

Abstract
T-helper cell programming and function is tightly regulated by complex biological networks to prevent excessive inflammatory responses and autoimmune disease. The importance of miRNAs in this process is highlighted by the preferential Th1 polarization of Dicer-deficient T cells that lack miRNAs. Using genetic knockouts, we demonstrate that loss of endogenous miR-29, derived from the miR-29ab1 genomic cluster, results in unrestrained T-bet expression and IFN-γ production. miR-29b regulates T-bet and IFN-γ via a direct interaction with the 3′UTRs, and IFN-γ itself enhances miR-29b expression, establishing a novel regulatory feedback loop. miR-29b is increased in memory CD4+ T cells from multiple sclerosis (MS) patients, which may reflect chronic Th1 inflammation. However, miR-29b levels decrease significantly upon T cell activation in MS patients, suggesting that this feedback loop is dysregulated in MS patients and may contribute to chronic inflammation. miR-29 thus serves as a novel regulator of Th1 differentiation, adding to the understanding of T cell-intrinsic regulatory mechanisms that maintain a balance between protective immunity and autoimmunity.
INTRODUCTION
Naïve CD4+ T cells are activated in response to antigen stimulation as part of the adaptive immune response. Depending on the cytokine and environmental cues present at the time of activation, T cells differentiate along specific developmental pathways characterized by unique cytokine and transcriptional profiles (1, 2). IFN-γ-producing T-helper type 1 cells (Th1 cells) are important mediators of protective immunity against intracellular pathogens, but uncontrolled Th1 inflammation can lead to tissue damage and autoimmune diseases such as multiple sclerosis (MS).
MS is an inflammatory demyelinating disease of the central nervous system (CNS) involving myelin-reactive IFN-γ - and IL-17-producing Th1 and Th17 cells (3–6). Many of the current MS therapies act by restoring the Th1/Th2 balance, and the effectiveness of therapeutics often correlates with reductions in Th1 markers such as T-bet (7, 8). Although therapeutic interventions can act to dampen Th1 responses, there are also endogenous immune mechanisms that serve to counteract Th1 inflammation. Activated T cells, including Th1 cells, are subject to cell extrinsic regulatory mechanisms such as regulatory T cell-mediated suppression (9). Cell intrinsic regulatory mechanisms can provide additional counter regulatory signals, such as induction of SOCS1 in response to IFN-γ (10, 11). Mechanisms such as these are critical in preventing prolonged and deleterious inflammatory T cell responses. Uncovering novel mechanisms that control Th1 responses will add to our understanding of the balance between immune homeostasis and dysregulation, and the relationship of these pathways to disease.
Recently, microRNAs (miRNAs) have emerged as critical regulators of immune homeostasis (12, 13). miRNAs are small non-coding RNAs that repress post-transcriptional target gene expression via an antisense RNA interaction (14, 15). miRNAs can influence expression of multiple target genes, often within the same gene network, making them powerful modulators of genetic programming (16). The first evidence that miRNAs may be important in controlling Th1 differentiation came from studies using Dicer-deficient T cells. Loss of the Dicer enzyme prevents miRNA maturation resulting in global miRNA deficiency. Dicer-deficient T cells polarize more readily to Th1 and produce increased levels of IFN-γ, implicating miRNAs in the regulation of Th1 differentiation and cytokine production (17). miRNA expression in regulatory T cells is also necessary to prevent pathological inflammation and autoimmunity in mice (18–21). miR-29 has recently been identified as a regulator of Th1 inflammation that acts by controlling T-box transcription factors and IFN-γ (22, 23). Despite evidence supporting the importance of miRNAs in controlling Th1 programming, the regulatory networks of Th1-associated miRNAs in the context of Th1-driven autoimmunity have not been explored.
Here we have identified an activation-associated miRNA, miR-29b, whose expression is elevated in T cells from mice with EAE and patients with MS. Using mice deficient in miR-29, we have implicated miR-29 derived specifically from one of two genomic miR-29 clusters, miR-29ab1, as critical in regulating the Th1 bias of activated T cells through repression of T-bet and IFN-γ. Further, IFN-γ contributes to miR-29 induction, demonstrating a novel regulatory pathway initiated through IFN-γ signaling. Elevated miR-29b in EAE mice and MS patients may therefore be reflective of chronic Th1 inflammation, adding to our understanding of T cell regulation and the relationship between regulatory molecules and autoimmune inflammation.
MATERIALS AND METHODS
Mice
All procedures were performed in accordance with OSU Institutional Animal Care and Use Committee-approved protocols. Adult C57Bl/6 mice (6–10 weeks of age) were purchased from Jackson Laboratories (Bar Harbor, ME) and myelin basic protein (MBP)-specific T cell receptor transgenic (TcR-tg) mice were obtained from Charles Janeway (Yale, New Haven, CT). Homozygous floxed miR29ab1 mice (C57BL6 strain) were generated as follows: For the targeting construct, two homologous recombination arms were amplified by PCR, on 129 SvJ/X1 genomic DNA, a 5′ one of 4171 bps and a 3′ one of 3857 bps. The genomic fragment to be deleted of 600 bps, containing the miR29a and miR29b1, was amplified the same way and cloned in between two loxP sites, in a pFlox vector. The recombination arms together with the floxed genes were all cloned into Gateway vectors and then assembled together into a destination vector that represented the targeting vector. 129SvJ/X1 ES cells were electroporated with the targeting vector, and clones were screened by Southern Blot. DNA was digested with SacI, and labeled with a 3′ probe. One positive clone was identified out of 336 screened. The mutant ES cell clone was injected into C57BL/6 blastocysts and agouti pups were screened by PCR to verify the generation of heterozygous floxed miR-29ab1 mice. Homozygous floxed miR-29ab1 mice were bred to EIIacre mice to induce ubiquitous deletion of the cluster. The portion of the targeting vector containing neoR is present in the knockout mice. miR-29ab1 and miR-29b2c knockout mice were used at 6–10 weeks of age.
Northern blot
Spleen and liver were dissociated between two frosted slides, and the lysate was washed in PBS, depleted of red cells by hypotonic lysis with ammonium chloride (NH4Cl), centrifuged, and resuspended in PBS. Total RNA was extracted with TRIzol (GIBCO, Invitrogen), loaded and denatured on SDS/PAGE, and blotted on a Hybond N+ membrane (Amersham Pharmacia). The membrane was hybridized with a γ-32P radioactive probe representing the antisense sequence of mature mmu-miR29a, incubated overnight, washed, and exposed to a PhosphorImager screen (Molecular Dynamics). The image was processed by using a Typhoon image processing system (Amersham Biosciences).
EAE immunization
Mice were immunized subcutaneously over four sites on the flank with 100 ml of an emulsion containing 200 mg of MOG 35–55 (CS Bio) in PBS and an equal volume of CFA (containing 200 mg of heat-killed Mycobacterium tuberculosis, Jamaica strain). Mice also received 200 ng pertussis toxin (List Biological Laboratories) injected i.p. at the time of immunization and 48 h later. Mice were monitored daily for clinical signs of disease and were scored as follows: 0, no signs; 1, limp tail or mild ataxia; 2, complete ataxia; 3, paralysis of one hindlimb; 4, complete hindlimb paralysis; 5, moribund or death.
Human subjects and memory CD4+ T cell isolation
All human samples were acquired under OSU Institutional Review Board-approved protocols in accordance with the guidelines of the Declaration of Helsinki. White blood cells were obtained by leukapheresis from healthy donors (HD) or MS patients who were in clinical remission and had not undergone immunomodulatory treatments (treatment-naïve). Memory CD4+CD45RO+ cells were isolated on an autoMACS Pro Separator with the Dead Cell Removal Kit (Miltenyi Biotec) followed by the negative selection Memory CD4+ T cell isolation kit (Miltenyi). ≥95% CD4+CD45RO+ pure samples were used for downstream analysis. The mean ± standard deviation percent purity of the CD4+CD45RO+ cells for the comparison groups was as follows: HD (98.08 ± 0.9), PPMS (98 ± 1.1), RRMS (97.7 ± 0.7) and SPMS (98.5 ± 0.5).
RNA isolation and miRNA quantification
Cells were collected by centrifugation and washed once with cold PBS, followed by RNA isolation using mirVana miRNA isolation kit according to the manufacturer’s instructions (Ambion). RNA concentration and quality were determined by NanoDrop prior to use in downstream assays. Expression of miR-29b in human memory T cells was quantified using the NanoString nCounter System as described (24). qRT-PCR analysis for miRNAs was performed in duplicate using TaqMan microRNA assays (Applied Biosystems, Foster City, CA). Small RNAs sno202 and RNU44 were used for normalization of murine and human miRNAs respectively, and results were analyzed using the comparative Ct method.
Murine T cell isolation and stimulation
For PCR analysis, naïve CD4+CD62L+ T cells were purified from myelin basic protein (MBP)-specific T cell receptor transgenic (TcR-tg) mice using MACS separation with the mouse CD4+CD62L+ T Cell Isolation Kit II (Miltenyi Biotec). Naïve T cells were stimulated with irradiated wild-type splenocytes (1:4 T cells:feeders) and 10 mg/ml MBPAc1–11 with the following cytokine/neutralizing antibody combinations: ThN (no exogenous cytokines or neutralizing antibodies), Th1 (0.5 ng/ml IL-12 and 5 mg/ml anti-IL-4), Th2 (10 ng/ml IL-4, 5 mg/ml anti-IFN-γ and 3 mg/ml anti-IL-12), Th17 (25 ng/ml IL-6, 1 ng/ml TGF-β, 2 mg/ml anti-IFN-γ, 2 mg/ml anti-IL-4 and 0.65 mg/ml anti-IL-12), and iTregs (5 ng/ml TGF-β). Cells were analyzed at 72 h after activation.
For Supplemental Figure 4, splenic CD4+ T cells were purified from myelin basic protein (MBP)-specific T cell receptor transgenic (TcR-tg) mice using MACS separation with the mouse CD4+ T Cell Isolation Kit II (Miltenyi Biotec) to obtain enriched naïve T cells. Purified CD4+ T cells were activated using the same T cell:feeder ratio, antigen dose, and cytokine/neutralizing antibody combinations as above for the first 72 h. After 72 h, cells were removed from stimulation and expanded in IL-2. RNA was obtained at 24 h increments up to 120 h, and cells were re-stimulated on day 7 with phorbol 12-myristate 13-acetate (PMA) and ionomycin for 8 h.
FACS analysis
For ex vivo cytokine secretion profiles, splenocytes and lymph node cells were stimulated for 4 h with PMA and ionomycin in the presence of brefeldin A (activation cocktail, BD Biosciences). For intracellular cytokine staining, cells were re-stimulated directly ex vivo or on d 6 after culture for 4.5 h with BD activation cocktail. All antibodies were purchased from BD Biosciences or eBioscience. Cell surface markers were labeled with directly conjugated antibodies diluted in PBS/10% FBS/sodium azide for 20 min at 4°C. Cells were fixed and permeabilized with BD Cytofix/Cytoperm (BD Biosciences) for 30 min, then stained for intracellular cytokines and T-bet for 40 minutes in Perm/Wash buffer at 4°C. Data were acquired with a BD FACSCanto II flow cytometer (BD Biosciences) using FACSDiva software and analyzed using FlowJo 9.0 (Tree Star).
Luciferase assay
The 3′ UTR segments containing the target sites for miR-29 from the human Tbx21 and Ifng gene were amplified from genomic DNA and inserted into the PGL3 control vector (Promega), using the XBA1 site immediately downstream from the luciferase stop codon. Mutant constructs were generated by mutating 4 bp in the miR-29b seed region using the QuickChange XL Site-Directed Mutagenesis kit (Stratagene). HEK-293 cells were transfected with 800 ng of firefly luciferase vector, 100 ng of Renilla luciferase control vector, and 200 nM of precursor miR-29b or scrambled oligonucleotides (negative control precursor, Ambion) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Cells were lysed in 1x Passive Lysis Buffer and assayed in triplicate using the Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity as measured by a Vertias microplate luminometer (Promega) and was then calculated relative to the scrambled control in each independent replicate.
Chromatin immunoprecipitation assay
T cells were stimulated with αCD3 and αCD28 for 48 h. The ChIP assay was performed as described previously (25). The cells were pulse sonicated for 5 sec at 40% amplitude to shear the DNA into 200–1000bp lengths. The primer set for the Stat1 site for PCR amplification of the miR-29ab1 promoters was as follows: CCTGCTGCATCTGGAGAGGGGT (forward) and CTCCCACCAAGGGGCTGAGT (reverse). PCR conditions were 94°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec for 35 cycles. Integrated density values were calculated using AlphaImager image analysis software (Alpha Innotech) and normalized to input DNA.
Statistical analysis
Parametric data was analyzed using an unpaired t-test for single comparisons involving unpaired data sets (a=0.05), paired t-test for single comparisons involving paired data sets (a=0.05), and two-way ANOVA with Tukey’s or Bonferroni’s post-hoc test for multiple comparisons (a adjusted according to number of comparisons). Nonparametric data was analyzed using a Wilcoxon signed rank test. Statistical analysis of miR-29b expression in human memory T cells included a technical normalization based on positive controls, then a geometric mean normalization using the top 50 highly expressed miRNAs, followed by a Student’s t-test for individual miRNA comparisons to calculate p-values.
RESULTS
miR-29b is associated with Th1 autoimmunity and directly interacts with the T-bet and IFN-γ 3′UTRs
There are over 1,400 unique miRNAs identified in humans, and over 700 in mice (miRBase v17). To narrow the field of candidate miRNAs related to Th1 autoimmunity, we used in silico target predictions to identify miRNAs predicted to target both T-bet and IFN-γ. T-bet mRNA contains an evolutionarily conserved miR-29 seed sequence at positions 245–251 of the 3′ UTR, and IFN-γ mRNA contains a similarly conserved seed match at positions 474–480 of the 3′ UTR. The predicted targets of miR-29b suggest that this miRNA may play an important role in Th1 inflammation, and thus the role of miR-29b in T cells was examined further.
T cells were purified from EAE and control immunized mice at 10 days post-immunization (dpi) and quantitative (q)PCR analysis revealed that miR-29b was significantly increased in CD4+ T cells from EAE mice (Fig. 1A). This presented a scenario in which miR-29b is induced in a model of Th1 inflammation, but, based on its’ predicted targets, would function to limit Th1 inflammation. Due to their regulatory nature, miRNAs may act as part of negative feedback loops. For example, miR-146a is induced downstream of Toll-like receptor (TLR) signaling and targets IL-1 receptor-associated kinase 1 (IRAK1) and TNF receptor associated factor 6 (TRAF6) (26). We therefore hypothesized that miR-29b may act in a similar negative feedback manner. As such, miR-29b would be induced in response to ongoing inflammation in EAE, and would in turn regulate Th1 inflammation by directly targeting T-bet and IFN-γ.
Figure 1. miR-29b is increased during EAE and directly interacts with the 3′UTRs of T-bet and IFN-γ.
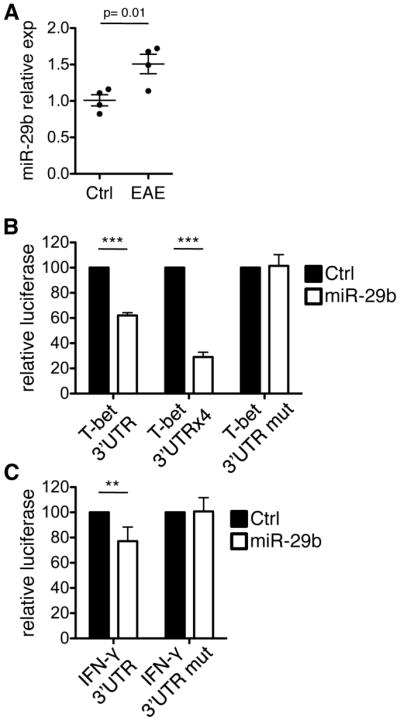
(A) CD4+ T cells were isolated from the spleens of mice with EAE or control mice receiving adjuvant alone (ctrl) at 10 dpi, and miR-29b was quantified by real-time PCR. Fold changes are expressed relative to the average of control mice. Data are representative of four biological replicates per group and values are means ± SEM. p=0.01; Student’s two-tailed t-test. (B) HEK-293 cells were co-transfected with dual luciferase reporters and miR-29b or control miRNA. Values represent luciferase activity of constructs with one or four copies of the human T-bet 3′ UTR, or a mutated miR-29 complementary site (T-bet 3′UTR mut). (C) Luciferase activity of a construct containing the human IFN-γ 3′ UTR or a mutated miR-29 complementary region (IFN-γ 3′UTR mut). Constructs were co-transfected as in (B). The ratio of firefly to renilla luciferase activity was normalized to the control miRNA within each experimental replicate. Data are means ± SD from three to six independent experiments, each performed in triplicate. **p<0.01 and ***p<0.001; Student’s two-tailed t-test.
To investigate this hypothesis, we sought to determine if T-bet and IFN-γ represent direct targets of miR-29b using luciferase assays. HEK-293 cells were co-transfected with miR-29b or a negative control miRNA, and a firefly luciferase gene linked to the target gene (IFN-γ or T-bet) 3′UTR. miR-29b repressed luciferase activity of the construct containing the human T-bet 3′UTR by 38% ± 2% relative to the negative control miRNA (Fig. 1B). This activity was copy number dependent, as shown by the additional repression of luciferase activity that was observed using a construct containing four T-bet UTR inserts (Fig. 1B). When the conserved site in the T-bet 3′UTR was mutated, luciferase expression was restored, demonstrating the direct interaction between miR-29b and the predicted site of hybridization. To validate a specific miRNA-target interaction between miR-29b and IFN-γ, we performed similar experiments assessing luciferase activity in cells co-transfected with miR-29b and luciferase linked to the human IFN-γ-3′UTR. miR-29b significantly reduced luciferase activity by 23% ±11% relative to a miRNA negative control, while mutation of the conserved miR-29 binding site abrogated this effect (Fig. 1C). These results indicate that miR-29b is able to interact with both the T-bet and IFN-γ 3′ UTRs.
Generation of miR-29-deficient mice
miR-29 is encoded in two intergenic genomic clusters, the miR-29ab1 cluster and the miR-29b2c cluster on human chromosomes 7 and 1, respectively. These two clusters encode three different miRNAs, miR-29a, -29b, and -29c, which share identical 5′ seed sequences and therefore have the capacity to regulate the same target genes. Indeed, we observed that miR-29a was also able to repress the T-bet luciferase construct (data not shown). This suggests that the homology amongst miR-29 family members may confer the ability to exert similar biological effects to varying degrees. We therefore aimed to dissect the relative contribution of each genomic cluster using a genetic knockout approach.
To determine the importance of endogenous miR-29 and the role of each genomic cluster, we genetically deleted the miR-29ab1 or the miR-29b2c cluster (Fig. S1 and S3). Mice with floxed alleles of miR-29ab1 were bred with EIIaCre mice to induce ubiquitous deletion, and deletion was confirmed by Northern blot and PCR (Fig. S1B–C). Mature miR-29a and miR-29b were undetectable in activated T cells, providing additional confirmation of genetic deletion (Fig. S1D). Although miR-29b is encoded in both miR-29 genomic clusters, deep sequencing results have shown that the miR-29ab1 cluster is the dominant source of miR-29b in T cells (22). These results confirm that miR-29b in activated T cells originates predominantly from the miR-29ab1 cluster.
miR-29ab1-deficient mice exhibit a shorter lifespan of 12 ± 2 months, compared to wild-type littermates that had an average lifespan of 24 ± 2 months. miR-29-deficient mice also have markedly reduced lymphoid organ cellularity, with a 50% reduction in total cell number in the spleen (Fig. S2A), lymph nodes and thymus (data not shown). Despite reduced overall cellularity, the cellular composition of the lymphoid organs was comparable between miR-29ab1-deficient mice and wild-type mice. Splenocytes had similar percentages of CD25+FoxP3+ Tregs in the CD4 population (Fig. S2B), and CD3-gated lymphocytes contained similar proportions of CD4+ (Fig. S2C) and CD8+ T cells (Fig. S2D), indicating that the relative proportion of T cells were not affected by loss of miR-29ab1.
Endogenous miR-29ab1 regulates T-bet and IFN-γ during T cell activation
To determine the role of endogenous miR-29 in regulating Th1 bias, purified CD4+ T cells from miR-29-deficient mice and wild-type littermates were activated with polyclonal αCD3/αCD28 stimulation. The percentage of T cells producing IFN-γ at 6 d was significantly increased from 5.3% ± 1% in wild-type T cells to 31.8% ± 3% in miR-29ab1-deficient T cells (Fig. 2A–B). There was a corresponding increase in IFN-γ MFI (Fig. 2C) and increased concentration of IFN-γ in the culture supernatants that accumulated over time (Fig. 2D). Similar effects were observed with T-bet expression. The percentage of T cells expressing T-bet was significantly increased from 27% ± 5% in wild-type T cells to 66% ± 8% in miR-29ab1-deficient T cells (Fig. 2E–F). There was also a significant increase in T-bet MFI (Fig. 2G), indicating that loss of miR-29ab1 results in higher T-bet expression on a per cell basis. Together these data demonstrate that miR-29 derived from the miR-29ab1 cluster is necessary to control the frequency of cells that commit to the Th1 lineage, and further regulates expression of T-bet and IFN-γ within the committed population. Using the miR-29b2c-deficient mice (Fig. S4A), we analyzed the contribution of this miR-29 cluster to Th1 regulation. miR-29b2c had no effect on the percentage of IFN-γ+ cells (Fig. 3A) or T-bet+ cells (Fig. 3C) following 6 d of ThN activation. IFN-γ MFI (Fig. 3B) and T-bet MFI (Fig. 3D) were also unaffected. Together these data indicate that the miR-29ab1 cluster is responsible for regulating IFN-γ and T-bet expression.
Figure 2. miR-29 derived from the miR-29ab1 genomic cluster is necessary to restrain Th1 programming.

(A) CD4+ T cells were purified from miR-29ab1 wild-type (ab1+/+), heterozygote (ab1+/−), and knockout (ab1−/−) mice, and activated under ThN conditions with no exogenous cytokines/neutralizing antibodies. IFN-γ expression was assessed on day 6 of culture by intracellular flow cytometry. (B–D) Additional quantification of IFN-γ expression following activation as in (A), including (B) percentage of IFN-γ-expressing cells, (C) mean fluorescence intensity (MFI) of IFN-γ+ cells, and (D) IFN-γ concentration in culture supernatants of knockout relative to wild-type cultures at 68 h and 6 d post-culture. Data are representative of two independent experiments and values are means ± SEM. *p<0.05; ANOVA Tukey’s post-hoc test (B–C) or Student’s one-sample t-test (D). (E) T-bet expression was assessed in T cell cultures from (A) using intracellular flow cytometry. (F–G) Additional quantification of T-bet expression including (F) percentage of T-bet-expressing cells and (G) MFI of T-bet+ cells. Data are representative of two independent experiments and values are means ± SEM. *p<0.05; ANOVA Tukey’s post-hoc test.
Figure 3. Effects of miR-29 derived from the miR-29b2c genomic cluster on Th1 programming.

T cells were purified from miR-29b2c wild-type (b2c+/+) and knockout (b2c−/−) mice and activated under ThN conditions. The percentage of cells expressing IFN-γ (A) and T-bet (C) was assessed after 6 d of culture by intracellular flow cytometry, and the MFI of cells expressing IFN-γ (B) and T-bet (D) was also quantified. Data are representative of two independent experiments and values are means ± SEM.
We next aimed to determine the effect of miR-29ab1 deficiency on the in vivo response to antigen challenge. Mice were immunized with myelin oligodendrocyte glycoprotein (MOG) peptide 35–55 in conjunction with Complete Freund’s adjuvant to induce experimental autoimmune encephalomyelitis (EAE), and cells from the draining lymph nodes were analyzed at 7 days post-immunization (dpi). CD4+ T cells from the draining lymph nodes of both wild-type and miR-29ab1-deficient mice expressed comparable percentages of the activation marker CD44 (Fig. 4A). We also observed comparable proliferation as measured by CFSE dilution during in vitro Th1 stimulation (Fig. 4B), indicating that miR-29ab1-deficient T cells are capable of becoming activated and proliferating to the same extent as wild-type T cells. However, the number of cells in the lymph nodes at 7 dpi is significantly decreased (Fig. 4C) compared to pre-immunization, suggesting that activation induced cell death is occurring. The decreased lymphoid organ cellularity in both naïve and EAE mice suggests that there is either a defect in the generation or survival of mature lymphocytes, which may interfere with the ability to generate an adaptive immune response. To this end, we observed a trend (p = 0.06) towards increased CD4+ T cell death as assessed by Annexin V/7-AAD staining (Fig. 4D), suggesting that miR-29ab1-deficient T cells undergo apoptosis at an increased frequency during activation. Not surprisingly, this failure of T cell expansion and/or survival results in reduced EAE severity (Fig. 4E). Consistent with the reduced EAE clinical severity, miR-29ab1-deficient mice had reduced CNS cellular infiltration. Isolation of CNS cells demonstrated similar percentages of CD45lo CNS resident cells, but a complete absence of CD45hi infiltrating cells in miR-29ab-deficient mice (Fig. 4F). This result was confirmed by H&E staining, which also showed an absence of cellular infiltration in the spinal cord of miR-29ab1-deficient mice at 17 dpi (Fig. 4G). This data demonstrates that systemic loss of miR-29ab1 results in protection from EAE due to an inability to generate a robust adaptive immune response, and further suggests that loss of miR-29ab1 may promote phenotypic and functional deficits beyond its’ role in Th1 regulation that may impair the development of EAE, as was shown in a recent publication demonstrating early thymic involution in miR-29ab1-deficient mice (27).
Figure 4. Effects of miR-29ab1 deficiency on EAE.

(A) Draining lymph nodes (inguinal and axillary) were obtained from MOG-immunized wild-type (ab1+/+) and miR-29ab1-deficient (ab1−/−) mice at 7 days post-immunization (dpi) and CD4+ T cell activation was assessed by flow cytometry for the activation marker CD44. (B) Splenocytes from wild-type (ab1+/+) and miR-29ab1-deficient (ab1−/−) mice were Th1-polarized in vitro and proliferation was assessed by CFSE dilution at 48 h post-activation. Data are means ± SEM from two biological replicates. (C) Cellularity of the lymph nodes pre- and post-induction of EAE mice 7 dpi. Data are means ± SEM from two biological replicates. **p<0.01; Student’s two-tailed t-test. (D) Splenocytes from wild-type and miR-29ab1-deficient mice were Th1-polarized as in (C), and apoptosis was analyzed by Annexin V/7-AAD staining at 48 h post-activation. Data are means ± SEM from two biological replicates. (E) EAE disease course in MOG-immunized wild-type and miR-29ab1-deficient mice. Data are representative of 6–8 biological replicates across two independent experiments. p=0.04; Mann-Whitney test. (F) CNS cells were isolated by density gradient from the spinal cords of EAE mice at 17 dpi, and different cellular subsets were distinguished by surface expression of CD45. CD45lo represents the CNS resident population, and CD45hi indicates CNS infiltrating cells. Cells were gated based on 7-AAD exclusion (live cells). (G) Histological assessment of inflammation in the lumbar spinal cord of wild-type and miR-29ab1-deficient mice at 17 dpi.
Given the role of miR-29 specifically within CD4+ T cells, we examined ex vivo T cells from naïve mice to determine if miR-29ab1-deficiency promotes an inherent inflammatory bias in the absence of immune challenge. T cells from both the lymph nodes (Fig. 5A) and spleen (Fig. 5B) of miR-29ab1-deficient mice produced significantly higher levels of IFN-γ relative to wild-type mice, and there was also a modest but consistent increase in IL-17 production. An increased percentage of CD4+ T cells expressing the activation marker CD44 was also observed in the spleen (Fig. 5C), suggesting that loss of miR-29ab1 promotes an activated, pro-inflammatory T cell phenotype in vivo. After establishing that miR-29 regulates inflammatory Th1 cell phenotypes in vitro and in vivo, we sought to identify what signaling pathway(s) engaged during T-helper polarization may regulate miR-29 expression.
Figure 5. T cells from miR-29ab1-deficient mice exhibit an activated, inflammatory phenotype ex vivo.

(A–B) Lymph node cells (A) and splenocytes (B) from miR-29ab1 wild-type (+/+) and knockout (−/−) mice were re-stimulated ex vivo with PMA and ionomycin, and IFN-γ and IL-17 expression was assessed by intracellular flow cytometry. Flow diagrams are gated on CD4+CD44hi cells. Data are representative of two independent experiments and values are means ± SEM. *p<0.05 and **p<0.01; Student’s one sample t-test. (C) Activation status of splenic CD4+ T cells was determined using flow cytometry for the surface marker CD44. Data are representative of two independent experiments and values are means ± SEM. *p<0.05; Student’s one sample t-test.
miR-29b is induced as a result of T cell activation and through IFN-γ-mediated Stat1 signaling
The dynamics of miR-29a and miR-29b expression in various T-helper subsets (ThN, Th1, Th2 and Th17) were examined over the course of 5 days and following a short re-stimulation on day 7 (Fig. S4). We observed maximal induction of miR-29a (Fig. S4A) and miR-29b (Fig. S4B) at 48–72 h, which corresponded with peak IFN-γ production in ThN and Th1 cells (Fig. S4C). Based on this kinetic analysis, we chose the 72 h time point to further analyze miR-29 induction.
Purified naïve CD4+CD62L+ T cells from myelin basic protein (MBP)-specific T cell receptor transgenic (TcR-tg) mice were activated and differentiated under ThN, Th1, Th2, Th17, or iTreg-inducing conditions, and miR-29 levels were assessed 72 h post-activation. miR-29b was significantly increased in all T-helper subsets relative to pre-culture levels, and miR-29a was elevated in Th1 and Th17 conditions (Fig. 6A). We also assessed the impact of miR-29ab1 deficiency on T-helper subsets, including Th1, Th2, Th17, and iTreg (Fig. S4D–G). Loss of miR-29ab1 resulted in a marked increase in the number of cells producing IFN-γ during Th1 polarization. miR-29ab1-deficiency had more modest effects on other T-helper lineages. This indicates that miR-29 may have a more global role in CD4 T cell activation, in addition to its effects on IFN-γ production. Elevated miR-29b expression is maintained in antigen-experienced murine memory CD4+ T cells, as they express higher levels of miR-29b than naïve CD4+ T cells ex vivo (Fig. 6B). miR-29b induction can also be extended to human T cell differentiation, as purified human naïve CD4+ T cells were found to up-regulate miR-29b following Th1 differentiation (Fig. 6C), indicating that miR-29 is induced during T cell activation and differentiation in both mouse and human.
Figure 6. miR-29 is induced as a result of T cell activation and IFN-γ signaling.

(A) Purified CD4+CD62L+ T cells from myelin basic protein (MBP)-specific T cell receptor transgenic (TcR-tg) mice were differentiated in vitro in unbiased (ThN), Th1, Th2, Th17 or iTreg conditions and miR-29 expression was quantified at 72 h post-differentiation. Fold induction of miR-29 post-culture is quantified relative to pre-culture. Data are means ± SEM from three independent experiments. *p< 0.05, **p<0.01, ***p<0.001; ANOVA Bonferroni post-hoc test. (B) Ex vivo qPCR analysis of miR-29b expression in naïve CD4+CD62L+ and memory CD4+CD62L− T cells from C57BL/6 mice. Data are normalized to average miR-29b expression in naïve T cells. Lines depict mean expression within each group. ***p<0.001; Student’s two-tailed paired t-test. (C) miR-29b expression in human naïve CD4+CD45RA+ T cells pre-culture and post-Th1 differentiation. Results are normalized to pre-culture expression within each experimental replicate. Data are means ± SEM from five independent experiments. *p< 0.05; Wilcoxon signed rank test. (D) Graphical representation of sequence conservation between the miR-29ab1 locus on human chromosome 7 and mouse chromosome 6. The genomic sequence was used to identify potential IFN-γ-activated site (GAS) elements (consensus sequence TTC/ANNNG/TAA) within 5 kb of the miR-29ab1 transcriptional start site (TSS). Predicted GAS consensus sites are delineated by an asterisk. (E) Murine T cells were activated under ThN conditions with neutralization of IFN-γ. Fold induction of miR-29b is quantified relative to pre-culture and a solid line connects paired samples. Data are from four independent experiments. *p<0.05; Student’s paired two-tailed t-test. (F) Murine T cells were stimulated with αCD3/αCD28 and exogenous IFN-γ, and miR-29b was quantified by qPCR. Percent increase was calculated relative to pre-stimulation miR-29b expression. Data are means ± SEM from three independent experiments. **p<0.01; Student’s paired two-tailed t-test. (G) T cells were activated in vitro with polyclonal TCR stimulation for 48 h and ChIP assays were performed with an Ab specific for Stat1. DNA bound to Stat1 was purified and used as a template for PCR analysis. Primers specific for a confirmed Stat1 binding site in the IFN-γ promoter were used a positive control (lanes 1–3). Stat1 was also bound to the miR-29ab1 promoter (lane 6). Lanes 1 and 4 represent 1% input DNA, and an isotype control Ab was used as a negative control in lanes 2 and 5. Numbers below each lane represent integrated density values relative to input DNA. Data are representative of two independent replicates. (H) Proposed miR-29 regulatory feedback loop, where IFN-γ-induced miR-29 represses T-bet and IFN-γ.
Although miR-29b is expressed at comparable levels in all T-helper subsets, we explored the specific induction signals that occur during Th1 polarization, given the increased expression in EAE (Fig. 1A) and the positive association between IFN-γ and miR-29 expression during in vitro polarization (Fig. S4). To identify potential Th1-associated transcriptional regulators of the miR-29ab1 cluster, we performed a promoter analysis on the conserved genomic sequence within 5 kb of the previously defined transcriptional start site (28). There were no predicted T-bet binding sites in the promoter region using a T-box consensus of CACNNNNGTG; however, there were three evolutionarily conserved IFN-γ-activated site (GAS) elements (Fig. 6D). To directly test the contribution of IFN-γ to miR-29b induction, T cells were activated under ThN conditions, or ThN with IFN-γ neutralization. miR-29b was significantly reduced when IFN-γ was neutralized (Fig. 6E), suggesting that IFN-γ contributes to miR-29b induction. However, miR-29b expression was still elevated relative to pre-stimulation levels, suggesting that T cell activation or other stimuli may also synergize to promote miR-29b expression. Stimulation of T cells with exogenous IFN-γ significantly increased miR-29b expression above that of polyclonal T cell activation alone (Fig. 6F), demonstrating that IFN-γ is able to amplify the induction of miR-29b. Additionally, we verified direct STAT1 binding to the miR-29ab1 promoter region. DNA from activated T cells was immunoprecipitated with a STAT1-specific antibody and analyzed by PCR. A region of the IFN-γ promoter was amplified from input DNA and STAT1-immunoprecipitated DNA (Fig. 6G), serving as a positive control (25). Importantly, a conserved region of the miR-29ab1 promoter was also amplified from input DNA and STAT1-immunoprecipitated DNA (Fig. 6G). These data support a role for IFN-γ-induced STAT1 in the transcriptional regulation of the miR-29ab1 cluster, establishing a regulatory feedback loop (Fig. 6H). Furthermore, these findings identify a mechanism whereby miR-29b may become elevated in response to ongoing Th1 autoimmune inflammation, including EAE.
MS is associated with increased miR-29b in memory CD4+ T cells
To validate the relevance of miR-29b in human autoimmune inflammation, miR-29b expression was quantified in memory CD4+CD45RO+ T cells from MS patients. We analyzed memory T cells since the memory T cell population of MS patients is enriched with autoreactive myelin-specific T cells (29). Memory T cells were purified to >95% purity using magnetic bead negative selection from seventeen healthy donors (HD) and nineteen MS patients of various subtypes including relapsing-remitting MS (RRMS, n=11), primary progressive MS (PPMS, n=4), and secondary progressive MS (SPMS, n=4). For quantification of miR-29b, we employed the NanoString nCounter System, which is an extremely sensitive and specific method of mature miRNA quantification that counts individual mature miRNAs and provides a digital readout of relative abundance (24). miR-29b was significantly increased by 2.25-fold in the MS population (p=1.1×10−6), as well as in each MS subtype (Fig. 7A). We confirmed the presence of an active Th1 response in the MS population, finding significantly elevated expression of T-bet in MS and RRMS compared to HD (Fig. 7B). Although we analyzed IFN-γ expression, we did not find any differences in expression in resting memory T cells of MS and HD (Fig. 7C). Together these data indicate that miR-29b is elevated in CD4+ T cells in both EAE and MS. The positive association between miR-29b and T-bet in MS patients’ memory T cells suggests that miR-29b may be induced in response to ongoing Th1 inflammation, as was observed in EAE (Fig. 1A) and consistent with a negative feedback model.
Figure 7. Memory CD4+ T cells expressed increased levels of miR-29b in MS patients.

(A) Memory CD4+CD45RO+ T cells were purified by negative selection (>95% purity) from healthy donors (HD) and MS patients. miR-29b levels were quantified using the NanoString nCounter System, which provides a digital readout of individual mature miR-29b copies in each sample. Data are categorized as all HD (n=17) and all MS (n=19), as well as stratified according to MS subtype including relapsing-remitting MS (RRMS, n=11), primary progressive MS (PPMS, n=4), and secondary progressive MS (SPMS, n=4). Lines represent mean expression. P-values were calculated by technical normalization based on positive controls, followed by a geometric mean normalization using the top 50 most highly expressed miRNAs, and a Student’s t-test for individual miRNA comparisons. (B–C) Quantification of T-bet (B) and IFN-γ (C) mRNA transcript in the purified memory T cells from (A). P-values were calculated using one-way ANOVA. (D) Resting memory T cells from a subset of HD (n=5) and MS (n=5) were reactivated with PMA and ionomycin. miR-29b levels were quantified using the NanoString nCounter System as in (A). Delta miR-29b was calculated by subtracting the activated miR-29b count from the resting memory miR-29b counts. P-value was determined using a Student’s two-tailed t-test.
In a subset of patients, we analyzed expression of miR-29b following re-activation of resting memory cells. Interestingly, there was a significant decrease in miR-29b expression in MS patients, while expression of miR-29b in HD remained stable (Fig. 7D). This data suggests that the miR-29 regulatory axis may be dysregulated in MS, such that re-activation represses miR-29b, which would facilitate robust expression of the miR-29 targets T-bet and IFN-γ and promote disease pathogenesis.
DISCUSSION
The transcription factor T-bet and cytokine IFN-γ act synergistically to coordinate type 1 inflammatory responses and are expressed by a variety of cell types, including antigen presenting cells and lymphocytes. Their expression is necessary for protective immunity; however, the timing and duration of expression must be tightly controlled to prevent inflammatory and autoimmune diseases. We have identified a specific miRNA, miR-29b, that is associated with MS and EAE and regulates Th1 differentiation. Despite the strong sequence homology amongst miR-29 family members and the presence of duplicate copies of miR-29b in two genomic loci, genetic deletion of the miR-29ab1 cluster preferentially regulates the Th1 phenotype. In theory, all miR-29 family members should be able to negatively regulate expression of T-bet and IFN-γ based on their high degree of sequence homology. Consistent with deep sequencing data (22), we have found that the miR-29ab1 cluster is the primary source of miR-29 in activated T cells, and loss of the miR-29ab1 cluster therefore exerts a dominant effect on the Th1 phenotype.
Our genetic knockout data implicates a role for the miR-29ab1 cluster in Th1 regulation, including both mature miR-29a and miR-29b, and others have reported similar roles for both miRNAs (22, 23), Ma et al. recently reported that miR-29a and miR-29b expression decreases during intracellular bacterial infection (23), while we have found that miR-29b is increased in MS patients’ memory T cells. These findings suggest that miR-29 may function differently within specific cellular subsets and during the course of infection as opposed to autoimmunity. Importantly, the data also emphasize that the expression and function of individual miRNAs within a homologous miRNA family can be both cell-type and context-dependent.
Despite the effect of miR-29ab1-deficiency on the Th1 phenotype in vitro and ex vivo, we observed that systemic loss of miR-29ab1 results in EAE resistance. There are several possibilities for this observation. First, peripheral lymphoid organs of miR-29ab1-deficient mice have significantly reduced cellularity. This global reduction in cell number suggests that miR-29 deficiency may affect the generation or survival of lymphocytes. In cancer, miR-29 acts as a tumor suppressor to repress expression of the anti-apoptotic molecules Mcl-1 (30, 31). However, we observed more apoptotic cell death during in vitro Th1 polarization. These results suggest that miR-29ab1 deficiency may render T cells susceptible to activation induced cell death, either directly through targeting molecules involved in apoptosis or indirectly by inducing a state of hyper-activation and over-production of IFN-γ. Lastly, miR-29 may have important functions in other cell types that impact the ability to develop EAE. Using a CD4 T cell-specific miR-29ab1 knockout may provide more conclusive information about the role of miR-29 in EAE specifically within this cellular subset. Nonetheless, our findings emphasize the important consideration that miRNAs are multi-functional, and miR-29 has other important roles in immune cells that remain to be elucidated.
miRNAs often act through repression of one central regulator or via repression of multiple target genes in the same pathway or gene network such as miR-181a targeting of multiple phosphatases downstream of TCR signaling (32). MiR-29b appears to operate in a similar fashion by targeting several components of the Th1 pathway. While Steiner et al. and Ma et al. (22, 23) have reported individual effects of miR-29 on T-box transcription factors and IFN-γ respectively, we have found that miR-29 is able to regulate expression of both T-bet and IFN-γ. In fact, Steiner et al. (22) observed a more dramatic repression of IFN-γ with miR-29 than with siRNA to T-bet, indicating that direct repression of T-bet alone can not fully recapitulate miR-29b-mediated IFN-γ repression. In the same way that T-bet and IFN-γ can act in a feed-forward manner to promote their expression, miR-29-mediated Th1 regulation may be amplified through repression of both T-bet and IFN-γ. The individual contribution of T-bet versus IFN-γ repression may depend on a variety of complex factors, including miR-29 target site accessibility within the target transcripts. For example, the miR-29 complementary region of the IFN-γ 3′UTR lies in close proximity to the AU-rich elements (AREs), and the complex in vivo crosstalk between these UTR elements may dictate overall IFN-γ mRNA stability. The relationship between miRNA binding sites and other UTR elements including AREs is undoubtedly complex. In some cases, miRNAs can antagonize ARE-mediated decay, while in other cases AU-rich motifs and miRNA machinery can cooperate to promote mRNA destabilization (33–35). Although miR-29b is capable of regulating both the T-bet and IFN-γ transcripts, the timing and cellular microenvironment may ultimately dictate the ability of miR-29 to repress their expression.
In this report, we have implicated Stat1 as a transcriptional regulator of the miR-29ab1 cluster. Stat1 stimulates transcription of IFN-γ-inducible genes through binding to the conserved consensus motif of GAS elements. The pro-inflammatory properties of IFN-γ are well established, but IFN-γ also has functions that limit inflammation such as induction of SOCS1 to attenuate cytokine signaling (10, 11, 36). The finding that IFN-γ promotes miR-29 expression identifies an additional regulatory function of IFN-γ. Although we have focused on transcriptional regulation of the miR-29ab1 cluster, which produces both mature miR-29a and miR-29b transcripts, the observation that mature miR-29b is preferentially up-regulated in MS/EAE and in most T-helper subsets indicates that the miR-29ab1 cluster may undergo additional post-transcriptional regulation. Several post-transcriptional mechanisms can affect mature miRNA biogenesis or stability, including crosstalk between the miRNA biogenesis pathway and intracellular signaling molecules, cis-acting modifications of mature miRNAs, and association of miRNAs with the RISC complex (37). Many factors can dictate the levels of mature miRNAs, and miR-29 expression may be determined by both transcriptional and post-transcriptional regulation.
We have validated a role for miR-29b in Th1 differentiation, however we also observed upregulation of miR-29b in other T-helper subsets. In contrast to Ma et al. (23), we found that miR-29b was induced under all T-helper conditions examined. It is important to note, however, that we utilized an antigen-driven differentiation system which could have differing effects on miR-29 expression. miR-29b may be induced in response to T cell activation alone, and lineage-specific cytokine and environmental cues may augment induction, similar to the coordinated regulation of T-bet expression by both TCR and cytokine signals (38, 39). In support of this conclusion, IFN-γ appears to play a role in miR-29b induction but miR-29b is still induced, albeit to a lesser extent, with IFN-γ neutralization. Further investigation into the function of miR-29b in other T cell subsets and the specific induction stimuli that synergize to promote expression will be critical to understanding miR-29b’s complex role in immune regulation.
Based on the ability of IFN-γ to augment miR-29b expression, the increased expression observed in MS patients may occur as a consequence of the altered inflammatory environment in MS. Previous studies in MS have found differential expression of specific miRNAs in CNS lesions, whole blood, CD4+ T cells or Tregs (40–44). We have specifically focused on miRNA expression in memory CD4+CD45RO+ T cells. Autoreactive myelin-specific T cells are present in healthy individuals as well as MS patients, however they assume an activated, proinflammatory phenotype in MS (3, 29, 45–48). Examination of miRNA expression in the memory T cell population likely captures the pathogenic cells contained within this subset and may reveal differences closely associated with disease pathogenesis. Using a highly specific and sensitive method of miRNA detection, NanoString nCounter System, we quantified miRNA expression and found that miR-29b was significantly upregulated in memory T cells from MS patients. Based on the role of Stat1 signaling in miR-29b induction, miR-29b may be elevated in MS due to the presence of ongoing Th1 inflammation, which is consistent with reports demonstrating that CD4+ T cells in MS express higher levels of Stat1α (49).
Despite elevated levels of miR-29b, which can negatively regulate T-bet, the MS population had higher expression of T-bet. Although miRNAs typically serve as negative regulators of target gene expression, positive correlations between miRNA:target pairs can also occur, as was recently demonstrated in Alzheimer’s disease (50). Another possibility is that disease-associated target 3′UTR variations such as alternative polyadenylation and SNPs are impacting the regulatory potential of miRNAs such as miR-29b. Alternative polyadenylation leading to shorter 3′UTRs can actively prevent miRNA-mediated regulation and occurs in cancer oncogenes and during T cell activation (51, 52). Additionally, polymorphisms in miRNA binding sites that render target transcripts insensitive to miRNAs have been associated with diseases such as cancer and hypertension (53, 54), and may also play a role in autoimmunity.
Supplementary Material
Acknowledgments
We thank I. Gienapp and T. Shawler (Ohio State) for technical assistance, and C. Janeway (Yale, New Haven, CT) for MBP TCR transgenic mice.
Footnotes
Supported by NIH grants R01 AI064320 (CCW), RO1 AI43376 (CCW), R01 NS037513 (MKR), K24 NS44250 (MKR), R21 NS067383 (AELR), RO1 NS067441 (AELR), National Multiple Sclerosis Society grant RG 3812 (AELR), T32 GM068412 (KMS), and NCRR TL1RR025753 (KMS)
References
- 1.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 2.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olsson T, Zhi WW, Hojeberg B, Kostulas V, Jiang YP, Anderson G, Ekre HP, Link H. Autoreactive T lymphocytes in multiple sclerosis determined by antigen-induced secretion of interferon-gamma. J Clin Invest. 1990;86:981–985. doi: 10.1172/JCI114800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voskuhl RR, Martin R, Bergman C, Dalal M, Ruddle NH, McFarland HF. T helper 1 (Th1) functional phenotype of human myelin basic protein-specific T lymphocytes. Autoimmunity. 1993;15:137–143. doi: 10.3109/08916939309043888. [DOI] [PubMed] [Google Scholar]
- 5.Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 6.Chitnis T. The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int Rev Neurobiol. 2007;79:43–72. doi: 10.1016/S0074-7742(07)79003-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drulovic J, Savic E, Pekmezovic T, Mesaros S, Stojsavljevic N, Dujmovic-Basuroski I, Kostic J, Vasic V, Mostarica Stojkovic M, Popadic D. Expression of Th1 and Th17 cytokines and transcription factors in multiple sclerosis patients: does baseline T-bet mRNA predict the response to interferon-beta treatment? J Neuroimmunol. 2009;215:90–95. doi: 10.1016/j.jneuroim.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 8.Schrempf W, Ziemssen T. Glatiramer acetate: mechanisms of action in multiple sclerosis. Autoimmun Rev. 2007;6:469–475. doi: 10.1016/j.autrev.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 10.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TW, Nicola NA, Hertzog PJ, Metcalf D, Hilton DJ. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 11.Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, Yoshimura A, Ihle JN. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. 1999;98:609–616. doi: 10.1016/s0092-8674(00)80048-3. [DOI] [PubMed] [Google Scholar]
- 12.O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 13.Taganov KD, Boldin MP, Baltimore D. MicroRNAs and immunity: tiny players in a big field. Immunity. 2007;26:133–137. doi: 10.1016/j.immuni.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 15.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 16.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 17.Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. 2005;202:261–269. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chong MM, Rasmussen JP, Rudensky AY, Littman DR. The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J Exp Med. 2008;205:2005–2017. doi: 10.1084/jem.20081219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cobb BS, Hertweck A, Smith J, O’Connor E, Graf D, Cook T, Smale ST, Sakaguchi S, Livesey FJ, Fisher AG, Merkenschlager M. A role for Dicer in immune regulation. J Exp Med. 2006;203:2519–2527. doi: 10.1084/jem.20061692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liston A, Lu LF, O’Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med. 2008;205:1993–2004. doi: 10.1084/jem.20081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT, Bluestone JA. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med. 2008;205:1983–1991. doi: 10.1084/jem.20080707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steiner DF, Thomas MF, Hu JK, Yang Z, Babiarz JE, Allen CD, Matloubian M, Blelloch R, Ansel KM. MicroRNA-29 Regulates T-Box Transcription Factors and Interferon-gamma Production in Helper T Cells. Immunity. 2011;35:169–181. doi: 10.1016/j.immuni.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, Hua M, Li N, Yao H, Cao X. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nature immunology. 2011;12:861–869. doi: 10.1038/ni.2073. [DOI] [PubMed] [Google Scholar]
- 24.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 25.Lovett-Racke AE, Rocchini AE, Choy J, Northrop SC, Hussain RZ, Ratts RB, Sikder D, Racke MK. Silencing T-bet defines a critical role in the differentiation of autoreactive T lymphocytes. Immunity. 2004;21:719–731. doi: 10.1016/j.immuni.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 26.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papadopoulou AS, Dooley J, Linterman MA, Pierson W, Ucar O, Kyewski B, Zuklys S, Hollander GA, Matthys P, Gray DH, et al. The thymic epithelial microRNA network elevates the threshold for infection-associated thymic involution via miR-29a mediated suppression of the IFN-α receptor. Nat Immunol. 2011;13:181–187. doi: 10.1038/ni.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lovett-Racke AE, Trotter JL, Lauber J, Perrin PJ, June CH, Racke MK. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients. A marker of activated/memory T cells. J Clin Invest. 1998;101:725–730. doi: 10.1172/JCI1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26:6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garzon R, Heaphy CE, Havelange V, Fabbri M, Volinia S, Tsao T, Zanesi N, Kornblau SM, Marcucci G, Calin GA, Andreeff M, Croce CM. MicroRNA 29b functions in acute myeloid leukemia. Blood. 2009;114:5331–5341. doi: 10.1182/blood-2009-03-211938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, Klein LO, Davis MM, Chen CZ. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 33.Sun G, Li H, Rossi JJ. Sequence context outside the target region influences the effectiveness of miR-223 target sites in the RhoB 3′UTR. Nucleic acids research. 2010;38:239–252. doi: 10.1093/nar/gkp870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–634. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 35.Ma F, Liu X, Li D, Wang P, Li N, Lu L, Cao X. MicroRNA-466l upregulates IL-10 expression in TLR-triggered macrophages by antagonizing RNA-binding protein tristetraprolin-mediated IL-10 mRNA degradation. Journal of immunology. 2010;184:6053–6059. doi: 10.4049/jimmunol.0902308. [DOI] [PubMed] [Google Scholar]
- 36.Morita Y, Naka T, Kawazoe Y, Fujimoto M, Narazaki M, Nakagawa R, Fukuyama H, Nagata S, Kishimoto T. Signals transducers and activators of transcription (STAT)-induced STAT inhibitor-1 (SSI-1)/suppressor of cytokine signaling-1 (SOCS-1) suppresses tumor necrosis factor alpha-induced cell death in fibroblasts. Proc Natl Acad Sci U S A. 2000;97:5405–5410. doi: 10.1073/pnas.090084797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kai ZS, Pasquinelli AE. MicroRNA assassins: factors that regulate the disappearance of miRNAs. Nat Struct Mol Biol. 2010;17:5–10. doi: 10.1038/nsmb.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schulz EG, Mariani L, Radbruch A, Hofer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 39.Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, Gadina M, Sher A, Paul WE, O’Shea JJ. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, Lassmann H, Wekerle H, Hohlfeld R, Meinl E. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain. 2009;132:3342–3352. doi: 10.1093/brain/awp300. [DOI] [PubMed] [Google Scholar]
- 41.Guerau-de-Arellano M, Lovett-Racke AE, Racke MK. miRNAs in multiple sclerosis: regulating the regulators. Journal of neuroimmunology. 2010;229:3–4. doi: 10.1016/j.jneuroim.2010.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cox MB, Cairns MJ, Gandhi KS, Carroll AP, Moscovis S, Stewart GJ, Broadley S, Scott RJ, Booth DR, Lechner-Scott J. MicroRNAs miR-17 and miR-20a inhibit T cell activation genes and are under-expressed in MS whole blood. PLoS One. 2010;5:e12132. doi: 10.1371/journal.pone.0012132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Santis G, Ferracin M, Biondani A, Caniatti L, Rosaria Tola M, Castellazzi M, Zagatti B, Battistini L, Borsellino G, Fainardi E, Gavioli R, Negrini M, Furlan R, Granieri E. Altered miRNA expression in T regulatory cells in course of multiple sclerosis. Journal of neuroimmunology. 2010;226:165–171. doi: 10.1016/j.jneuroim.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 44.Lindberg RL, Hoffmann F, Mehling M, Kuhle J, Kappos L. Altered expression of miR-17-5p in CD4+ lymphocytes of relapsing-remitting multiple sclerosis patients. European journal of immunology. 2010;40:888–898. doi: 10.1002/eji.200940032. [DOI] [PubMed] [Google Scholar]
- 45.Giegerich G, Pette M, Meinl E, Epplen JT, Wekerle H, Hinkanen A. Diversity of T cell receptor alpha and beta chain genes expressed by human T cells specific for similar myelin basic protein/major histocompatibility complexes. European journal of immunology. 1992;22:1331. doi: 10.1002/eji.1830220533. [DOI] [PubMed] [Google Scholar]
- 46.Allegretta M, Nicklas JA, Sriram S, Albertini RJ. T cells responsive to myelin basic protein in patients with multiple sclerosis. Science. 1990;247:718–721. doi: 10.1126/science.1689076. [DOI] [PubMed] [Google Scholar]
- 47.Balashov KE, Smith DR, Khoury SJ, Hafler DA, Weiner HL. Increased interleukin 12 production in progressive multiple sclerosis: induction by activated CD4+ T cells via CD40 ligand. Proc Natl Acad Sci U S A. 1997;94:599–603. doi: 10.1073/pnas.94.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hellings N, Baree M, Verhoeven C, D’Hooghe B, Medaer MR, Bernard CC, Raus J, Stinissen P. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J Neurosci Res. 2001;63:290–302. doi: 10.1002/1097-4547(20010201)63:3<290::AID-JNR1023>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 49.Oliver-Martos B, Orpez T, Pinto-Medel MJ, Mayorga C, Garcia-Leon JA, Maldonado-Sanchez R, Suardiaz M, Guerrero M, Luque G, Leyva L, Fernandez O. Gene expression in IFNss signalling pathway differs between monocytes, CD4 and CD8 T cells from MS patients. Journal of neuroimmunology. 2011;230:153–159. doi: 10.1016/j.jneuroim.2010.10.033. [DOI] [PubMed] [Google Scholar]
- 50.Nunez-Iglesias J, Liu CC, Morgan TE, Finch CE, Zhou XJ. Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS One. 2010;5:e8898. doi: 10.1371/journal.pone.0008898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ceolotto G, Papparella I, Bortoluzzi A, Strapazzon G, Ragazzo F, Bratti P, Fabricio AS, Squarcina E, Gion M, Palatini P, Semplicini A. Interplay between miR-155, AT1R A1166C polymorphism, and AT1R expression in young untreated hypertensives. Am J Hypertens. 2011;24:241–246. doi: 10.1038/ajh.2010.211. [DOI] [PubMed] [Google Scholar]
- 54.Nicoloso MS, Sun H, Spizzo R, Kim H, Wickramasinghe P, Shimizu M, Wojcik SE, Ferdin J, Kunej T, Xiao L, Manoukian S, Secreto G, Ravagnani F, Wang X, Radice P, Croce CM, Davuluri RV, Calin GA. Single-nucleotide polymorphisms inside microRNA target sites influence tumor susceptibility. Cancer Res. 2010;70:2789–2798. doi: 10.1158/0008-5472.CAN-09-3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.