

Abstract
Background
Hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels generate the pacemaking current, Ih, which regulates neuronal excitability, burst firing activity, rhythmogenesis, and synaptic integration. The physiological consequence of HCN activation depends on regulation of channel gating by endogenous modulators and stabilization of the channel complex formed by principal and ancillary subunits. KCNE2 is a voltage-gated potassium channel ancillary subunit that also regulates heterologously expressed HCN channels; whether KCNE2 regulates neuronal HCN channel function is unknown.
Methodology/Principal Findings
We investigated the effects of Kcne2 gene deletion on Ih properties and excitability in ventrobasal (VB) and cortical layer 6 pyramidal neurons using brain slices prepared from Kcne2 +/+ and Kcne2 −/− mice. Kcne2 deletion shifted the voltage-dependence of Ih activation to more hyperpolarized potentials, slowed gating kinetics, and decreased Ih density. Kcne2 deletion was associated with a reduction in whole-brain expression of both HCN1 and HCN2 (but not HCN4), although co-immunoprecipitation from whole-brain lysates failed to detect interaction of KCNE2 with HCN1 or 2. Kcne2 deletion also increased input resistance and temporal summation of subthreshold voltage responses; this increased intrinsic excitability enhanced burst firing in response to 4-aminopyridine. Burst duration increased in corticothalamic, but not thalamocortical, neurons, suggesting enhanced cortical excitatory input to the thalamus; such augmented excitability did not result from changes in glutamate release machinery since miniature EPSC frequency was unaltered in Kcne2 −/− neurons.
Conclusions/Significance
Loss of KCNE2 leads to downregulation of HCN channel function associated with increased excitability in neurons in the cortico-thalamo-cortical loop. Such findings further our understanding of the normal physiology of brain circuitry critically involved in cognition and have implications for our understanding of various disorders of consciousness.
Introduction
The pacemaker current Ih, which is generated by hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels, regulates intrinsic excitability, synaptic integration and rhythmic oscillatory activity [1]–[3]. There are four Hcn genes, each coding for a distinct isoform (HCN1-4) (reviewed by Biel et al. 2009 [3]), which are variably distributed in the brain [4]. Although permeable to both Na+ and K+, HCN channels are members of the voltage-gated potassium channel superfamily. HCN channels are not inhibited by the inwardly rectifying K+ channel blockers Ba2+ or tetraethlylammonium, nor the voltage-gated K+ channel blocker 4-aminopyrindine, although they are inhibited by several different organic blockers, including ZD7288 [1], [3].
KCNE2, originally named MinK-related protein 1 (MiRP1), is a single transmembrane-spanning protein that acts as an ancillary (β) subunit for a number of potassium channel pore-forming α subunits, regulating channel conductance, voltage dependence, gating kinetics, trafficking and pharmacology [5]–[9] (for review see [10]). Studies using heterologous or over-expression systems have shown that co-expression of KCNE2 with HCN1, 2 or 4 significantly alters the amplitude and kinetics of Ih with variable effects on voltage-dependent gating [11]–[14]. KCNE2 also increases HCN1, HCN2, and HCN4 single channel conductance, further suggesting a direct interaction [14]. Despite these observations, however, the impact of KCNE2 expression on brain HCN channel function is unknown.
Kcne2 mRNA is present in many brain regions [15] where HCN isoforms are strongly expressed [16]–[18], raising the possibility that KCNE2 could directly influence the function of HCN channels in central neurons. KCNE2 is also highly expressed in the apical membrane of the choroid plexus epithelium, where it influences cerebrospinal fluid composition by regulating KCNQ1 and Kv1.3 K+ channel α subunits [19], potentially also indirectly influencing neuronal excitability. Thalamic neurons express HCN2 and HCN4, with HCN2 being the major functional isoform [20], [21] while cortical pyramidal neurons strongly express HCN1 [4].
Dysregulation of HCN channel function is strongly implicated in various experimental seizure models [22], [23] as well as in human epilepsy [24]. Changes in cellular excitability within corticothalamic circuits can result in seizure activity [25]–[27]. The corticothalamocortical circuit consists of reciprocal connections between the cortex and thalamus such that thalamic VB neurons project to layer 4 and 6 of the somatosensory cortex [28], and layer 6 pyramidal neurons in turn send axons to thalamic neurons, including those in VB [28], [29]. Thus, the thalamus and cortex are ideal regions to study the effects of KCNE2 on HCN channel function.
Here, using Kcne2 +/+ and Kcne2 −/− mice, we have discovered that targeted Kcne2 deletion alters Ih properties and neuronal excitability in VB and somatosensory cortex layer 6 neurons and reduces HCN1 and HCN2 protein expression in the brain. Preliminary results have been previously reported [30].
Methods
Ethics statement
All experiments were performed following approval by, and in accordance with, Weill Cornell Medical College, University of California, and US federal guidelines.
Generation of Kcne2−/− mice
Kcne2 +/+ and Kcne2 −/− C57BL/6 mice used in this study were generated by breeding Kcne2 +/− pairs and genotyped as described previously [31], [32].
Electrophysiology
A total of 78 mice of either sex (P60–96) were used for preparation of brain slices. Electrophysiological experiments in either current or voltage clamp configuration were performed as previously described [33], [34], and methods were slightly modified for this study. Briefly, thalamocortical slices (200–300 µm) were prepared using ice-cold slicing solution containing (in mM): 2 KCl, 26 NaHCO3, 1.25 NaH2PO4, 240 sucrose, 12 glucose, 2 MgSO4, 1 MgCl2, and 1 CaCl2. Whole-cell patch-clamp recordings were made from visually identified neurons in thalamic ventrobasal (VB) complex and somatosensory cortex layer 6. Slices were perfused with carbogenated normal artificial cerebrospinal fluid (ACSF), which contained (in mM): 126 NaCl, 26 NaHCO3, 3.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2 CaCl2, and 17 glucose. To isolate Ih currents in voltage-clamp recordings, an “Ih isolation solution” was used with the following compounds added to the ACSF (in mM): 0.001 tetrodotoxin (TTX, from Alomone Labs, Jerusalem, Israel), 2 4-aminopyridine (4-AP), 1 BaCl2 and 0.1 NiCl2; in some cases (n = 10), 0.02 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX: Tocris Bioscience, Ellisville, MO) and 0.04 DL-2-amino-5-phosphonopentanoic acid (AP5; Tocris) were included. For recordings of Ih and firing activity, the intracellular solution contained (in mM): 135 K+-gluconate, 5 NaCl, 10 HEPES, 0.5 EGTA, 3 K2-ATP, 0.2 Na-GTP, and 10 Na2-phosphocreatine, pH adjusted to 7.3 with KOH. For recordings of excitatory postsynaptic currents (EPSCs), the intracellular solution contained (in mM): 130 CH3SO3Cs, 10 CH3SO3Na, 5 NaCl, 1 CaCl2, 10 EGTA, 2 Mg2-ATP, 0.3 Na-GTP, and 10 HEPES, pH adjusted to 7.2 with CsOH. To obtain miniature EPSCs (mEPSCs), bicuculline (20 µM) and TTX (1 µM) were included in normal ACSF; neurons were clamped at −80 mV. CNQX (20 µM) and AP5 (40 µM) were used to identify EPSCs. All chemicals and drugs were purchased from Sigma unless otherwise noted.
Access resistance and capacitance were compensated after a whole-cell configuration was established, and were monitored throughout recordings; data were discarded if either of the two parameters changed by >20% of the original values. Liquid junction potentials were calculated and corrected off-line [35]. For recordings of Ih, neurons were voltage-clamped at −50 mV; 10- and 5-s hyperpolarizing voltage steps respectively were applied to VB and cortical neurons from −50 to −120 mV (10 mV/step).
Protein biochemistry
Co-immunoprecipitations (co-IPs) and associated western blots were performed as previously described [36] using brains from Kcne2 +/+ and Kcne2 −/− mice (∼P 90). For western blot analysis of whole-brain HCN protein expression (independent of the co-IP experiments), brain tissue was obtained from Kcne2 +/+ and Kcne2 −/− mice (∼P 90), homogenized in microcentrifuge tubes using motorized disposable pestles, then solubilized in PBS containing 20% (w/v) sodium dodecyl sulfate (SDS). Lysates were heated to 42°C for 30 minutes with multiple vortex mixing steps, centrifuged at 12,000 g for 15 minutes, and the supernatant retained. Supernatants (40 µg protein/lane) were size-fractionated on 8–12% Bis-Tris gels (Invitrogen, Grand Island, NY) with MES running buffer (HCN2, GAPDH; glyceraldehyde-3-phosphate dehydrogenase) or 3–8% Tris-Acetate gels (Invitrogen) with Tris-Acetate SDS running buffer (Invitrogen) (HCN1, GAPDH, Kv2.1, KCC1), transferred onto polyvinylidene difluoride (PVDF) membranes, and probed with mouse monoclonal antibodies against HCN1, HCN2 or HCN4 (UC Davis/NIH NeuroMab Facility), and GAPDH loading control (Sigma), or rabbit polyclonal antibodies raised against Kv2.1 (Sigma) or KCC1 (Chemicon/Millipore, Temecula, CA). Horseradish peroxidase (HRP)-conjugated goat anti-mouse or anti-rabbit immunoglobulin G (IgG) secondary antibody (Bio-Rad Labs, Hercules, CA) was used for visualization with chemiluminescence (ECL Plus; Amersham Biosciences, Piscataway, NJ, USA). Band intensities were compared directly from PVDF membrane chemiluminescence using the GBox imager (Syngene, Frederick, MD) after normalization for total protein concentration using the Bicinchoninic Acid (BCA) assay.
Data Analysis
Data processing and analysis including the construction of Ih activation curves, fit of time constants, and measurements of temporal summation, burst and tonic spike firing were performed using MiniAnalysis (Synaptosoft, Decatur, GA) or Clampfit 10 (Molecular Devices, Foster City, CA) as previously described [34]. Steady-state activation curves of Ih currents were determined from the tail current (for VB neurons, tail currents were analyzed upon returning to −50 mV while in cortical neurons, the tail current was measured at −60 mV); normalized tail current values were plotted as a function of the voltage steps, and were fitted with the Boltzmann function. Activation time constants were determined by fitting 4-s and 1-s segments of the current trace using a double exponential function [18], while tail current traces were fitted with a single exponential to obtain deactivation kinetics. Data are presented as mean ± SEM; statistical significance was determined using Student's t test or one-way ANOVA with pairwise comparisons, as appropriate.
Results
Kcne2 deletion impairs HCN channel function in VB neurons
VB neurons predominantly express HCN2 (and to a lesser extent HCN4) subunits in the soma and generate a large Ih current which slowly activates [20], [21], making the VB an excellent region in which to gain insights into whether KCNE2 influences native HCN function. We therefore compared properties of Ih currents recorded from VB neurons in brain slices prepared from Kcne2 +/+ and Kcne2 −/− mice. Families of Ih current traces were elicited by a series of 10 s hyperpolarizing voltage steps (Fig. 1A). Other voltage-gated ion channels (Na+, Ca2+ and K+) were blocked by concomitant application of tetrodotoxin, Ni2+, Ba2+ and 4-aminopyridine (see Methods). To determine the dependence of HCN channel activation on voltage, we measured tail current amplitudes at −50 mV following application of hyperpolarizing voltage steps. Tail currents were normalized to maximum amplitude and normalized values were fitted with the Boltzmann function (Fig. 1B) to construct activation curves. Group data demonstrate that there was a significant difference in the mid-point voltage of steady-state activation (V1/2 in mV): −84.3±0.7 for Kcne2 +/+ and −91.8±0.9 for Kcne2 −/− (P<0.001, t-test, n = 37/genotype); slope (mV) was not altered by the deletion.
Figure 1. Deletion of Kcne2 results in a hyperpolarizing shift in HCN channel activation in VB neurons.

A, Representative families of current traces recoded in VB neurons from Kcne2+/+ and Kcne2−/− mice; the voltage protocol is shown below. The tail currents from the same neurons are shown on an expanded time scale for comparison. Arrow indicates the trace elicited at −90 mV. B, Normalized tail currents (Itail) are plotted as a function of voltage steps and are fit with the Boltzmann equation. V1/2 values (in mV) were significantly shifted by the deletion to more hyperpolarized potentials. C, Comparison of Ih density in the two genotypes. *, P<0.01, one-way ANOVA, vs. Kcne2+/+. D, Deletion of Kcne2 did not alter Ih reversal potential.
All VB neurons recorded demonstrated a robust Ih (up to 5,500 pA) in both genotypes. Currents were normalized to cell capacitance to obtain Ih density (pA/pF, Fig. 1C). Although the maximum Ih density at −120 mV was not different between Kcne2 +/+ and Kcne2 −/− mice, Kcne2 deletion reduced Ih density in Kcne2 −/− neurons at physiological potentials (−80 and −90 mV, Fig. 1C) by virtue of the hyperpolarizing shift in the voltage dependence of activation (Fig. 1 B). Instantaneous Ih currents were not significantly changed by Kcne2 deletion (not shown).
To examine whether Kcne2 deletion altered the ion selectivity of VB HCN channels, Ih reversal potential was measured as previously described [33] from tail currents elicited by a series of voltage steps and plotted as a function of pre-pulse voltage. Ih reversal potential, and therefore HCN ion selectivity, was not significantly changed by Kcne2 deletion (Kcne2 +/+, −34.5±1.2 mV; Kcne2 −/−, −33.4±0.8 mV, n = 3 cells/genotype) (Fig. 1D).
We also analyzed the kinetics of channel activation by using fits of traces elicited at −120 mV with two exponential components. The fast time component (Taufast) was 0.35±0.026 s for Kcne2 +/+ and 0.707±0.05 s for Kcne2 −/−, and the slow time component (Tauslow) was 1.84±0.21 s for Kcne2 +/+ and 3.07±0.29 for Kcne2 −/− (Fig. 2A). Tail currents measured at −50 mV following a voltage step to −120 mV were fitted with a single exponential to quantify deactivation kinetics. Kcne2 deletion also increased the deactivation time constant (0.898±0.02 s vs. 1.97±0.045 s, Fig. 2B). Thus, Kcne2 deletion slowed the kinetics of both activation and deactivation, doubling the time constants for both processes.
Figure 2. Deletion of Kcne2 slows the activation and deactivation of HCN channels in VB neurons.

A, Overlay of current traces showing the effect of Kcne2 deletion on the activation time course of Ih at −120 mV. The activation time constant (Tauactivation) is determined by a two-exponential function, yielding fast (Taufast) and slow (Tauslow) components. Bar graph comparing the activation time constants. *, P<0.01, t-test, n = 26/genotype. B, Overlay of current traces showing the differences in the deactivation time of Ih; the Kcne2 −/− current trace (gray) is scaled to that of the Kcne2 +/+ trace (black) so as to better observe the kinetic differences in the currents. The dotted line indicates 0 pA. Bar graph summarizing the effect of Kcne2 deletion on the deactivation time constant (Taudeactivation). * P<0.05, t-test, n = 26/genotype.
To support the hypothesis that the altered VB currents in Kcne2 −/− mice arose from shifts in Ih rather than other currents, we compared the functional attributes of VB currents pharmacologically isolated using the Ih blocker ZD7288 in Kcne2 +/+ and Kcne2 −/− mice. Thus, “net” Ih, was calculated by subtracting current traces obtained in the presence of ZD7288 (50 µM) from “control” traces (no ZD7288). Comparison of control Ih with net Ih demonstrated no significant difference in HCN channel properties between these two groups within a given genotype, yet the genotype-dependent differences we observed using control Ih, i.e., a shift in voltage dependence (Fig. 1B) were recapitulated with net Ih, Thus, V1/2 values for control and net Ih, respectively, were (in mV) −85.2±2 vs. −83.3±2.6 for Kcne2 +/+ and −91.7±2 vs. −90.7±1.8 for Kcne2 −/− (Fig. 3C). The relationship of current-voltage curves was also the same for control versus net Ih within a given genotype (Fig. 3D). The data support the hypothesis that changes in Ih properties arising from Kcne2 deletion result from altered HCN channel function.
Figure 3. Kcne2 deletion does not alter Ih sensitivity to the HCN channel blocker ZD7288 in VB neurons.

A and B, Examplar families of current traces in the absence (control) and presence of ZD7288 (50 µM). Data were obtained using the same protocol as in Fig. 1A. Net Ih = control – ZD7288. C, Bar graph summarizing the effect of Kcne2 deletion on V1/2. *, P<0.001, t-test, n = 27 for control (no ZD7288) data and n = 16 for data with ZD7288. D, Comparison of net Ih density as a function of voltage. *, P<0.05, one-way ANOVA, vs. Kcne2+/+; the number of neurons is the same as in C.
Kcne2 deletion markedly down-regulates Ih in cortical pyramidal neurons
Kcne2 mRNA is detected at relatively high density in cortical dendritic regions [15], where HCN1 is also predominantly expressed [4], [37]. We therefore examined the potential influence of Kcne2 deletion on Ih properties in cortical layer 6 pyramidal (corticothalamic) neurons. Families of Ih current traces were obtained (Fig. 4A) using the same Ih isolation solution as for VB neurons. Analysis of steady-state activation curves (Fig. 4B) revealed that the deletion markedly shifted voltage dependence to more hyperpolarized potentials by 10.2±0.8 mV, with the V1/2 (in mV) being −81.2±0.6 for Kcne2 +/+ pyramidal neurons and −91.4±0.7 for Kcne2 −/− neurons (P<0.001, t-test, n = 10/genotype). There was no significant change in slope (9.2±0.6 vs. 8.9±0.5 mV, respectively). Unlike the scenario in VB, Ih current density (pA/pF) measured in these pyramidal neurons was small, and the maximal density detected at −120 mV was 2.8±0.26 pA/pF for Kcne2 +/+ and 1.9±0.15 pA/pF for Kcne2 −/−. The low Ih density in either genotype is consistent with uneven distribution of HCN subunits along the somatodendritic axis, with very low density over the somata and extremely high density in distal dendrites [4], [37], [38].
Figure 4. Loss of Kcne2 markedly down-regulates HCN channel function in layer 6 pyramidal neurons.

A, Families of Ih current traces were recorded in layer 6 neurons from both genotypes using the voltage protocol shown. Tail currents are shown on an expanded time scale for better comparison. Arrow indicates the trace elicited at −90 mV. B, Steady-state activation curves show that loss of Kcne2 resulted in a significant negative shift in voltage dependence (V1/2 in mV: −81.2±0.6 for Kcne2+/+ vs. −91.4±0.7 for Kcne2−/−, t-test, n = 15/genotype), with no apparent change in slope. C, Comparison of Ih density, *, P<0.05, one way ANOVA, vs. Kcne2+/+. D, Bar graphs summarizing the effects of Kcne2 deletion on the kinetics of HCN channel activation and deactivation. *, P<0.01.
Unlike VB neurons, Kcne2 −/− pyramidal neurons displayed a significant decrease in Ih current density across the −70 to −120 mV range (Fig. 4C). While activation time constants for Ih in pyramidal cells were fast in both genotypes compared to those in VB neurons, Kcne2 deletion again increased time constants in pyramidal neurons (Kcne2 +/+: Taufast, 0.06±0.004 s, Tauslow, 0.41±0.02 s; Kcne2 −/−: Taufast, 0.097±0.007, Tauslow, 0.71±0.07). The kinetics of both activation and deactivation for Ih were slowed in Kcne2 −/− pyramidal neurons (Fig. 4D). The kinetic data strongly suggest that Ih recorded from cortical pyramidal neurons is primarily generated by the HCN1 isoform [39]–[42] and slowed by Kcne2 deletion.
Kcne2 deletion enhances excitability and susceptibility to 4-AP in VB neurons
Down-regulation of HCN channel function by Kcne2 deletion would be predicted to alter intrinsic and synaptic excitability, and this possibility was investigated here. The resting membrane potential (RMP) was −68.6±0.7 mV for Kcne2 +/+ VB neurons compared to −72.1±0.8 mV for Kcne2 −/− VB neurons (n = 20). A small voltage response was elicited for measurement of input resistance by intracellular injection of a hyperpolarizing current pulse (−30 pA, 500 ms); Kcne2 deletion significantly increased input resistance from 182±14 to 288±12 MΩ (n = 12/genotype; Fig. 5A). Intrinsic temporal summation of subthreshold voltage response was evoked by intracellular injection of an EPSC-shaped current train [35], [43]. As shown in Fig. 5B, temporal summation (%) was 204±12 in Kcne2 +/+ neurons, and was significantly increased to 288±18 (P<0.01, n = 8/genotype) in neurons from Kcne2 −/− mice. Thus, Kcne2 deletion increased intrinsic excitability in VB neurons, raising the question as to whether excitatory synaptic transmission was altered.
Figure 5. Deletion of Kcne2 increases sensitivity of VB neurons to the convulsant 4-AP.

A, Comparison of input resistance (Rin) between genotypes. Voltage traces were elicited by intracellular injection of a hyperpolarizing current pulse (−30 pA, 500 ms) at holding potential of −70 mV. *, P<0.01. B, Comparison of intrinsic temporal summation of the voltage response in representative VB neurons from each genotype. Voltage traces were elicited by intracellular injection of an EPSC-shared train (5 pulses, 200 pA, 33 Hz). *, P<0.01, t-test. C, Voltage traces showing spontaneous activity in the absence (control) and presence of 4-aminopyridine (4-AP). The first burst from each genotype is expanded for better view of fast spikes. Circles indicate single fast action potentials; a single spike in the Kcne2+/+ neuron is shown for comparison. D. Bar graph summarizing burst firing data. *, P<0.05, t-test, n = 12/genotype.
Spontaneous EPSPs were evident in both genotypes (Fig. 5C, and see below for additional information) and could be blocked by CNQX and AP5 (not shown). As very few spontaneous bursts were observed in either genotype under control conditions, we used a low concentration of 4-aminopyridine (4-AP; 0.1 mM) to study burst firing and neuronal sensitivity to convulsant challenge. Bath-application of 4-AP induced low-threshold bursts in both genotypes. Fast single action potentials occurred between bursts in Kcne2 +/+ neurons but few appeared in Kcne2 −/− cells. Group data indicate that the burst frequency was significantly higher in Kcne2 −/− neurons than that in Kcne2 +/+ neurons (Fig. 5 D) although no significant change in burst duration or spikes/burst was observed. These data indicate that Kcne2 deletion increases intrinsic excitability and facilitates low-threshold burst firing in thalamocortical VB neurons.
Loss of kcne2 produces hypersusceptibility to 4-AP in layer 6 pyramidal neurons
Since Ih density was significantly reduced across the voltage range in Kcne2−/− layer 6 pyramidal neurons (Fig. 4C), we also examined whether excitability in these cells was altered. Input resistance significantly increased in Kcne2 −/− pyramidal neurons compared to those from Kcne2 +/+ mice (395±18 vs. 172±11 MΩ, n = 8/genotype, P<0.001, Fig. 6C). Temporal summation was tested in both genotypes using the same protocol for VB neurons (traces not shown). Kcne2 deletion significantly increased summation by 45±3.5% (220±14% vs. 320±16%, n = 8). As was the case with VB neurons, spontaneous EPSPs were evident in both genotypes in the absence (control) of 4-AP; however, the frequency was higher in Kcne2 −/− than in Kcne2 +/+ pyramidal neurons (Fig. 6 A–B). Bath application of 4-AP (0.1 mM) induced rhythmic low threshold Ca2+ spike (LTS) burst firing patterns in both genotypes; such bursting could last for more than 90 min. Group data for cortical burst properties are summarized in Fig. 6D. Compared to Kcne2 +/+, the burst frequency in Kcne2 −/− pyramidal neurons was much higher (0.18±0.01 vs. 0.06±0.008 Hz), burst duration was longer (3.4±0.3 s vs. 1.2±0.02 s), and the number of fast spikes per burst larger (88.2±7.2 vs. 18.5).
Figure 6. Loss of Kcne2 results in hypersusceptibility to 4-AP in cortical layer 6 pyramidal neurons of somatosensory cortex.

A and B, Representative voltage traces in the absence (control) and presence of 4-aminopyridine (4-AP) in both genotypes. The first burst (marked by arrow) is shown on an expanded time scale to better view fast spikes. Kcne2 −/− pyramidal neurons exhibited prolonged bursts (increased burst duration), as compared to wild-types. C, Voltage responses were elicited to determine input resistance (Rin) by intracellular injections of a hyperpolarizing current pulse (−30 pA, 500 ms). +/+, Kcne2+/+; −/−, Kcne2−/−. Bar graph showing comparison of Rin values in the two genotypes. *, P<0.00, t-test. D, Bar graphs summarizing burst firing properties. *, P<0.05, t-test.
In Kcne2 −/− mice, cortical burst duration was also much longer than that seen in VB counterparts (3.4±0.3 s vs. 0.6 s±0.04, P<0.001), a 5.6-fold increase. The magnitude of the effects on voltage responses in Kcne2 −/− neurons was much larger in corticothalamic pyramidal neurons than that observed in thalamocortical VB neurons, implying that corticothalamic excitatory transmission was increased in brain slices from Kcne2 −/− mice.
Kcne2 deletion had little effect on glutamate release
As noted above, the frequency of spontaneous EPSPs in layer 6 pyramidal neurons in the absence of 4-AP was increased by Kcne2 deletion, suggesting a possible alteration in glutamate release machinery. To investigate this possibility, miniature excitatory postsynaptic currents (mEPSCs) were recorded in both genotypes in the absence of 4-AP but the presence of TTX (500 nM) and bicuculline (20 µM), thereby blocking spike-driven events and fast GABAergic transmission. Fast mEPSCs were readily detected in pyramidal neurons from both genotypes and could be blocked by co-application of CNQX and AP-5 (Fig. 7A–B), confirming that they were mediated by ionotropic glutamate receptors. mEPSC frequency (8.3±1.1 vs. 9.8±1.5 Hz, Kcne2 +/+ and Kcne2 −/−, respectively, n = 6 cells/genotype), and amplitude (29.8±1.3 vs. 31.2±1.4 pA) were similar between the two genotypes. mEPSC decay time, however, was significantly prolonged in Kcne2 −/− neurons (1.58±0.2 vs. 0.54±0.05 ms) (Fig. 7C–D), and the prolongation in the decay time was consistent with the observed increase in Rin (cf. [44]). The data suggest that Kcne2 deletion does not alter glutamate release from presynaptic glutamatergic neurons, and that the enhanced excitability observed above is likely due to the reduction of shunting produced by Ih, and appears similar to what has been described for HCN1-null cortical pyramidal neurons [45].
Figure 7. Kcne2 deletion has little effect on glutamate release.

A and B, Miniature EPSCs (mEPSCs) were recorded in cortical layer 6 pyramidal neurons from both genotypes in the presence of bicuculline (20 µM) and TTX (1 µM) at holding potential of −80 mV. Synaptic currents in both genotypes could be blocked by co-application of CNQX 20 µM and AP5 40 µM. C, Overlay of averaged mEPSCs for the two genotypes. D, Bar graphs comparing mEPSC frequency, decay time and amplitude (n = 5/genotype).
Kcne2 deletion reduces brain HCN1 and HCN2 protein expression
Co-IP experiments were employed to determine whether KCNE2 forms protein complexes with HCN1 and HCN2 in mouse brain. Western blots using HCN1 or HCN2 antibodies to probe KCNE2 antibody-precipitated fractions did not yield specific signal (data not shown), suggesting either these complexes do not form in mouse brain, that the amount of complex formation is below our detection limit, or our co-IP protocol did not preserve native complexes.
As Kcne2 deletion reduced native VB and pyramidal neuron Ih, we also quantified HCN1, HCN2 and HCN4 protein in Kcne2 +/+ and Kcne2 −/− whole-brain lysates to determine if Kcne2 deletion altered HCN1, HCN2 and HCN4 protein expression. Strikingly, neural expression of HCN2 protein was significantly reduced in Kcne2-deleted mice compared to Kcne2 +/+ mice (P = 0.02) and there was a trend toward reduction of HCN1 protein expression (P = 0.07) (Fig. 8A–B). This change appears to be specific as HCN4 protein expression was unchanged (Fig. 8A–B) and whole-brain expression of two other membrane proteins, the Kv2.1 K+ channel α subunit and the KCC1 K+/Cl− co-transporter, was unaffected by Kcne2 deletion (data not shown).
Figure 8. Kcne2 deletion down-regulates HCN1 and HCN2 protein expression in the brain.
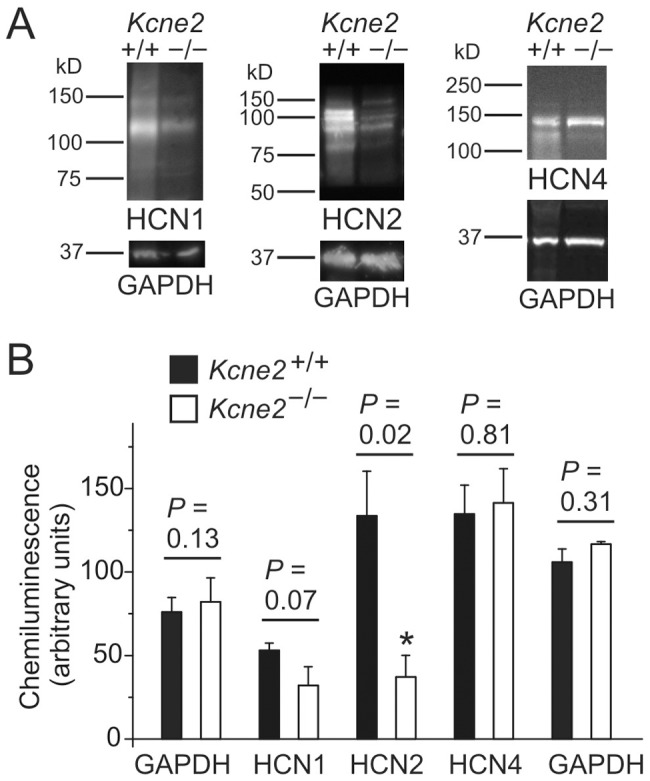
A, Exemplar chemiluminescence signals from western blots of whole brain lysates from Kcne2 +/+ and Kcne2 −/− mice, normalized to total protein concentration and probed with antibodies raised against HCN1, HCN2, HCN4 or GAPDH, as indicated. B, Mean chemiluminescence intensities for bands corresponding to known molecular weights for HCN1, HCN2, HCN4 and GAPDH from blots as in panel A, n = 3–4 mice per genotype. The cumulative GAPDH data on the left were obtained concomitantly with the HCN1 and HCN2 samples while the comparable GAPDH data on the right were obtained concomitantly with the HCN4 samples. *Significant difference between genotypes at 95% confidence interval. Error bars indicate SEM.
Discussion
KCNE2 is a voltage-gated potassium channel ancillary subunit that also regulates heterologously expressed HCN channels. We present novel data which demonstrate that KCNE2 is required for normal HCN channel activity in central neurons.
Kcne2 deletion impairs neuronal Ih
Previous studies have provided evidence that the potassium channel β subunit KCNE2 regulates heterologously expressed HCN1, 2 and 4 channels to modulate the activation kinetics and amplitude of Ih [11], [14]. Previous investigations of the ability of KCNE2 to modulate HCN gating in heterologous expression systems have yielded variable results. Thus, in Xenopus oocytes expressing HCN1 or HCN2, KCNE2 co-expression produces a small (4 mV) depolarizing shift in V1/2 [11] while co-expression of KCNE2 with HCN4 produced a significant hyperpolarizing shift (8 mV) in V1/2 [12]. In Chinese Hamster Ovary (CHO) cells, KCNE2 co-expression with HCN1, 2, or 4 had no effect on the V1/2, although other Ih properties were markedly altered [14]. Similarly, over-expression of KCNE2 with HCN2 failed to alter HCN gating in neonatal ventricular myocytes [13]. Given the contradictory results obtained using different expression systems it is therefore necessary, as previously noted [3], to study the effects of native KCNE proteins on native HCN channel function.
Kcne2 acceleration of current activation
The activation time constants for Ih in Kcne2+/+ VB neurons was much slower than that of layer 6 pyramidal neurons (the fast component being 5.6-fold slower and the slow component being 4.5-fold slower), consistent with previous reports of predominant expression of HCN2 in VB and HCN1 in the cortex [16], [17], [41], [42], [46]–[48]. Deletion of Kcne2 led to a significant slowing of activation time constants in both VB and cortical neurons (Figs. 2 and 4). Our data are consistent with previous studies using heterologous expression models showing that the presence of KCNE2 accelerates the activation kinetics of all HCN channel subtypes (HCN1, 2 and 4) [13], [14], although there is one report indicating that KCNE2 co-expression with HCN4 slows activation [12].
A comparison of neuronal and recombinant channel properties raises an important question with respect to direct regulation of gating kinetics. When HCN channels are heterologously expressed, the presence of KCNE2 accelerates the kinetics of current activation resulting from homomeric HCN channel expression, and this likely reflects a direct protein-protein interaction [12]. The kinetics of Ih in CNS neurons, however, appear to be strongly influenced by HCN channel composition in different cell populations, and expression of heteromultimeric channels may also contribute to differences in the kinetics of neuronal Ih [18]. VB neurons primarily express HCN2 channels, with a smaller population of HCN4 channels also present [20], [21]. Given that KCNE2 accelerates Ih activation in heterologous expression systems (as discussed above), the slower kinetics of Ih observed in kcne2−/− VB neurons may be due to loss of direct regulation by KCNE2, but could also be due to alterations in HCN subunit expression. As shown in Fig. 8, there is a significant decrease in overall HCN2 expression in the brain when Kcne2 is knocked out, while overall brain HCN4 expression remains constant. This could theoretically increase the relative contribution of the more slowly activating HCN4 (compared to HCN2) to native Ih [48], [49], thereby giving rise to a slowly activating Ih current in VB neurons.
KCNE2 protein has been detected in a range of tissues, and its deletion impacts the function of the stomach, heart and thyroid, all of which normally express KCNE2 in mice and humans [10], [19], [31], [32], [50]. While Kcne2 mRNA has been detected in a range of neural tissues [15], in a recent study we did not observe specific KCNE2 protein staining by immunohistochemistry in neuronal populations of mouse brain. In contrast, robust, highly specific KCNE2 protein staining was apparent in the apical membrane of the choroid plexus epithelium, which lines the fourth and lateral ventricles of the brain and secretes cerebrospinal fluid [19]. It is possible, then, that Kcne2 deletion alters neuronal Ih characteristics indirectly by, for example, altering the cerebrospinal fluid composition in a manner that leads to electrical remodeling such as the reduction in HCN2 protein expression we observed here. This could not only reduce Ih density, but also lead to slower-activating Ih because of a shift in the balance between HCN2 and HCN4.
Kcne2 critically regulates excitability and burst firing
Alteration of Ih is associated with a marked change in intrinsic excitability in thalamic neurons [33], [34], [43]. Here we found that downregulation of HCN channel function by Kcne2 deletion resulted in the increase of input resistance and temporal summation of subthreshold voltage response in both thalamic and cortical neurons, indicating an increase in intrinsic excitability (Figs. 5 and 6). As a result, burst firing also increased in Kcne2-null brain slices. Previous studies have shown that genetic downregulation or pharmacological diminishment of the Ih conductance is tightly linked to the occurrence of seizure activity [20], [51]–[54] and the decrease of seizure threshold during convulsant challenge [20], [45]. We found that a low concentration of 4-AP induced long-lasting burst firing, which is similar to epileptiform activity [55]–[58]. The burst firing frequency was significantly increased by Kcne2 deletion in both thalamocortical and layer 6 pyramidal neurons, suggesting increased susceptibility to the convulsant challenge. Intriguingly, the duration of bursts in Kcne2-null pyramidal, but not thalamocortical, neurons was much longer, with a marked increase in fast action potentials riding on bursting calcium spikes. Such augmented excitability did not seem to result from a direct alteration of the glutamatergic release machinery as neither mEPSC amplitude nor frequency were changed by the deletion (Fig. 7), but was likely mediated through shunting reduction caused by downregulation of Ih (Fig. 6).
HCN1 channels show a 60-fold increase from somatic to distal dendritic membrane in layer 5 pyramidal neurons [37] and not surprisingly, dendritic Ih plays a pivotal role in controlling pyramidal cell excitability [59]. A recent study has shown that cortical layer 6 pyramidal dendrites also possess membrane properties similar to those of other layer pyramidal neurons [38]. Hence, downregulation of cortical dendritic Ih leads to reduction of dendritic shunting and consequently constraints on local spike propagation to the soma, thereby facilitating synaptic integration and excitatory synaptic transmission. This effect may account for the occurrence of cortical hypersusceptibility to 4-AP observed here, and suggests that Kcne2-null mice may be more susceptible to chemical-induced seizures because of dysregulation of HCN channel function. Since 4-AP-induced synchronized oscillations (epileptiform activity) originate in cortical layer 6 and propagate to thalamus through corticothalamic projections [56], the increase in apparent EPSP frequency and burst firing in thalamocortical VB neurons (Fig. 5) may result in part from increased corticothalamic excitatory synaptic input [56], which in turn leads to an enhancement of thalamocortical output to layer 4 and 6 [28], creating an imbalance between excitation and inhibition in the cortico-thalamo-cortical loop. Our results (Fig. 4 and 6) support the notion that dendritic HCN1 plays a critical role for the regulation of cortical pyramidal excitability [45].
KCNE2 can modulate the function of other ion channels [10], [60], including the 4-aminopyridine (4-AP)-sensitive rapidly activated, transient outward potassium current IA, which is mediated by the KCND3 gene-product, Kv4.3 [61]. KCNE2 is a β subunit for Kv4.3 [62], and Kv4.3 mRNA is present in the cortex and thalamus [63]; in a thalamic relay neuron model, IA slows the rate of rise and reduces the peak amplitude of the low-threshold Ca2+ spike and reducing IA to 0 results in a decrease in the inter-spike interval and in increase in the response of the cell to a depolarizing current injection [64]. Those observations raise the theoretical possibility that the observed changes in excitability observed in thalamic (Fig. 5) and cortical (Fig. 6) neurons resulted from 4-AP block of IA.
We do not think that the enhanced excitability in Kcne2 −/− mice is the result of changes in Kv4.3 function for the following reasons. Firstly, at membrane potentials less than −65 mV, Kcne2 deletion by itself does not produce spontaneous spike firing in either thalamic (Fig. 5C, top current trace) or cortical (Fig. 6A, top current trace) neurons, indicating that IA-dependent control of spontaneous spike firing is not altered by the deletion. This is not entirely unexpected as IA and Ih co-vary and have opposing and complementary effects [65]–[67]. Secondly, there is little if any measurable IA at membrane potentials less than −60 mV, as its V1/2 is quite positive (∼−36 mV) [68], and the excitability experiments in the present study were performed at membrane potentials more negative than −65 mV. Thirdly, 4-AP blocks neuronal IA in the low millimolar range, with an IC50 of 1 to 2 mM [68], [69] (similar to the IC50 of ∼1.5 mM reported in HEK293 cells expressing Kv4.3 channels [70]), and the concentration used to induce burst firing in the present study was 0.1 mM; at this concentration, IA is only blocked by 7% [69]. Finally, absence of IA (as would occur in the presence of full block) produces a very different pattern of activity (at least in a model thalamic cell [64]) than what we observed. In total, these studies strongly indicate that the burst firing patterns observed in the present study did not result from 4-AP modulation of Kv4.3 mediated IA currents.
Limitations of the present study
Our Ih data from VB and pyramidal neurons demonstrate that native HCN gating is KCNE2-dependent inasmuch as Kcne2 deletion alters native HCN gating, but the lack of native co-immunoprecipitation between KCNE2 and HCN1 or HCN2 from the brain in our study leaves the door open for at least several potential mechanisms for this functional dependence. First, KCNE2 mRNA is clearly detected in cortical and thalamic neurons [15], and it is possible that KCNE2 directly regulates HCN channels in the brain but our biochemical studies did not provide sufficient resolution to detect this, or the necessary conditions to preserve complex stability. Thus, the slowed gating and reduced current density of Ih we observe upon Kcne2 deletion could stem from the loss of KCNE2 from HCN channel complexes, which would be predicted to slow gating and reduce current density via reduced single channel conductance [11]–[14]. Second, KCNE2 could function as an ancillary subunit and regulate HCN trafficking to specific regions of the cell membrane in a manner analogous to that of TRIP8B [71]–[74], so in the absence of KCNE2 perhaps HCN surface expression is impaired. Third, it is possible that KCNE2 indirectly impacts neuronal HCN function due to modulatory effects on other ionic currents, including those mediated by a number of voltage-gated potassium channels [10], [60], and that those changes indirectly alter the HCN-mediated current. Fourth, we previously observed that KCNE2 levels were highest in the choroid plexus [19], and KCNE2 is present in many other non-neuronal tissues [50], [75], so the observed effects could be indirect from one or more of a variety of sources within the mouse. These caveats notwithstanding, Kcne2 deletion is the root cause of the observed effects reported here.
Conclusion
In summary, voltage-dependent gating of native HCN channels in CNS neurons appears to be controlled by multiple factors, including in some shape or form the β subunit KCNE2, as shown here. The present findings have revealed for the first time that KCNE2 exerts an important role in the maintenance of brain pacemaking function at physiological membrane potentials. Loss of KCNE2 leads to downregulation of HCN channel function associated with increased excitability in neurons in the cortico-thalamo-cortical loop. Thus, KCNE2 strongly influences HCN channel activity crucial for homeostatic regulation of a dynamic balance of excitation and inhibition [76] in the interconnected circuitry. In future work, the specific mechanisms for this functional link will be further investigated.
Funding Statement
This work was supported by the Dept. of Anesthesiolgy and by the National Institutes of Health (R01 HL079275 and R01HL101190, both to GWA). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Robinson RB, Siegelbaum SA (2003) Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol 65: 453–480. [DOI] [PubMed] [Google Scholar]
- 2. Wahl-Schott C, Biel M (2009) HCN channels: structure, cellular regulation and physiological function. Cell Mol Life Sci 66: 470–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Biel M, Wahl-Schott C, Michalakis S, Zong X (2009) Hyperpolarization-activated cation channels: from genes to function. Physiol Rev 89: 847–885. [DOI] [PubMed] [Google Scholar]
- 4. Notomi T, Shigemoto R (2004) Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J Comp Neurol 471: 241–276. [DOI] [PubMed] [Google Scholar]
- 5. Tinel N, Diochot S, Borsotto M, Lazdunski M, Barhanin J (2000) KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J 19: 6326–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang M, Jiang M, Tseng GN (2001) minK-related peptide 1 associates with Kv4.2 and modulates its gating function: potential role as β subunit of cardiac transient outward channel Circ Res 88: 1012–1019. [DOI] [PubMed] [Google Scholar]
- 7. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, et al. (1999) MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97: 175–187. [DOI] [PubMed] [Google Scholar]
- 8. Kanda VA, Lewis A, Xu X, Abbott GW (2011) KCNE1 and KCNE2 provide a checkpoint governing voltage-gated potassium channel α-subunit composition. Biophys J 101: 1364–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kanda VA, Lewis A, Xu X, Abbott GW (2011) KCNE1 and KCNE2 inhibit forward trafficking of homomeric N-type voltage-gated potassium channels. Biophys J 101: 1354–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McCrossan ZA, Abbott GW (2004) The MinK-related peptides. Neuropharmacology 47: 787–821. [DOI] [PubMed] [Google Scholar]
- 11. Yu H, Wu J, Potapova I, Wymore RT, Holmes B, et al. (2001) MinK-related peptide 1: A β subunit for the HCN ion channel subunit family enhances expression and speeds activation. Circ Res 88: E84–87. [DOI] [PubMed] [Google Scholar]
- 12. Decher N, Bundis F, Vajna R, Steinmeyer K (2003) KCNE2 modulates current amplitudes and activation kinetics of HCN4: influence of KCNE family members on HCN4 currents. Pflugers Arch 446: 633–640. [DOI] [PubMed] [Google Scholar]
- 13. Qu J, Kryukova Y, Potapova IA, Doronin SV, Larsen M, et al. (2004) MiRP1 modulates HCN2 channel expression and gating in cardiac myocytes. J Biol Chem 279: 43497–43502. [DOI] [PubMed] [Google Scholar]
- 14. Brandt MC, Endres-Becker J, Zagidullin N, Motloch LJ, Er F, et al. (2009) Effects of KCNE2 on HCN isoforms: distinct modulation of membrane expression and single channel properties. Am J Physiol Heart Circ Physiol 297: H355–363. [DOI] [PubMed] [Google Scholar]
- 15. Tinel N, Diochot S, Lauritzen I, Barhanin J, Lazdunski M, et al. (2000) M-type KCNQ2-KCNQ3 potassium channels are modulated by the KCNE2 subunit. FEBS Lett 480: 137–141. [DOI] [PubMed] [Google Scholar]
- 16. Monteggia LM, Eisch AJ, Tang MD, Kaczmarek LK, Nestler EJ (2000) Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Brain Res Mol Brain Res 81: 129–139. [DOI] [PubMed] [Google Scholar]
- 17. Moosmang S, Biel M, Hofmann F, Ludwig A (1999) Differential distribution of four hyperpolarization-activated cation channels in mouse brain. Biol Chem 380: 975–980. [DOI] [PubMed] [Google Scholar]
- 18. Santoro B, Chen S, Lüthi A, Pavlidis P, Shumyatsky GP, et al. (2000) Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci 20: 5264–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roepke TK, Kanda VA, Purtell K, King EC, Lerner DJ, et al. (2011) KCNE2 forms potassium channels with KCNA3 and KCNQ1 in the choroid plexus epithelium. FASEB J 25: 4264–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, et al. (2003) Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J 22: 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abbas SY, Ying SW, Goldstein PA (2006) Compartmental distribution of hyperpolarization-activated cyclic-nucleotide-gated channel 2 and hyperpolarization-activated cyclic-nucleotide-gated channel 4 in thalamic reticular and thalamocortical relay neurons. Neuroscience 141: 1811–1825. [DOI] [PubMed] [Google Scholar]
- 22. Lewis AS, Chetkovich DM (2011) HCN channels in behavior and neurological disease: too hyper or not active enough Mol Cell Neurosci 46: 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Noam Y, Bernard C, Baram TZ (2011) Towards an integrated view of HCN channel role in epilepsy. Curr Opin Neurobiol 21 6 873–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wierschke S, Lehmann TN, Dehnicke C, Horn P, Nitsch R, et al. (2010) Hyperpolarization-activated cation currents in human epileptogenic neocortex. Epilepsia 51: 404–414. [DOI] [PubMed] [Google Scholar]
- 25. Timofeev I, Steriade M (2004) Neocortical seizures: initiation, development and cessation. Neuroscience 123: 299–336. [DOI] [PubMed] [Google Scholar]
- 26. Blumenfeld H (2005) Cellular and network mechanisms of spike-wave seizures. Epilepsia 46 Suppl 9: 21–33. [DOI] [PubMed] [Google Scholar]
- 27. Huguenard JR, McCormick DA (2007) Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci 30: 350–356. [DOI] [PubMed] [Google Scholar]
- 28. Jones EG (2009) Synchrony in the interconnected circuitry of the thalamus and cerebral cortex. Ann N Y Acad Sci 1157: 10–23. [DOI] [PubMed] [Google Scholar]
- 29. Lam YW, Sherman SM (2010) Functional organization of the somatosensory cortical layer 6 feedback to the thalamus. Cereb Cortex 20: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ying SW, Kanda VA, King EC, Purtell K, Abbott GW, et al. (2011) Kcne2 modulates HCN channel function in thalamic neurons. In: Daily Books, 40th Annual Meeting of the Society for Neuroscience, Neuroscience 2010. [Google Scholar]
- 31. Roepke TK, Anantharam A, Kirchhoff P, Busque SM, Young JB, et al. (2006) The KCNE2 potassium channel ancillary subunit is essential for gastric acid secretion. J Biol Chem 281: 23740–23747. [DOI] [PubMed] [Google Scholar]
- 32. Roepke TK, Kontogeorgis A, Ovanez C, Xu X, Young JB, et al. (2008) Targeted deletion of kcne2 impairs ventricular repolarization via disruption of IK,slow1 and Ito,f . FASEB J 22: 3648–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ying SW, Abbas SY, Harrison NL, Goldstein PA (2006) Propofol block of Ih contributes to the suppression of neuronal excitability and rhythmic burst firing in thalamocortical neurons. Eur J Neurosci 23: 465–480. [DOI] [PubMed] [Google Scholar]
- 34. Ying SW, Jia F, Abbas SY, Hofmann F, Ludwig A, et al. (2007) Dendritic HCN2 channels constrain glutamate-driven excitability in reticular thalamic neurons. J Neurosci 27: 8719–8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ying SW, Goldstein PA (2005) Propofol-block of SK channels in reticular thalamic neurons enhances GABAergic inhibition in relay neurons. J Neurophysiol 93: 1935–1948. [DOI] [PubMed] [Google Scholar]
- 36. McCrossan ZA, Lewis A, Panaghie G, Jordan PN, Christini DJ, et al. (2003) MinK-related peptide 2 modulates Kv2.1 and Kv3.1 potassium channels in mammalian brain. J Neurosci 23: 8077–8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lörincz A, Notomi T, Tamás G, Shigemoto R, Nusser Z (2002) Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci 5: 1185–1193. [DOI] [PubMed] [Google Scholar]
- 38. Ledergerber D, Larkum ME (2010) Properties of layer 6 pyramidal neuron apical dendrites. J Neurosci 30: 13031–13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen S, Wang J, Siegelbaum SA (2001) Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol 117: 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ulens C, Tytgat J (2001) Functional heteromerization of HCN1 and HCN2 pacemaker channels. J Biol Chem 276: 6069–6072. [DOI] [PubMed] [Google Scholar]
- 41. Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, et al. (1998) Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell 93: 717–729. [DOI] [PubMed] [Google Scholar]
- 42. Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M (1998) A family of hyperpolarization-activated mammalian cation channels. Nature 393: 587–591. [DOI] [PubMed] [Google Scholar]
- 43. Ying SW, Tibbs GR, Picollo A, Abbas SY, Sanford RL, et al. (2011) PIP2-Mediated HCN3 Channel Gating Is Crucial for Rhythmic Burst Firing in Thalamic Intergeniculate Leaflet Neurons. J Neurosci 31: 10412–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang Z, Lujan R, Kadurin I, Uebele VN, Renger JJ, et al. (2011) Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat Neurosci 14: 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang Z, Walker MC, Shah MM (2009) Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J Neurosci 29: 10979–10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Santoro B, Grant SG, Bartsch D, Kandel ER (1997) Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc Natl Acad Sci U S A 94: 14815–14820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Biel M, Ludwig A, Zong X, Hofmann F (1999) Hyperpolarization-activated cation channels: a multi-gene family. Rev Physiol Biochem Pharmacol 136: 165–181. [DOI] [PubMed] [Google Scholar]
- 48. Seifert R, Scholten A, Gauss R, Mincheva A, Lichter P, et al. (1999) Molecular characterization of a slowly gating human hyperpolarization-activated channel predominantly expressed in thalamus, heart, and testis. Proc Natl Acad Sci U S A 96: 9391–9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ludwig A, Zong X, Stieber J, Hullin R, Hofmann F, et al. (1999) Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J 18: 2323–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roepke TK, King EC, Reyna-Neyra A, Paroder M, Purtell K, et al. (2009) Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis. Nat Med 15: 1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Budde T, Caputi L, Kanyshkova T, Staak R, Abrahamczik C, et al. (2005) Impaired regulation of thalamic pacemaker channels through an imbalance of subunit expression in absence epilepsy. J Neurosci 25: 9871–9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kole MH, Bräuer AU, Stuart GJ (2007) Inherited cortical HCN1 channel loss amplifies dendritic calcium electrogenesis and burst firing in a rat absence epilepsy model. J Physiol 578: 507–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jung S, Bullis JB, Lau IH, Jones TD, Warner LN, et al. (2010) Downregulation of dendritic HCN channel gating in epilepsy is mediated by altered phosphorylation signaling. J Neurosci 30: 6678–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kanyshkova T, Meuth P, Bista P, Liu Z, Ehling P, et al. (2012) Differential regulation of HCN channel isoform expression in thalamic neurons of epileptic and non-epileptic rat strains. Neurobiol Dis 45: 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hoffman SN, Prince DA (1995) Epileptogenesis in immature neocortical slices induced by 4-aminopyridine. Brain Res Dev Brain Res 85: 64–70. [DOI] [PubMed] [Google Scholar]
- 56. Golshani P, Jones EG (1999) Synchronized paroxysmal activity in the developing thalamocortical network mediated by corticothalamic projections and “silent” synapses. J Neurosci 19: 2865–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gonzalez-Sulser A, Wang J, Motamedi GK, Avoli M, Vicini S, et al. (2011) The 4-aminopyridine in vitro epilepsy model analyzed with a perforated multi-electrode array. Neuropharmacology 60: 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Salah A, Perkins KL (2011) Persistent ictal-like activity in rat entorhinal/perirhinal cortex following washout of 4-aminopyridine. Epilepsy Res 94: 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Berger T, Larkum ME, Lüscher HR (2001) High Ih channel density in the distal apical dendrite of layer V pyramidal cells increases bidirectional attenuation of EPSPs. J Neurophysiol 85: 855–868. [DOI] [PubMed] [Google Scholar]
- 60. Pongs O, Schwarz JR (2010) Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev 90: 755–796. [DOI] [PubMed] [Google Scholar]
- 61. Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, et al. (1999) Molecular diversity of K+ channels. Ann N Y Acad Sci 868: 233–285. [DOI] [PubMed] [Google Scholar]
- 62. Deschênes I, Tomaselli GF (2002) Modulation of Kv4.3 current by accessory subunits. FEBS Lett 528: 183–188. [DOI] [PubMed] [Google Scholar]
- 63. Tsaur ML, Chou CC, Shih YH, Wang HL (1997) Cloning, expression and CNS distribution of Kv4.3, an A-type K+ channel α subunit. FEBS Lett 400: 215–220. [DOI] [PubMed] [Google Scholar]
- 64. McCormick DA, Huguenard JR (1992) A model of the electrophysiological properties of thalamocortical relay neurons. J Neurophysiol 68: 1384–1400. [DOI] [PubMed] [Google Scholar]
- 65. MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM (2003) Activity-independent homeostasis in rhythmically active neurons. Neuron 37: 109–120. [DOI] [PubMed] [Google Scholar]
- 66. MacLean JN, Zhang Y, Goeritz ML, Casey R, Oliva R, et al. (2005) Activity-independent coregulation of IA and Ih in rhythmically active neurons. J Neurophysiol 94: 3601–3617. [DOI] [PubMed] [Google Scholar]
- 67. Amendola J, Woodhouse A, Martin-Eauclaire MF, Goaillard JM (2012) Ca2+/cAMP-sensitive covariation of IA and IH voltage dependences tunes rebound firing in dopaminergic neurons. J Neurosci 32: 2166–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Huguenard JR, Coulter DA, Prince DA (1991) A fast transient potassium current in thalamic relay neurons: kinetics of activation and inactivation. J Neurophysiol 66: 1304–1315. [DOI] [PubMed] [Google Scholar]
- 69. Whyment AD, Coderre E, Wilson JM, Renaud LP, O'Hare E, et al. (2011) Electrophysiological, pharmacological and molecular profile of the transient outward rectifying conductance in rat sympathetic preganglionic neurons in vitro . Neuroscience 178: 68–81. [DOI] [PubMed] [Google Scholar]
- 70. Faivre JF, Calmels TP, Rouanet S, Javre JL, Cheval B, et al. (1999) Characterisation of Kv4.3 in HEK293 cells: comparison with the rat ventricular transient outward potassium current. Cardiovasc Res 41: 188–199. [DOI] [PubMed] [Google Scholar]
- 71. Santoro B, Wainger BJ, Siegelbaum SA (2004) Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. J Neurosci 24: 10750–10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Santoro B, Piskorowski RA, Pian P, Hu L, Liu H, et al. (2009) TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channels in the brain. Neuron 62: 802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zolles G, Wenzel D, Bildl W, Schulte U, Hofmann A, et al. (2009) Association with the auxiliary subunit PEX5R/Trip8b controls responsiveness of HCN channels to cAMP and adrenergic stimulation. Neuron 62: 814–825. [DOI] [PubMed] [Google Scholar]
- 74. Lewis AS, Schwartz E, Chan CS, Noam Y, Shin M, et al. (2009) Alternatively spliced isoforms of TRIP8b differentially control h channel trafficking and function. J Neurosci 29: 6250–6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dedek K, Waldegger S (2001) Colocalization of KCNQ1/KCNE channel subunits in the mouse gastrointestinal tract. Pflügers Arch 442: 896–902. [DOI] [PubMed] [Google Scholar]
- 76. Le Roux N, Amar M, Baux G, Fossier P (2006) Homeostatic control of the excitation-inhibition balance in cortical layer 5 pyramidal neurons. Eur J Neurosci 24: 3507–3518. [DOI] [PubMed] [Google Scholar]