein substrate to facilitate phosphorylation. This is poorly analogous to NO signaling, as NOS is not thought to be a catalyst of S-nitrosation but is rather the source of a necessary substrate. The localization of effects that depend on the chemical biology of reactive species is much harder to rationalize. The hydroxyl radical, with a half life in nanoseconds, has long been considered a local oxidant due to its inherently high reactivity with almost all biological molecules (27). In this case, damage has to be local to the site of oxidant formation. Peroxynitrite, a highly reactive oxidant with a millisecond biological half life, is thought to be able to react across the diameter of a cell (76). NO, in comparison, is extremely stable with a biological half life in seconds. It is clear that NO itself cannot be localized at the sub-cellular level. The evidence for this is somewhat overwhelming, as extracellular oxyhemoglobin (oxyHb) is a potent vasoconstrictor and antagonist of EDRF (29). The only plausible mechanism for this activity is the ability of oxyHb in the lumen of a blood vessel to destroy NO. This means that NO should be freely and widely diffusible, and NO-dependent activities can be inhibited by an extracellular scavenger (59). We have previously published that the formation of S-nitrosothiols in activated macrophages can be antagonized by oxyHb (105), as can the ability of extracellular S-nitrosothiols to activate sGC (82). In fact, there is a long tradition of antagonizing NO-dependent effects in organs or cell culture systems by oxyHb, and increased plasma hemoglobin, and consequent EDRF antagonism, has been thought of as a pathological event in hemolytic disorders (81). The packaging of hemoglobin in red cells has been shown to reduce its ability to scavenge endothelium-derived NO (66). All this points to the fact that NO is not locally constrained at the sub-cellular level, even when the target reacts with NO at near-diffusion controlled rates (i.e., sGC). This raises the paradox of how localized NO production can result in localized S-nitrosation (or perhaps more accurately protein thiol modification) in a hemoglobin-insensitive manner, as recently described (50). One logical response to this paradox is that it is the localization of some unknown catalytic activity and not the localization of NO formation that is crucial to such targeting, and that eNOS somehow provides or attracts this activity. The problem with this explanation is that it makes S-nitrosation somewhat NO-concentration independent, in the same way that phosphorylation is somewhat ATP-concentration independent. There are currently no data to support such possibilities, and since the detection of localized S-nitrosothiol formation is necessarily indirect, these speculations may be premature.
In total, our knowledge of the mechanisms of S-nitrosothiol formation in vivo is tenuous. It is not clear that we need to invoke “unknown mechanisms” which explain the levels of S-nitrosothiol detected in vivo, and it may be that the currently established chemistry of NO is sufficient. However, in order to explain the specificity of S-nitrosation that is inherent in many S-nitrosothiol-dependent signaling pathways, novel and currently unknown targeted pathways of protein S-nitrosation need to be invoked.
Protein S-Nitrosation Independent of NOS
S-nitrosothiols may be formed in cells after the addition of nitroso and nitro species. In some cases, this conversion is thought to require enzymatic activity. The transnitrosation to GSH from both n-butyl nitrite and amyl nitrite was shown to be catalyzed by glutathione-S-transferase (69), and the transfer of the nitroso group of a nitroso cyanoguanidine to GSH was inhibited by glutathione-S-transferase inhibitors (53). The effect of administering glycerol trinitrate, a nitric acid ester, on tissue nitroso and nitrosyl compounds has been shown to be complex and tissue specific (52). In other cases, the ability of an exogenous agent to stimulate S-nitrosation may be triggered by a change in condition. For example, inorganic nitrite does not increase S-nitrosothiol formation in cells under normoxic conditions (105), but rather promotes S-nitrosation under hypoxia (86). Such S-nitrosation may occur, in part, via NO formation, but there is also evidence for NO-independent S-nitrosation from nitrite in hypoxia (25). Since low levels of nitrite are present in all tissues, such a mechanism could have important physiological sequelae.
Levels of S-Nitrosothiols in Cells and Tissues
The formation of protein S-nitrosothiols has been postulated as encompassing a significant contribution to NO-dependent signaling and has been suggested as representing a redox-based signaling mechanism. Despite the numerous reports of novel protein targets being regulated via nitrosation, there is a surprising paucity of studies focusing on the levels of the S-nitroso species found in cells or tissues under physiological or pathological conditions.
The detection methods for S-nitrosothiols can be arbitrarily divided into qualitative and quantitative categories. Qualitative methods involve multiple switch techniques, ranging from the original biotin-switch (51) and the modifications thereof (28, 46, 107) to the approaches designed to suit proteomic analysis (20, 39, 42, 80). Quantitative methods appropriate for the detection of physiologically relevant levels of S-nitrosothiols are largely fluorescence or chemiluminescence based and allow for the detection of low pmol levels of S-nitrosothiols. Unfortunately, there are very few studies correlating the nitrosation of a specific protein target with the levels of S-nitrosothiols found under given conditions. In other words, the increased level of modification of a protein target is not assessed in the context of total cellular S-nitrosation changes. Additionally, for a proper cause-and-effect conclusion, the quantitative extent of a specific thiol S-nitrosation should be quantitatively related to the observed signaling effect.
The levels of S-nitrosothiols that are detected under basal conditions are within 0.5–2 pmol/mg protein in rat tissues (12) and 5–6 pmol/mg protein in RAW 264.7 macrophages (105). An S-nitrosothiol level of 3 pmol/mg protein was measured in pre-ischemic hearts by 2, 3-diaminonaphthalene fluorescence (92). The plasma levels of S-nitroso species were reported to range between 1 and 2 nM for rats and monkeys and 25 nM for guinea pigs, highlighting the variability among various species (24). The measured levels of S-nitrosothiols in human plasma have varied widely over 3–4 orders of magnitude [see (33)] depending on the method of measurement. On inflammation, the plasma levels of S-nitrosated albumin increased from about 120 to 400 nM in LPS-treated rats (57). In the case of LPS-stimulated macrophages in culture, the levels of S-nitrosothiols increased to ∼25 pmol/mg protein (105).
There are multiple reports in the literature where S-nitrosothiol levels are expressed in concentration units and, due to lack of normalization, are difficult to compare across studies. The levels of S-nitrosothiols in rat tissues were reported to range from 1 nM for plasma, 13 nM for heart, 22 nM for brain, to 36 nM for kidney (13). Recently, Dyson et al. (21) reported the basal levels of NO metabolites in rat tissues: The highest concentration of S-nitrosothiols was detected in red blood cells (0.31 μM) with much lower levels for brain, heart, liver, kidney, and lung (within 10–50 nM range). Similar levels for red cells S-nitrosothiols were previously reported (97). During endotoxemia, S-nitrosothiol formation is greatly enhanced, achieving 6 μM for red blood cells and 0.5–2 μM for the other organs (21). A similar trend, with preferred S-nitrosation in erythrocytes, was observed in mice breathing air supplemented with NO. Whole-body S-nitrosation increased from 0.06 μM in control animals to 0.5 μM on NO inhalation, and the accumulation of nitrosothiols was the highest in the red blood cells, followed by lungs, liver, heart, and brain (73). S-nitrosothiol levels in human bronchoalveolar lavage fluid were found to be about 300 nM, but increase to more than 4 μM in patients with pneumonia (31).
In total, these studies show that tissue levels of S-nitrosothiols are low but measurable and increase under severe experimental or pathological conditions, such as endotoxemia (inducible nitric oxide synthase induction) or ischemia. What has not been observed is a quantitative increase in cellular or tissue levels of S-nitrosothiols on the stimulation of eNOS.
Current Working Model of Cellular S-Nitrosation
Figure 5 represents our current working model of cellular S-nitrosation based on work done by us and many other groups. NO, as a freely diffusible hydrophobic molecule, is able to directly access intracellular compartments where it has effectively no ability to directly S-nitrosate thiols. The leading pathways for thiol S-nitrosation from NO are as follows (Fig. 5A): (i) The reaction of NO with another radical, oxidant, or enzyme that generates an NO-derived oxidant which is able to oxidize a thiol to a thiyl radical, with the subsequent addition of NO to form an S-nitrosothiol (alternatively, the NO-derived oxidant could react with NO to form a nitrosating agent such as N2O3). Concomitant with, and perhaps dominant over, S-nitrosation is thiol oxidation, mixed disulfide formation, and possibly irreversible protein modifications due to a reaction of the thiyl radical with the protein backbone. (ii) A more concerted mechanism of S-nitrosothiol formation through the action of a metal center as exemplified via cytochrome c and DNIC formation. Whether these mechanisms form GSNO or directly form protein S-nitrosothiols is almost moot as transnitrosation (perhaps catalyzed by thioredoxin) can distribute the nitroso functional group among available thiols. S-nitrosothiols may also be introduced into cells via transport from the extracellular space (Figure 5B). In most cell studies, this predominantly occurs through the amino-acid transport system L (L-AT) (65, 106), though other mechanisms have also been implicated. The L-AT-dependent mechanism is specific for S-nitrosothiols based on simple amino acids such as CysNO or S-nitrosohomocysteine (11). Once inside the cell, transnitrosation can again distribute the nitroso function group to other thiols. The S-nitrosation of a protein thiol is most often reversible (as is the case with caspase 3), but in some cases, it may lead to irreversible inhibition of the enzyme (as observed with glyceraldehyde-3 phosphate dehydrogenase) (9). S-nitrosothiol metabolism occurs mainly through an NADH-dependent process catalyzed by glutathione-dependent formaldehyde dehydrogenase (now sometimes referred to as GSNO reductase) (53, 67), but other less well-defined pathways exist that lead to NO formation (82). The cellular export of S-nitrosothiols is an area that has not been investigated in any detail.
FIG. 5.

Model for NO and S-nitrosothiol-dependent cellular effects. (A) NO can oxidatively modify intracellular thiols (e.g., by forming thiyl radical) after reacting with O2, O2•−, or through its interaction with peroxidases. Cellular-reducing machinery reacts with the free radical, thus prohibiting further damage to the protein (e.g., glutathionation). Glutathionation is a transient modification that can be reduced back to the parent thiol. When protein thiyl radical is formed, it can react with NO to form S-nitrosothiol or lead to irreversible protein damage due to the oxidation of adjacent amino acids. S-nitrosothiol may also be formed by a more concerted mechanism involving NO, a metal center, and a thiol. (B) CysNO is transported into the cell through L-AT in an L-leucine-inhibitable process. Inside the cell, CysNO can undergo transnitrosation with protein thiols and GSH. The formation of protein-associated S-nitrosothiols can be either reversible (e.g., caspase-3) or facilitate further reactions, leading to the irreversible inhibition of a given protein (e.g., GAPDH). CysNO can release NO in a process that can be inhibited by DPI, an inhibitor of flavin-containing enzymes. GSNO, formed from GSH on transnitrosation with CysNO or protein S-nitrosothiol, is metabolized by GSNO reductase. DPI, diphenyleneiodonium chloride; GAPDH, glyceraldehyde-3 phosphate dehydrogenase. (To see this illustration in color the reader is referred to the web version of this article at www.liebertpub.com/ars).
Conclusions
This review has attempted to highlight the chemical and biophysical characterization of S-nitrosothiols and S-nitrosation and the variety of biological activities that are reported to be controlled by S-nitrosation. While we are no doubt continuing to gain insights into the mechanisms of S-nitrosation and into the modulation of biological processes by this post-translational modification, the paradigm that alteration in NO formation leads to a plethora of altered physiological responses through protein S-nitrosation still has many challenges. Not the least of which is the lack of quantitative correlation between the extent of protein modification and the observed biological end-point. The explosion of studies using the qualitative “switch” techniques has created many candidates for control by S-nitrosation, but in most cases, absolute quantitative evidence that protein nitrosation is the definitive event is missing. It is pertinent to point out that protein S-nitrosation occurs in conjunction with other NO-dependent modifications, such as protein oxidation, nitration, heme nitrosylation, and iron association, which may contribute to NO-dependent effects. S-nitrosation is clearly an important NO-dependent event, and improved methodology may be required to address some of the more challenging questions concerning its role in NO-dependent signaling.
Abbreviations Used
- CP
ceruloplasmin
- D-CysNO
S-nitroso-D-cysteine
- DNIC
dinitrosyl iron complexes
- EDRF
endothelium-derived relaxing factor
- eNOS
endothelial nitric oxide synthase
- GSH
glutathione
- GSNO
S-nitrosoglutathione
- L-CysNO
S-nitroso-l-cysteine
- NOS
nitric oxide synthase
- oxyHb
oxyhemoglobin
- RSH
thiol
- sGC
soluble guanylyl cyclase
- SNAP
S-nitroso-N-acetyl penicillamine
Acknowledgments
The authors would like to thank Dr. Agnes Keszler and Dr. Anne Diers for their contributions in preparing this review.
References
- 1.Arnelle DR. Stamler JS. NO+, NO, and NO- donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch Biochem Biophys. 1995;318:279–285. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 2.Ascenzi P. Colasanti M. Persichini T. Muolo M. Polticelli F. Venturini G. Bordo D. Bolognesi M. Re-evaluation of amino acid sequence and structural consensus rules for cysteine-nitric oxide reactivity. Biol Chem. 2000;381:623–627. doi: 10.1515/BC.2000.081. [DOI] [PubMed] [Google Scholar]
- 3.Basu S. Keszler A. Azarova NA. Nwanze N. Perlegas A. Shiva S. Broniowska KA. Hogg N. Kim-Shapiro DB. A novel role for cytochrome c: efficient catalysis of S-nitrosothiol formation. Free Radic Biol Med. 2010;48:255–263. doi: 10.1016/j.freeradbiomed.2009.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bateman RL. Rauh D. Tavshanjian B. Shokat KM. Human carbonyl reductase 1 is an S-nitrosoglutathione reductase. J Biol Chem. 2008:M807125200. doi: 10.1074/jbc.M807125200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayliss NS. Watts DW. The spectra and equilibria of nitrosonium ion, nitroacidium ion, and nitrous acid in solutions of sulphuric, hydrochloric, and phosporic acids. Aust J Chem. 1956;9:319–332. [Google Scholar]
- 6.Benhar M. Forrester MT. Hess DT. Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boese M. Mordvintcev P. Vanin AF. Busse R. Mülsch A. S-Nitrosation of serum albumin by dinitrosyl-iron complex. J Biol Chem. 1995;270:29244–29249. doi: 10.1074/jbc.270.49.29244. [DOI] [PubMed] [Google Scholar]
- 8.Bosworth CA. Toledo JC. Zmijewski JW. Li Q. Lancaster JR. Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proc Natl Acad Sci U S A. 2009;106:4671–4676. doi: 10.1073/pnas.0710416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broniowska KA. Hogg N. Differential mechanisms of inhibition of Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by S-nitrosothiols and NO in cellular and cell-free conditions. Am J Physiol Heart Circ Physiol. 2010;299:H1212–H1219. doi: 10.1152/ajpheart.00472.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broniowska KA. Keszler A. Basu S. Kim-Shapiro DB. Hogg N. Cytochrome c-mediated formation of S-nitrosothiol in cells. Biochem J. 2012;442:191–197. doi: 10.1042/BJ20111294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broniowska KA. Zhang Y. Hogg N. Requirement of transmembrane transport for S-nitrosocysteine-dependent modification of intracellular thiols. J Biol Chem. 2006;281:33835–33841. doi: 10.1074/jbc.M603248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bryan NS. Rassaf T. Maloney RE. Rodriguez CM. Saijo F. Rodriguez JR. Feelisch M. Cellular targets and mechanisms of nitros(yl)ation: an insight into their nature and kinetics in vivo. Proc Natl Acad Sci U S A. 2004;101:4308–4313. doi: 10.1073/pnas.0306706101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bryan NS. Rassaf T. Rodriguez J. Feelisch M. Bound NO in human red blood cells: fact or artifact? Nitric Oxide. 2004;10:221–228. doi: 10.1016/j.niox.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Bulaj G. Kortemme T. Goldenberg DP. Ionization-Reactivity Relationships for Cysteine Thiols in Polypeptides. Biochemistry. 1998;37:8965–8972. doi: 10.1021/bi973101r. [DOI] [PubMed] [Google Scholar]
- 15.Burner U. Obinger C. Transient-state and steady-state kinetics of the oxidation of aliphatic and aromatic thiols by horseradish peroxidase. FEBS Lett. 1997;411:269–274. doi: 10.1016/s0014-5793(97)00713-8. [DOI] [PubMed] [Google Scholar]
- 16.Chan NL. Kavanaugh JS. Rogers PH. Arnone A. Crystallographic analysis of the interaction of nitric oxide with quaternary-T human hemoglobin. Biochemistry. 2004;43:118–132. doi: 10.1021/bi030172j. [DOI] [PubMed] [Google Scholar]
- 17.Czapski G. Goldstein S. The role of the reactions of. NO with superoxide and oxygen in biological systems: a kinetic approach. Free Radic Biol Med. 1995;19:785–794. doi: 10.1016/0891-5849(95)00081-8. [DOI] [PubMed] [Google Scholar]
- 18.Dahm CC. Moore K. Murphy MP. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J Biol Chem. 2006;281:10056–10065. doi: 10.1074/jbc.M512203200. [DOI] [PubMed] [Google Scholar]
- 19.Dasgupta TP. Aquart DV. Transfer of nitric oxide from nitrovasodilators to free thiols—evidence of two distinct stages. Biochem Biophys Res Commun. 2005;335:730–733. doi: 10.1016/j.bbrc.2005.07.126. [DOI] [PubMed] [Google Scholar]
- 20.Doulias PT. Greene JL. Greco TM. Tenopoulou M. Seeholzer SH. Dunbrack RL. Ischiropoulos H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci U S A. 2010;107:16958–16963. doi: 10.1073/pnas.1008036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dyson A. Bryan NS. Fernandez BO. Garcia-Saura MF. Saijo F. Mongardon N. Rodriguez J. Singer M. Feelisch M. An integrated approach to assessing nitroso-redox balance in systemic inflammation. Free Radic Biol Med. 2011;51:1137–1145. doi: 10.1016/j.freeradbiomed.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 22.Dyson HJ. Jeng MF. Tennant LL. Slaby I. Lindell M. Cui DS. Kuprin S. Holmgren A. Effects of buried charged groups on cysteine thiol ionization and reactivity in Escherichia coli thioredoxin: structural and functional characterization of mutants of Asp 26 and Lys 57ΓÇá. Biochemistry. 1997;36:2622–2636. doi: 10.1021/bi961801a. [DOI] [PubMed] [Google Scholar]
- 23.Fang M. Jaffrey SR. Sawa A. Ye K. Luo X. Snyder SH. Dexras1: a G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron. 2000;28:183–193. doi: 10.1016/s0896-6273(00)00095-7. [DOI] [PubMed] [Google Scholar]
- 24.Feelisch M. Rassaf T. Mnaimneh S. Singh N. Bryan NS. Jourd'heuil D. Kelm M. Concomitant S-, N-, and heme-nitros(yl)ation in biological tissues and fluids: implications for the fate of NO in vivo. FASEB J. 2002;16:1775–1785. doi: 10.1096/fj.02-0363com. [DOI] [PubMed] [Google Scholar]
- 25.Feelisch M. Fernandez BO. Bryan NS. Garcia-Saura MF. Bauer S. Whitlock DR. Ford PC. Janero DR. Rodriguez J. Ashrafian H. Tissue processing of nitrite in hypoxia: an intricate interplay of nitric oxide-generating and -scavenging systems. J Biol Chem. 2008;283:33927–33934. doi: 10.1074/jbc.M806654200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernhoff NB. Derbyshire ER. Marletta MA. A nitric oxide/cysteine interaction mediates the activation of soluble guanylate cyclase. Proc Natl Acad Sci U S A. 2009;106:21602–21607. doi: 10.1073/pnas.0911083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forman HJ. Torres M. Fukuto J. Redox signaling. Mol Cell Biochem. 2002;234–235:49–62. [PubMed] [Google Scholar]
- 28.Forrester MT. Foster MW. Benhar M. Stamler JS. Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic Biol Med. 2009;46:119–126. doi: 10.1016/j.freeradbiomed.2008.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furchgott RF. Cherry PD. Zawadzki JV. Jothianandan D. Endothelial cells as mediators of vasodilation of arteries. J Cardiovasc Pharmacol. 1984;6(Suppl 2):S336–S343. doi: 10.1097/00005344-198406002-00008. [DOI] [PubMed] [Google Scholar]
- 30.Furter R. Furter-Graves EM. Wallimann T. Creatine kinase: the reactive cysteine is required for synergism but is nonessential for catalysis. Biochemistry. 1993;32:7022–7029. doi: 10.1021/bi00078a030. [DOI] [PubMed] [Google Scholar]
- 31.Gaston B. Reilly J. Drazen JM. Fackler J. Ramdev P. Arnelle D. Mullins ME. Sugarbaker DJ. Chee C. Singel DJ. Loscalzo J. Stamler JS. Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc Natl Acad Sci U S A. 1993;90:10957–10961. doi: 10.1073/pnas.90.23.10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 33.Gladwin MT. Wang X. Hogg N. Methodological vexation about thiol oxidation versus S-nitrosation—A commentary on “An ascorbate-dependent artifact that interferes with the interpretation of the biotin-switch assay”. Free Radic Biol Med. 2006;41:557–561. doi: 10.1016/j.freeradbiomed.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 34.Glover RE. Koshkin V. Dunford HB. Mason RP. The reaction rates of NO with horseradish peroxidase compounds I and II. Nitric Oxide. 1999;3:439–444. doi: 10.1006/niox.1999.0256. [DOI] [PubMed] [Google Scholar]
- 35.Goldstein S. Czapski G. Kinetics of nitric oxide autoxidation in aqueous solution in the absence and presence of various reductants. The nature of the oxidizing intermediates. J Am Chem Soc. 1995;117:12078–12084. [Google Scholar]
- 36.Goldstein S. Czapski G. Mechanism of the nitrosation of thiols and amines by oxygenated NO solutions: the nature of the nitrosating intermediates. J Am Chem Soc. 1996;118:3419–3425. [Google Scholar]
- 37.Gow AJ. Buerk DG. Ischiropoulos H. A novel reaction mechanism for the formation of S-nitrosothiol in vivo. J Biol Chem. 1997;272:2841–2845. doi: 10.1074/jbc.272.5.2841. [DOI] [PubMed] [Google Scholar]
- 38.Gow AJ. Luchsinger BP. Pawloski JR. Singel DJ. Stamler JS. The oxyhemoglobin reaction of nitric oxide. Proc Natl Acad Sci U S A. 1999;96:9027–9032. doi: 10.1073/pnas.96.16.9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greco TM. Hodara R. Parastatidis I. Heijnen HFG. Dennehy MK. Liebler DC. Ischiropoulos H. Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc Natl Acad Sci U S A. 2006;103:7420–7425. doi: 10.1073/pnas.0600729103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han TH. Hyduke DR. Vaughn MW. Fukuto JM. Liao JC. Nitric oxide reaction with red blood cells and hemoglobin under heterogeneous conditions. Proc Natl Acad Sci U S A. 2002;99:7763–7768. doi: 10.1073/pnas.122118299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansen SK. Cancilla MT. Shiau TP. Kung J. Chen T. Erlanson DA. Allosteric Inhibition of PTP1B Activity by Selective Modification of a Non-Active Site Cysteine ResidueΓÇá. Biochemistry. 2005;44:7704–7712. doi: 10.1021/bi047417s. [DOI] [PubMed] [Google Scholar]
- 42.Hao G. Derakhshan B. Shi L. Campagne F. Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci U S A. 2006;103:1012–1017. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hess DT. Matsumoto A. Kim SO. Marshall HE. Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 44.Hogg N. The kinetics of S-Transnitrosation—a reversible second-order reaction. Anal Biochem. 1999;272:257–262. doi: 10.1006/abio.1999.4199. [DOI] [PubMed] [Google Scholar]
- 45.Hogg N. Singh RJ. Kalyanaraman B. The role of glutathione in the transport and catabolism of nitric oxide. FEBS Lett. 1996;382:223–228. doi: 10.1016/0014-5793(96)00086-5. [DOI] [PubMed] [Google Scholar]
- 46.Huang B. Chen C. Detection of protein S-nitrosation using irreversible biotinylation procedures (IBP) Free Radic Biol Med. 2010;49:447–456. doi: 10.1016/j.freeradbiomed.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 47.Huang KT. Azarov I. Basu S. Huang J. Kim-Shapiro DB. Lack of allosterically controlled intramolecular transfer of nitric oxide from the heme to cysteine in the beta subunit of hemoglobin. Blood. 2006;107:2602–2604. doi: 10.1182/blood-2005-10-4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ignarro LJ. Edwards JC. Gruetter DY. Barry BK. Gruetter CA. Possible involvement of S-nitrosothiols in the activation of guanylate cyclase by nitroso compounds. FEBS Lett. 1980;110:275–278. doi: 10.1016/0014-5793(80)80091-3. [DOI] [PubMed] [Google Scholar]
- 49.Inoue K. Akaike T. Miyamoto Y. Okamoto T. Sawa T. Otagiri M. Suzuki S. Yoshimura T. Maeda H. Nitrosothiol formation catalyzed by ceruloplasmin. Implication for cytoprotective mechanism in vivo. J Biol Chem. 1999;274:27069–27075. doi: 10.1074/jbc.274.38.27069. [DOI] [PubMed] [Google Scholar]
- 50.Iwakiri Y. Satoh A. Chatterjee S. Toomre DK. Chalouni CM. Fulton D. Groszmann RJ. Shah VH. Sessa WC. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci U S A. 2006;103:19777–19782. doi: 10.1073/pnas.0605907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaffrey SR. Erdjument-Bromage H. Ferris CD. Tempst P. Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 52.Janero DR. Bryan NS. Saijo F. Dhawan V. Schwalb DJ. Warren MC. Feelisch M. Differential nitros(yl)ation of blood and tissue constituents during glyceryl trinitrate biotransformation in vivo. Proc Natl Acad Sci U S A. 2004;101:16958–16963. doi: 10.1073/pnas.0406075101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jensen DE. Belka GK. Du Bois GC. S-Nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem J. 1998;331:659–668. doi: 10.1042/bj3310659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jia L. Bonaventura C. Bonaventura J. Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 55.Joshi MS. Ferguson TB., Jr. Han TH. Hyduke DR. Liao JC. Rassaf T. Bryan N. Feelisch M. Lancaster JR., Jr. Nitric oxide is consumed, rather than conserved, by reaction with oxyhemoglobin under physiological conditions. Proc Natl Acad Sci U S A. 2002;99:10341–10346. doi: 10.1073/pnas.152149699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jourd'heuil D. Jourd'heuil FL. Feelisch M. Oxidation and nitrosation of thiols at low micromolar exposure to nitric oxide. Evidence for a free radical mechanism. J Biol Chem. 2003;278:15720–15726. doi: 10.1074/jbc.M300203200. [DOI] [PubMed] [Google Scholar]
- 57.Jourd'heuil D. Gray L. Grisham MB. S-Nitrosothiol Formation in Blood of Lipopolysaccharide-Treated Rats. Biochem Biophys Res Commun. 2000;273:22–26. doi: 10.1006/bbrc.2000.2892. [DOI] [PubMed] [Google Scholar]
- 58.Keszler A. Zhang Y. Hogg N. The reaction between nitric oxide, glutathione and oxygen in the presence and absence of protein: how are S-nitrosothiols formed? Free Radic Biol Med. 2009;48:55–64. doi: 10.1016/j.freeradbiomed.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kilbourn RG. Joly G. Cashon B. DeAngelo J. Bonaventura J. Cell-free hemoglobin reverses the endotoxin-mediated hyporesponsivity of rat aortic rings to [alpha].-adrenergic agents. Biochem Biophys Res Commun. 1994;199:155–162. doi: 10.1006/bbrc.1994.1208. [DOI] [PubMed] [Google Scholar]
- 60.Kim SF. Huri DA. Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 61.Kobayashi M. Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 62.Lakshmi VM. Nauseef WM. Zenser TV. Myeloperoxidase potentiates nitric oxide-mediated nitrosation. J Biol Chem. 2005;280:1746–1753. doi: 10.1074/jbc.M411263200. [DOI] [PubMed] [Google Scholar]
- 63.Lancaster JR., Jr. Hibbs JB., Jr. EPR demonstration of iron-nitrosyl complex formation by cytotoxic activated macrophages. Proc Natl Acad Sci U S A. 1990;87:1223–1227. doi: 10.1073/pnas.87.3.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lancaster JR., Jr. Protein cysteine thiol nitrosation: maker or marker of reactive nitrogen species-induced nonerythroid cellular signaling? Nitric Oxide. 2008;19:68–72. doi: 10.1016/j.niox.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 65.Li S. Whorton AR. Identification of Stereoselective Transporters for S-Nitroso-L-cysteine: role of LAT1 and LAT2 in biological activity of S-nitrosothiols. J Biol Chem. 2005;280:20102–20110. doi: 10.1074/jbc.M413164200. [DOI] [PubMed] [Google Scholar]
- 66.Liao JC. Hein TW. Vaughn MW. Huang K-T. Kuo L. Intravascular flow decreases erythrocyte consumption of nitric oxide. Proc Natl Acad Sci U S A. 1999;96:8757–8761. doi: 10.1073/pnas.96.15.8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu L. Hausladen A. Zeng M. Que L. Heitman J. Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 68.McMahon TJ. Stone AE. Bonaventura J. Singel DJ. Stamler JS. Functional coupling of oxygen binding and vasoactivity in S-nitrosohemoglobin. J Biol Chem. 2000;275:16738–16745. doi: 10.1074/jbc.M000532200. [DOI] [PubMed] [Google Scholar]
- 69.Meyer DJ. Kramer H. Ketterer B. Human glutathione transferase catalysis of the formation of S-nitrosoglutathione from organic nitrites plus glutathione. FEBS Lett. 1994;351:427–428. doi: 10.1016/0014-5793(94)00904-x. [DOI] [PubMed] [Google Scholar]
- 70.Meyer DJ. Kramer H. Ozer N. Coles B. Ketterer B. Kinetics and equilibria of S-nitrosothiol-thiol exchange between glutathione, cysteine, penicillamines and serum albumin. FEBS Lett. 1994;345:177–180. doi: 10.1016/0014-5793(94)00429-3. [DOI] [PubMed] [Google Scholar]
- 71.Mitchell DA. Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 72.Moller MN. Li Q. Vitturi DA. Robinson JM. Lancaster JR. Denicola A. Membrane “lens” effect: focusing the formation of reactive nitrogen oxides from the NO/O2 reaction. Chem Res Toxicol. 2007;20:709–714. doi: 10.1021/tx700010h. [DOI] [PubMed] [Google Scholar]
- 73.Nagasaka Y. Fernandez BO. Garcia-Saura MF. Petersen B. Ichinose F. Bloch KD. Feelisch M. Zapol WM. Brief periods of nitric oxide inhalation protect against myocardial ischemia-reperfusion injury. Anesthesiology. 2008;109:675–682. doi: 10.1097/ALN.0b013e318186316e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nedospasov A. Rafikov R. Beda N. Nudler E. An autocatalytic mechanism of protein nitrosylation. Proc Natl Acad Sci U S A. 2000;97:13543–13548. doi: 10.1073/pnas.250398197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oae S. Shinhama K. Organic thionitrites and related substances. Org Prep Proc Int. 1983;15:165–198. [Google Scholar]
- 76.Pacher P. Beckman JS. Liaudet L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Patel RP. Hogg N. Spencer NY. Kalyanaraman B. Matalon S. Darley-Usmar VM. Biochemical characterization of human S-nitrosohemoglobin: effects on oxygen binding and and transnitrosation. J Biol Chem. 1999;274:15487–15492. doi: 10.1074/jbc.274.22.15487. [DOI] [PubMed] [Google Scholar]
- 78.Pryor WA. Church DF. Govindan CK. Crank G. Oxidation of thiols by nitric oxide and nitrogen dioxide: synthetic utility and toxicological implications. J Org Chem. 1982;47:159–161. [Google Scholar]
- 79.Rafikova O. Rafikov R. Nudler E. Catalysis of S-nitrosothiols formation by serum albumin: the mechanism and implication in vascular control. Proc Natl Acad Sci U S A. 2002;99:5913–5918. doi: 10.1073/pnas.092048999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raju K. Doulias PT. Tenopoulou M. Greene JL. Ischiropoulos H. Strategies and tools to explore protein S-nitrosylation. Biochim Biophys Acta. 2012;1820:684–688. doi: 10.1016/j.bbagen.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reiter CD. Wang X. Tanos-Santos JE. Hogg N. Cannon RO., III Schechter A. Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavalability in sickle-cell disease. Nat Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 82.Riego JA. Broniowska KA. Kettenhofen NJ. Hogg N. Activation and inhibition of soluble gunylyl cyclase by S-nitrosocysteine: involvement of amino acid transport system L. Free Radic Biol Med. 2009;47:269–274. doi: 10.1016/j.freeradbiomed.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rossi R. Lusini L. Giannerini F. Giustarini D. Lungarella G. Di Simplicio P. A method to study kinetics of transnitrosation with nitrosoglutathione: reactions with hemoglobin and other thiols. Anal Biochem. 1997;254:215–220. doi: 10.1006/abio.1997.2424. [DOI] [PubMed] [Google Scholar]
- 84.Salavej P. Spalteholz H. Arnhold J. Modification of amino acid residues in human serum albumin by myeloperoxidase. Free Radic Biol Med. 2006;40:516–525. doi: 10.1016/j.freeradbiomed.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 85.Scharfstein JS. Keaney JFJ. Slivka A. Welch GN. Vita JA. Stamler JS. Loscalzo J. In vivo transfer of nitric oxide between a plasma protein-bound reservoir and low molecular weight thiols. J Clin Invest. 1994;94:1432–1439. doi: 10.1172/JCI117480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shiva S. Sack MN. Greer JJ. Duranski M. Ringwood LA. Burwell L. Wang X. Macarthur PH. Shoja A. Raghavachari N. Calvert JW. Brookes PS. Lefer DJ. Gladwin MT. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–2102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shiva S. Wang X. Ringwood LA. Xu X. Yuditskaya S. Annavajjhala V. Miyajima H. Hogg N. Harris ZL. Gladwin MT. Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat Chem Biol. 2006;2:486–493. doi: 10.1038/nchembio813. [DOI] [PubMed] [Google Scholar]
- 88.Sliskovic I. Raturi A. Mutus B. Characterization of the S-denitrosation activity of protein disulfide isomerase. J Biol Chem. 2005;280:8733–8741. doi: 10.1074/jbc.M408080200. [DOI] [PubMed] [Google Scholar]
- 89.Stamler JS. Jaraki O. Osborne J. Simon DI. Keaney J. Vita J. Singel D. Valeri CR. Loscalzo J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci U S A. 1992;89:7674–7677. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stamler JS. Toone EJ. Lipton SA. Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 91.Stubauer G. Giuffre A. Sarti P. Mechanism of S-Nitrosothiol Formation and Degradation Mediated by Copper Ions. J Biol Chem. 1999;274:28128–28133. doi: 10.1074/jbc.274.40.28128. [DOI] [PubMed] [Google Scholar]
- 92.Sun J. Morgan M. Shen RF. Steenbergen C. Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 93.van der Vliet A. Eiserich JP. Halliwell B. Cross CE. Formation of reactive nitrogen species during peroxidase-catalyzed oxidation of nitrite. A potential additional mechanism of nitric oxide-dependent toxicity. J Biol Chem. 1997;272:7617–7625. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 94.Vanin AF. On the stability of the dinitrosyl-iron-cysteine complex, a candidate for the endothelium-derived relaxation factor. Biochemistry (Mosc) 1995;60:225–230. [Google Scholar]
- 95.Vanin AF. Dinitrosyl iron complexes and S-nitrosothiols are two possible forms for stabilization and transport of nitric oxide in biological systems. Biochemistry (Mosc) 1998;63:782–793. [PubMed] [Google Scholar]
- 96.Vanin AF. Malenkova IV. Serezhenkov VA. Iron catalyzes both decomposition and synthesis of S-nitrosothiols: optical and electron paramagnetic resonance studies. Nitric Oxide. 1997;1:191–203. doi: 10.1006/niox.1997.0122. [DOI] [PubMed] [Google Scholar]
- 97.Wang X. Bryan NS. MacArthur PH. Rodriguez J. Gladwin MT. Feelisch M. Measurement of nitric oxide levels in the red cell: validation of tri-iodide-based chemiluminescence with acid-sulfanilamide pretreatment. J Biol Chem. 2006;281:26994–27002. doi: 10.1074/jbc.M603953200. [DOI] [PubMed] [Google Scholar]
- 98.Wink DA. Beckman JS. Ford PC. Kinetics of nitric oxide reaction in liquid and gas phase. In: Feelisch M, editor; Stamler JS, editor. Methods in Nitric Oxide Research. New York: John Wiley & Sons Ltd.; 1996. pp. 29–37. [Google Scholar]
- 99.Wink DA. Nims RW. Darbyshire JF. Christodoulou D. Hanbauer I. Cox GW. Laval F. Laval J. Cook JA. Krishna MC. Reaction kinetics for nitrosation of cysteine and glutathione in aerobic nitric oxide solutions at neutral pH. Insights into the fate and physiological effects of intermediates generated in the NO/O2 reaction. Chem Res Toxicol. 1994;7:519–525. doi: 10.1021/tx00040a007. [DOI] [PubMed] [Google Scholar]
- 100.Wolzt M. MacAllister RJ. Davis D. Feelisch M. Moncada S. Vallance P. Hobbs AJ. Biochemical characterization of S-nitrosohemoglobin: mechanisms underlying synthesis, NO release and biological activity. J Biol Chem. 1999;274:28983–28990. doi: 10.1074/jbc.274.41.28983. [DOI] [PubMed] [Google Scholar]
- 101.Xu X. Cho M. Spencer NY. Patel N. Huang Z. Shields H. King SB. Gladwin MT. Hogg N. Kim-Shapiro DB. Measurements of nitric oxide on the heme iron and β-93 thiol of heman hemoglobin during cycles of oxygenation and deoxygenation. Proc Natl Acad Sci U S A. 2003;100:11303–11308. doi: 10.1073/pnas.2033883100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang H. Andrekopoulos C. Xu Y. Joseph J. Hogg N. Feix J. Kalyanaraman B. Decreased S-Nitrosation of peptide thiols in the membrane interior. Free Radic Biol Med. 2009;47:962–968. doi: 10.1016/j.freeradbiomed.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang H. Xu Y. Joseph J. Kalyanaraman B. Intramolecular electron transfer between tyrosyl radical and cysteine residue inhibits tyrosine nitration and induces thiyl radical formation in model peptides treated with myeloperoxidase, H2O2, and NO2-: EPR spin trapping studies. J Biol Chem. 2005;280:40684–40698. doi: 10.1074/jbc.M504503200. [DOI] [PubMed] [Google Scholar]
- 104.Zhang Y. Hogg N. Mixing artifacts from bolus addition of nitric oxide to oxymyoglobin: implications for S-nitrosohtiol formation. Free Radic Biol Med. 2002;32:1212–1219. doi: 10.1016/s0891-5849(02)00829-8. [DOI] [PubMed] [Google Scholar]
- 105.Zhang Y. Hogg N. The Formation and Stability of S-Nitrosothiols in RAW 264.7 Cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L467–L474. doi: 10.1152/ajplung.00350.2003. [DOI] [PubMed] [Google Scholar]
- 106.Zhang Y. Hogg N. The mechanism of transmembrane S-nitrosothiol transport. Proc Natl Acad Sci U S A. 2004;101:7891–7896. doi: 10.1073/pnas.0401167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Y. Keszler A. Broniowska KA. Hogg N. Characterization and application of the biotin-switch assay for the identification of S-nitrosated proteins. Free Radic Biol Med. 2005;38:874–881. doi: 10.1016/j.freeradbiomed.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 108.Zhao YL. Houk KN. Thionitroxides, RSNHO: the structure of the SNO moiety in “S-Nitrosohemoglobin”, a possible NO reservoir and transporter. J Am Chem Soc. 2006;128:1422–1423. doi: 10.1021/ja057097f. [DOI] [PMC free article] [PubMed] [Google Scholar]