

Abstract
Suppressor of cytokine signalling (SOCS) 3 is an essential regulator of cytokine signalling, and in turn its expression is tightly regulated. Data from overexpression studies in cell lines suggest that SOCS2 regulates SOCS3 protein degradation, by forming a molecular bridge to an E3 ubiquitin-ligase complex. Whether this regulation is relevant in primary cells is unknown. In this study, we utilized Socs2−/− mice to examine the role of SOCS2 in modulating SOCS3 expression and degradation, and its impact on IL-2 and IL-6 signalling in primary haemopoietic cells. Both biochemical and biological analyses demonstrated unperturbed SOCS3 expression and cytokine signalling in the absence of SOCS2. Our results suggest that SOCS2 is not a physiological regulator of SOCS3 expression and action in primary haemopoietic cells.
Keywords: SOCS2, SOCS3, IL-6, IL-2, STAT, macrophages, lymphocytes
Introduction
Cytokines initiate a spectrum of biological effects through activation of a range of intracellular signalling cascades. JAK-STAT signalling downstream of cytokine receptors is tightly regulated by members of the suppressor of cytokine signalling (SOCS) protein family (Croker et al. 2008; Murray 2007). The inhibitory effects of SOCS proteins on cytokine signalling have been definitively demonstrated through analyses of mice harboring loss of functional alleles. In the absence of SOCS1 or SOCS3, mice exhibited profound pathologies including multiple haemopoietic and immunological defects as a consequence of prolonged STAT activation (Alexander et al. 1999; Cornish et al. 2003; Croker et al. 2003; Croker et al. 2004; Lang et al. 2003; Naka et al. 1998; Starr et al. 1998; Yasukawa et al. 2003).
Mice that lack SOCS3 die at midgestation due to placental defects consequent upon unrestrained LIF signalling (Robb et al. 2005; Roberts et al. 2001). Subsequent studies using conditional alleles have revealed the importance of SOCS3 in haemopoietic cells, with absence of SOCS3 leading to inflammatory disorders in young mice and hypersensitivity to IL-6, G-CSF and IL-1 stimulation (Croker et al. 2003; Croker et al. 2004; Wong et al. 2006).
SOCS3 contains an N-terminal region, a central SH2 domain and a C-terminal SOCS box motif (Hilton et al. 1998). Each of these modules contributes to attenuation of the duration and intensity of JAK-STAT signalling. The SH2 domain is responsible for SOCS3 binding to both the phosphotyrosine residues within cytoplasmic domain of the receptor and JAK kinase following cytokine-mediated activation (Dunn et al. 2005; Hortner et al. 2002a; Hortner et al. 2002b; Nicholson et al. 2000; Sasaki et al. 1999). As a consequence, SOCS3 is thought to inhibit signal transduction by directly inhibiting JAK activity through the kinase inhibitory region (KIR) located in the SOCS3 N-terminus (Sasaki et al. 1999; Yasukawa et al. 1999). In addition, the SOCS box motif recruits an E3 ubiquitin-ligase complex that targets SOCS3 binding partners for ubiquitin-mediated proteasomal and/or lysosomal degradation to reinforce inhibition of signalling (Boyle et al. 2008; Irandoust et al. 2007; Zhang et al. 1999).
Being a regulator itself, SOCS3 expression is also tightly controlled at multiple levels. Transcriptionally, STATs are responsible for upregulation of Socs3 transcripts following cytokine stimulation. (Auernhammer et al. 1999; Gatto et al. 2004; He et al. 2003; Lejeune et al. 2001; Naka et al. 1997). Recently, it has been demonstrated that other factors such as Specificity protein 3 (Sp3), c-Fos, c-Jun and FOXO3a and coactivators CREB-binding protein (CBP) and p300 are also involved in the transcriptional activation of Socs3 in a cytokine- and cell type-dependent manner (Barclay et al. 2007; Ehlting et al. 2005; Qin et al. 2007). On the other hand, proto-oncoprotein growth factor independence-1B (GFI-1B) and hepatocyte nuclear factor-1β (HNF-1β) have been identified as repressors of Socs3 transcription (Jegalian and Wu 2002; Ma et al. 2007). At the post-transcriptional level, Socs3 mRNA stability can be affected by TNFα-mediated activation of the MAPK Kinase 6 (MKK6)/p38MAPK cascade. MAPK-activated protein kinase 2 (MK2), a downstream effector of the cascade, is an important facilitator of this process (Ehlting et al. 2007).
At the protein level, SOCS3 can be targeted for both non-proteasomal and proteasomal-mediated degradation. Modification of SOCS box, Lys6 and PEST sequences of the SOCS3 protein have all been reported to influence the stability of the protein (Babon et al. 2006; Haan et al. 2003; Sasaki et al. 2003; Zhang et al. 1999). However, the exact molecular mechanisms by which SOCS3 protein stability is influenced remain unclear. It has been proposed that SOCS2 regulates SOCS3 stability by forming a molecular bridge between an E3 ubiquitin-ligase complex and other SOCS proteins, targeting them for degradation (Piessevaux et al. 2008). Acceleration of SOCS3 degradation has been observed in cell lines overexpressing SOCS2, and was associated with deregulated IL-2 and IL-3 signalling (Piessevaux et al. 2006; Tannahill et al. 2005). Such data suggest that SOCS2 is a negative regulator of SOCS3 protein levels, and thus a positive modulator of SOCS3-inhibited cytokine signalling cascades.
A corollary of this model is that, in the absence of SOCS2, enhanced or prolonged SOCS3 expression would be expected, resulting in excessive inhibition of signalling. To examine the physiological requirement for SOCS2 in the regulation of SOCS3, we have analyzed SOCS3 protein levels and SOCS3-dependent signal transduction in cells from Socs2−/− mice.
Materials and Methods
Generation and maintenance of mice
Generation of Socs2−/− mice on a C57BL/6 background has been described previously (Metcalf et al. 2000). For conditional deletion of Socs3, a new floxed allele of the Socs3 locus was generated via homologous recombination in ES cells. The targeted allele incorporated three loxP sites allowing initial removal of the hygromycin (hygro) selectable marker via transient expression of Cre in targeted ES cells, followed by generation of mice bearing the floxed allele (fl) (Fig. S1A). Socs3−/fl vavCre+ mice with a conditional deletion of SOCS3 in haemopoietic cells (hereafter referred to as Socs3−/Δvav) were then generated by crossing Socs3fl/fl mice with Socs3+/− vavCre+ mice (generated as described previously (Croker et al. 2003)). Efficient deletion of Socs3 was verified by Southern and absence of protein expression was monitored by Western blot analysis (Fig. S1B and C). Prolonged IL-6 signalling, which is characteristic of absence of SOCS3 (Croker et al. 2003), was confirmed in Socs3−/Δvav bone marrow derived macrophages (BMM) cells by Western blot analysis (Fig. S1C). Mice were housed at the Walter and Eliza Hall Institute of Medical Research (WEHI) and all animal experiments were performed with the approval of the Animal Ethics Committees of the Melbourne Health Research Directorate or WEHI.
Generation of Monoclonal Antibody
SOCS2 monoclonal antibodies (mAb) were generated by immunization of mice with NusA SOCS2-SH2 fusion protein (Greenhalgh et al. 2002) three times with Freund’s complete and incomplete adjuvant, before spleens were removed and fused. Antibodies were screened for SOCS2 specific binding before being cloned and grown for use.
Bone marrow-derived macrophages (BMM) culture
Bone marrow cells were cultured overnight at 37 °C in a 10% (v/v) CO2 incubator in 10-cm tissue culture-treated plates (2 × 107/dish) in 20 mL of Dulbecco’s modified Eagle’s medium with 10% (v/v) fetal calf serum (DMEM/10% FCS) and 20% (v/v) L-cell-conditioned medium (LCM), a source of macrophage colony-stimulating factor. 10 mL of cells and media were then transferred to 10 cm Petri dishes and incubated for a further 4 days, with 1 mL of LCM being added to the cells on day 3. On day 5 adherent cells, comprising a pure population of macrophages, were washed in mouse-tonicity phosphate-buffered saline (MT-PBS) and resuspended in 4 mL of cell dissociation buffer (Invitrogen, Carlsbad, CA). After incubation at 37 °C for 10 min, cells were harvested and resuspended in DMEM/10% FCS.
Cytokine stimulation of BMM
BMM were plated into 6-well tissue culture treated plates (1 × 106 cells/well), and deprived of LCM by incubation in DMEM/10% FCS for 1h. Cells were then pulse stimulated with mouse IL-6 (mIL-6) (100 ng/mL, a gift generously given by Prof. Richard Simpson, Ludwig Institute for Cancer Research, Melbourne) for 0.5 h, then the media was removed and replaced with DMEM/10% FCS for up to 8 h. In some experiments, BMM were pre-incubated with mouse IL-4 (mIL-4) (10 ng/mL, a gift generously given by the Division of Immunology, WEHI) for 3.5 h.
BMM proliferation assay
Cells were plated into 96-well plates (1 × 104 cells/well) in 100 μL of DMEM/15% FCS, 103 U/mL of human macrophage-colony stimulating factor (hM-CSF) (Cetus, Emeryville, CA) and various concentrations of mIL-6. Cells were cultured at 37 °C, 10% CO2 for 36 h followed by addition of 1 μCi/well of [3H]thymidine for 16 h. Cell associated radioactivity was then harvested onto glass fibre filters using an automatic cell harvester, and measured using a scintillation counter (MicroBeta TriLux, Waltham, MA). Data were normalized to the [3H]thymidine incorporation of cells cultured in the same media without mIL-6.
Culture and stimulation of lymph node (LN) cells
A single-cell suspension was prepared from pooled cervical, brachial, axillary, inguinal, and mesenteric lymph nodes in MT-RPMI supplemented with 10% (v/v) heat inactivated (HI) FCS and 40 μM β-mercaptoethanol (RPMI/HI FCS/β-Me) and incubated at 37 °C in a 10% CO2 incubator for 1 h. For biochemistry experiments, cells were either stimulated with 10 ng/mL of mouse IL-2 (mIL-2) (Peprotech, Rocky Hill, NJ) for up to 8 h or stimulated with anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) mAb and human IL-2 (hIL-2) (10 ng/mL) (Peprotech) in the presence of anti-mIL-2 neutralizing antibody (2 μg/mL) (R&D Systems, Minneapolis, MN) for 4 h. For the latter stimulating condition, the media was removed and replaced with RPMI/HI FCS/β-Me in the presence of anti-mIL-2 neutralizing antibody for up to a further 8 h incubation with or without cycloheximide (15μg/mL) (Sigma, St. Louis, MO).
T-cell purification
Lymph node T cells were enriched by negative selection using Mouse T cell Enrichment Columns (R&D Systems) according to the manufacturer’s instructions. Purity was assessed by flow cytometry with anti-CD3 antibody (BD Pharmingen, San Jose, CA) and was routinely >95% (data not shown).
T-cell proliferation assay
5 × 104 purified T-cells were incubated in 100 μL of RPMI/HI FCS/β-Me, 2 μg/mL anti-mIL-2 (R&D Systems) and various concentrations of human IL-2 (hIL-2) (Peprotech) in the presence or absence of soluble anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) mAb. Cells were cultured at 37 °C in a 10% CO2 incubator for 3 days followed by addition of 1 μCi/well of [3H]thymidine for 17h. Cell associated radioactivity was then harvested onto glass fiber filters using an automatic cell harvester, and measured using a scintillation counter (MicroBeta TriLux).
Western blot Analysis
LN cells or BMM were lysed in 1% (v/v) Triton X-100, 1 mM EGTA, 1 mM EDTA, 10 mM Na pyrophosphate, 1 mM Na3VO4, 10 mM Na β-glycerophosphate, 150 mM NaCl, 50 mM NaF, 50 mM Tris-HCl pH 7.4, containing protease inhibitors (Complete Mixture tablets, Roche Applied Science, Indianapolis, IN). Lysates were subjected to SDS-PAGE Western blotting with antibodies specific for either phospho-STAT3 Y705, total STAT3 (Cell Signaling, Danvers, MA), phospho-STAT5A/B Y694/677 (Upstate Biotechnology, Lake Placid, NY), total STAT5 (Zymed, San Francisco, CA), phospho-STAT1 Y701 (Cell Signaling), total STAT1 (Cell signaling), total SOCS3 (IBL, Gunma, Japan) and total SOCS2 (in-house mAb raised against NusA-SOCS2 SH2 fusion protein) proteins. Antibody binding was visualized with either horseradish peroxidase-conjugated anti-rabbit (Chemicon, Temecula, CA) or anti-mouse-IgG (GE Health, Piscataway, NJ) and the ECL Western blotting detection reagent (Amersham, Uppsala, Sweden). Protein expression was quantified by densitometric analyses (Bio-Rad GS-800 Calibrated Densitometer, Bio-Rad Laboratories, Hercules, CA) of Western blots.
RNA extraction and real-time PCR Analysis
DNase-treated RNA was extracted from BMM or LN cells using RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. RNA was then reverse-transcribed into complementary DNA (cDNA) with an Oligo-dT primer (Promega, Madison, WI). cDNA was mixed with SYBR Green PCR master mix (Qiagen) and primers then amplified by PCR using ABI7900 Sequence Detector to quantify Socs2 and Socs3 mRNA levels. PCR conditions were 15 sec at 94°C, 30 sec at 60°C and 30 sec at 72°C with 45 cycles. The forward and reverse primers used for Socs2, Socs3 and Gapdh were (Socs2) TCCAGATGTGCAAGGATAAACG and AGGTACAGGTGAACAGTCCCATT; (Socs3) ATTTCGCTTCGGGACTAGCTC and AGCTGTCGCGGATAAGAAAGG; (Gapdh) TTGTCAAGCTCATTTCCTGGT and TTACTCCTTGGAGGCCATGTA. The expression level of each target gene was normalized to Gapdh expression. Data represent mean ± standard deviation of the mean (SD) from 3–4 independent experiments.
Statistical Analysis
To evaluate changes in gene expression over time, data were log-transformed and one-way analysis of variance (ANOVA) was performed (GraphPad Prism Version 5.0, GraphPad Software, San Diego, CA). Variation in expression between WT and Socs2−/− at given timepoints were analysed by both t-test with equal variance assumption and paired t-test (Microsoft Excel, Microsoft Corporation, Redwood, WA, USA) on the log-transformed data.
Results
SOCS3 expression and IL-6-mediated responses in WT and Socs2−/− BMM
Previous work identified SOCS3 as a negative regulator of IL-6 signalling in macrophages (Croker et al. 2003). We utilized this system to assess whether the presence or absence of SOCS2 affected the level of SOCS3 expression and/or perturbed STAT3 activation by IL-6. In primary macrophages, expression of Socs2 mRNA was induced from a low baseline upon stimulation with mIL-6 for 30 min (Fig. 1A). No differences in transcript levels were observed between wildtype (WT) and Socs3−/− cells. Even with stimulation, the level of SOCS2 protein expression remained below the level of detection by Western blotting with available antibodies (data not shown). In WT (Socs2+/+) cells, SOCS3 protein expression was undetectable prior to pulse stimulation with mIL-6, then peaked at 1 h and was downregulated by 2 h. In the absence of SOCS2, this pattern of mIL-6-induced SOCS3 expression was unaltered (Fig. 1B). Consistent with maintenance of normal regulation of SOCS3 expression, the induction and duration of mIL-6-dependent STAT3 activation in Socs2−/− BMM were identical to that of the WT cells (Fig. 1B). As expected, Socs3−/− BMM displayed prolonged activation of STAT3 (Fig. 1B).
Figure 1.

Normal SOCS3 expression, STAT3 phosphorylation and SOCS3-dependent inhibition of proliferation in IL-6 stimulated Socs2−/− macrophages. (A) Expression of Socs2 transcript relative to Gapdh transcript. BMM from WT, Socs2−/− and Socs3−/Δvav mice were stimulated with mIL-6 (100 ng/mL) for 0.5 h, then incubated in cytokine-free media for up to 8 hours. Quantification of Socs2 mRNA was performed by Real-Time PCR after RNA extraction and reverse transcription. Data represent mean ± standard error of the mean (SEM) of replicates from 4 independent experiments. (B) Western blot analyses of cell lysates of BMM cells treated as described for A. Indicative example of 3 independent experiments. (C) BMM from WT, Socs2−/− and Socs3−/Δvav mice were cultured in presence of hM-CSF (103 U/mL) and various concentrations of mIL-6. Cell proliferation was determined by measuring [3H]thymidine incorporation after 52 h, and normalized to [3H]thymidine incorporation of cells cultured without mIL-6. Data represent mean ± SEM of 4 independent experiments.
To examine IL-6 action in a biological context, we monitored its recognized inhibitory effect on M-CSF-induced BMM proliferation in WT macrophages (Takeda et al. 1999) and those lacking either SOCS2 or SOCS3. Consistent with the unaltered STAT3 phosphorylation observed in Fig. 1B, following mIL-6 stimulation of Socs2−/− BMM, Socs2−/− and WT cells displayed equivalent inhibition of proliferation when incubated with the cytokine. As expected, greater inhibition was seen in Socs3−/Δvav cells due to unrestrained IL-6 signalling (Fig. 1C). Thus, in BMM, SOCS3 expression and regulation of signal transduction and biological responses to mIL-6 appear independent of SOCS2. Certainly there was no evidence for aberrant SOCS3 activity in the absence of SOCS2.
Effect of SOCS2 upregulation on SOCS3 expression and IL-6 signalling in WT and Socs2−/− BMM
As IL-6 is a relatively weak stimulus of SOCS2, we sought to explore any regulatory role of SOCS2 at higher levels of expression. BMM were pre-incubated with mIL-4. SOCS2 expression was markedly upregulated and was readily detectable by Western blot (Fig. 2). Despite this higher level of SOCS2 expression in WT control cells, the kinetics and magnitude of STAT3 activation and the regulation of SOCS3 expression induced by subsequent mIL-6 stimulation remained indistinguishable from those observed in similarly treated SOCS2-deficient cells (Fig. 2).
Figure 2.

SOCS3 expression and STAT3 phosphorylation are unaltered by IL-4-induced expression of SOCS2 in macrophages. BMM from WT and Socs2−/− mice were preincubated with mIL-4 (10 ng/mL) for 3.5 h to induce SOCS2 expression, then stimulated with mIL-6 (100 ng/mL) for 0.5 h, and subsequently incubated in cytokine-free media for up to 8 hours. Cell lysates were collected for Western blot analyses. Indicative example of 3 independent experiments.
Expression of SOCS3 and response to IL-2 in lymphocytes in the absence of SOCS2
Previously, it was demonstrated that enforced expression of SOCS2 in BaF/3 cells enhanced IL-2 signalling by accelerating SOCS3 degradation (Tannahill et al. 2005). Since IL-2 is an essential proliferative cytokine for primary lymphocytes, we examined signal transduction and SOCS3 expression in Socs2−/− lymphocytes stimulated with mIL-2. In WT lymphocytes from lymph nodes, both SOCS2 and SOCS3 mRNA and protein were detectable and did not alter substantially after mIL-2 stimulation (Fig. 3A and B). No differences in the intensity of SOCS3 expression nor the duration of mIL-2 activated STAT5 were observed between WT and Socs2−/− LN cells (Fig. 3A). Identical results were also seen in purified T cells (data not shown).
Figure 3.

SOCS3 expression, STAT5 phosphorylation and IL-2 induced proliferation are unaltered in Socs2−/− lymphocytes stimulated with IL-2. (A) WT, Socs2−/− and Socs3−/Δvav LN cells were stimulated with mIL-2 (10 ng/mL) for up to 8 h. Indicative Western blot of cell lysates from one of three experiments. SOCS2 and SOCS3 are indicated by white arrow and black arrows, respectively. The grey arrow indicates non-specific bands (light chains). “C” indicates SOCS3 positive control from LPS stimulated BMM. (B) Expression of Socs3 mRNA in WT and Socs2−/− cells in response to mIL-2 (10 ng/mL) quantified by real-time PCR analysis, and expressed relative to Gapdh expression. Data represent mean ± SEM of 3–4 independent experiments. The variation in SOCS3 expression level between the time points for each genotype was not statistically significant as judged by one-way analysis of variance (ANOVA) on log-transformed data (p=0.6192 for WT and p=0.7782 for Socs2−/−). (C) Purified LN T-cells from WT and Socs2−/− mice were cultured with various concentrations of hIL-2 for 3.5 days in the presence of anti-mIL-2. Cell proliferation was determined by measuring [3H]thymidine incorporation. Data represent mean ± SEM of 5 independent experiments.
To further examine whether the presence or absence of SOCS2 influences SOCS3-regulated cellular responses, we monitored T cell proliferation in response to hIL-2. Given that Socs2−/− mice display a normal distribution of T-cell subsets in various lymphoid compartments (data not shown), LN T cell proliferation in response to hIL-2 treatment was measured by [3H]thymidine incorporation. Endogenous IL-2 was neutralized with anti-mIL-2 antibody. Consistent with unaltered IL-2 signalling in Socs2−/− cells, proliferation of Socs2−/− T cells in response to hIL-2 was indistinguishable from WT (Fig. 3C).
SOCS3 expression by, and proliferation of, anti-CD3/CD28 activated Socs2−/− LN cells
To examine regulation of SOCS3 in an inducible system, T cells were stimulated for 4 h with anti-CD3 and anti-CD28 mAb, and the level of SOCS3 expression monitored without stimulation for a further 8 h. In both WT and Socs2−/− LN cells, SOCS3 expression was induced to maximal levels at 4.5 h and downregulated at equivalent rates in the absence of further stimuli (Fig. 4A and B). Similarly, no difference between genotypes was observed in SOCS3 expression when cycloheximide was added to prevent further protein synthesis after removal of the stimuli at 4 h (Fig. S2). Consistent with normal regulation of SOCS3 expression, anti-CD3 and anti-CD28 mAb activated Socs2−/− T-cells displayed a normal proliferative response to hIL-2 (Fig. 4C).
Figure 4.
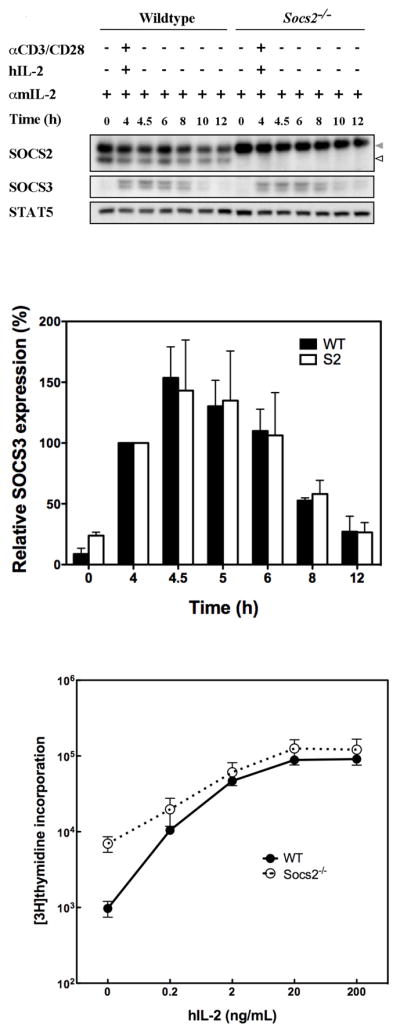
Induction and decay of SOCS3 expression and proliferation in activated LN cells is independent of SOCS2 expression. (A) LN cells collected from WT and Socs2−/− mice were incubated with anti-CD3 (5μg/mL), anti-CD28 (2 μg/mL) and hIL-2 (10ng/mL) for 4 h. Thereafter, cells were washed and incubated in media for a further 8 h. Throughout the experiment cells were exposed to anti-mIL-2 (2 μg/mL). Cell lysates were collected for Western blot analyses. SOCS2 and non-specific bands (light chains) are indicated by white arrow and grey arrows, respectively. Indicative example of 3 independent experiments. (B) SOCS3 expression in above experiments was quantified by densitometry. Expression levels were normalized to the 4 h time point for each genotype and presented as relative SOCS3 expression (%). Data represent mean ± standard deviation (SD) of 3 independent experiments. (C) Purified LN T-cells from WT and Socs2−/− mice were cultured with anti-CD3 (5μg/mL), anti-CD28 (2 μg/mL) and hIL-2 or hIL-2 alone at various concentrations for 2.5 days in the presence of anti-mIL-2. Cell proliferation was determined by measuring [3H]thymidine incorporation. Data represent mean ± SD of 2 independent experiments.
Discussion
Although SOCS3 is recognized as a key suppressor of cytokine signalling, our understanding of the regulation of SOCS3 protein levels is still inexact. Both transcriptional and post-translational mechanisms have been proposed for controlling the rapid induction of SOCS3 following cytokine stimulation, as well as the timely decay of protein levels to allow ongoing cellular responses to cytokines where appropriate. Within this context, SOCS2 has been proposed to interact with, and target SOCS3 for proteasomal-mediated degradation. In a cell line-based ectopic expression model, SOCS2 was shown to accelerate SOCS3 degradation, leading to prolonged IL-2 and IL-3 signalling (Piessevaux et al. 2006; Tannahill et al. 2005). If this model reflected physiology, we would predict that in the absence of SOCS2, enhanced or prolonged SOCS3 protein expression would ensue, resulting in curtailed duration and/or intensity of cytokine signalling pathways normally inhibited by SOCS3. To formally explore a physiological role for SOCS2 in the regulation of SOCS3 we analyzed primary cells from Socs2−/− mice for regulation of SOCS3 protein levels and control of signalling pathways known to be regulated by SOCS3 under physiological conditions (IL-6) (Croker et al. 2003) as well as cytokine pathways sensitive to SOCS3 when overexpressed (IL-2) (Cohney et al. 1999).
Our data revealed that SOCS3 protein expression and STAT3 activation upon mIL-6 stimulation was indistinguishable in Socs2−/− BMM from that in WT cells (Fig. 1B and 2). Consistent with this, the absence of SOCS2 did not desensitize cells to IL-6-induced growth inhibition (Fig. 1C). We also observed similar phenomena in lymphocytes. Both SOCS3 expression and mIL-2-dependent signal transduction in SOCS2-deficient cells remained identical to the WT patterns (Fig. 3A) and the decline in cellular SOCS3 protein levels following induction in activated T-cells was also comparable between the two genotypes (Fig. 4A and B and Fig S2). In line with this, biological assays revealed that loss of SOCS2 made no difference to the proliferation of T cells in response to hIL-2 in either the quiescent or activated states (Fig. 3C and 4C). Taken together, the data presented here suggest that any interaction between SOCS2 and SOCS3 leading to targeting of SOCS3 for degradation does not play a significant role in regulation of SOCS3 expression in primary macrophages and T cells.
Thus, our data do not support there being a physiological role for SOCS2 in the regulation of SOCS3 expression and activity in the haemopoietic cells. Since the previously reported actions of SOCS2 in this regard were described following ectopic expression in cells lines, the regulatory action of SOCS2 on SOCS3 may require levels of SOCS2 not normally achieved in vivo. There are examples in the literature where ectopic or over-expression of SOCS proteins revealed activities that are not physiological. For example, ectopic expression of SOCS3 effectively inhibits interferon-γ (IFNγ) signalling (Karlsen et al. 2001; Song and Shuai 1998; Woldman et al. 2001), but SOCS3 appears dispensable for normal regulation of IFNγ in vivo (Croker et al. 2003; Lang et al. 2003) and similar observations have been made for SOCS1 and IL-6 signalling (Croker et al. 2003; Lang et al. 2003; Starr et al. 1997). It may be possible that crosstalk between SOCS2 and SOCS3 occurs in pathological states if levels of cytokines and SOCS proteins are extreme. Our data cannot exclude a role for SOCS2 in modulating SOCS3 regulation of other signalling systems, such as has been proposed for growth hormone (Piessevaux et al. 2006; Piessevaux et al. 2008), although our data do suggest that assessment of such models under physiological conditions is warranted.
Supplementary Material
Acknowledgments
We are grateful to Janelle Lochland for technical assistance and Jian-Guo Zhang and Sandra Nicholson for invaluable discussion and advice. This work was supported by a grant from the National Institutes of Health (CA022556); Fellowships (CJG, DJH, WSA, NN and AWR), a Program Grant from the National Health and Medical Research Council (NHMRC) of Australia (461291); NHMRC Independent Research Institutes Infrastructure Support Scheme (361646); and Victorian State Government Operational Infrastructure Support grant. HK is a recipient of a Biomedical (Dora Lush) Postgraduate Research Scholarship from the NHMRC.
References
- Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TW, Nicola NA, Hertzog PJ, Metcalf D, Hilton DJ. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- Auernhammer CJ, Bousquet C, Melmed S. Autoregulation of pituitary corticotroph SOCS-3 expression: characterization of the murine SOCS-3 promoter. Proc Natl Acad Sci U S A. 1999;96:6964–9. doi: 10.1073/pnas.96.12.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babon JJ, McManus EJ, Yao S, DeSouza DP, Mielke LA, Sprigg NS, Willson TA, Hilton DJ, Nicola NA, Baca M, Nicholson SE, Norton RS. The structure of SOCS3 reveals the basis of the extended SH2 domain function and identifies an unstructured insertion that regulates stability. Mol Cell. 2006;22:205–16. doi: 10.1016/j.molcel.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Barclay JL, Anderson ST, Waters MJ, Curlewis JD. Regulation of suppressor of cytokine signaling 3 (SOC3) by growth hormone in pro-B cells. Mol Endocrinol. 2007;21:2503–15. doi: 10.1210/me.2006-0498. [DOI] [PubMed] [Google Scholar]
- Boyle K, Zhang JG, Nicholson SE, Trounson E, Babon JJ, McManus EJ, Nicola NA, Robb L. Deletion of the SOCS box of suppressor of cytokine signaling 3 (SOCS3) in embryonic stem cells reveals SOCS box-dependent regulation of JAK but not STAT phosphorylation. Cell Signal. 2008;21:394–404. doi: 10.1016/j.cellsig.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohney SJ, Sanden D, Cacalano NA, Yoshimura A, Mui A, Migone TS, Johnston JA. SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol Cell Biol. 1999;19:4980–8. doi: 10.1128/mcb.19.7.4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish AL, Chong MM, Davey GM, Darwiche R, Nicola NA, Hilton DJ, Kay TW, Starr R, Alexander WS. Suppressor of cytokine signaling-1 regulates signaling in response to interleukin-2 and other gamma c-dependent cytokines in peripheral T cells. J Biol Chem. 2003;278:22755–61. doi: 10.1074/jbc.M303021200. [DOI] [PubMed] [Google Scholar]
- Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19:414–22. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–5. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- Croker BA, Metcalf D, Robb L, Wei W, Mifsud S, DiRago L, Cluse LA, Sutherland KD, Hartley L, Williams E, Zhang JG, Hilton DJ, Nicola NA, Alexander WS, Roberts AW. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity. 2004;20:153–65. doi: 10.1016/s1074-7613(04)00022-6. [DOI] [PubMed] [Google Scholar]
- Dunn SL, Bjornholm M, Bates SH, Chen Z, Seifert M, Myers MG., Jr Feedback inhibition of leptin receptor/Jak2 signaling via Tyr1138 of the leptin receptor and suppressor of cytokine signaling 3. Mol Endocrinol. 2005;19:925–38. doi: 10.1210/me.2004-0353. [DOI] [PubMed] [Google Scholar]
- Ehlting C, Haussinger D, Bode JG. Sp3 is involved in the regulation of SOCS3 gene expression. Biochem J. 2005;387:737–45. doi: 10.1042/BJ20041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlting C, Lai WS, Schaper F, Brenndorfer ED, Matthes RJ, Heinrich PC, Ludwig S, Blackshear PJ, Gaestel M, Haussinger D, Bode JG. Regulation of Suppressor of Cytokine Signaling 3 (SOCS3) mRNA Stability by TNF-{alpha} Involves Activation of the MKK6/p38MAPK/MK2 Cascade. J Immunol. 2007;178:2813–26. doi: 10.4049/jimmunol.178.5.2813. [DOI] [PubMed] [Google Scholar]
- Gatto L, Berlato C, Poli V, Tininini S, Kinjyo I, Yoshimura A, Cassatella MA, Bazzoni F. Analysis of SOCS-3 promoter responses to interferon gamma. J Biol Chem. 2004;279:13746–54. doi: 10.1074/jbc.M308999200. [DOI] [PubMed] [Google Scholar]
- Greenhalgh CJ, Metcalf D, Thaus AL, Corbin JE, Uren R, Morgan PO, Fabri LJ, Zhang JG, Martin HM, Willson TA, Billestrup N, Nicola NA, Baca M, Alexander WS, Hilton DJ. Biological evidence that SOCS-2 can act either as an enhancer or suppressor of growth hormone signaling. J Biol Chem. 2002;277:40181–4. doi: 10.1074/jbc.C200450200. [DOI] [PubMed] [Google Scholar]
- Haan S, Ferguson P, Sommer U, Hiremath M, McVicar DW, Heinrich PC, Johnston JA, Cacalano NA. Tyrosine phosphorylation disrupts elongin interaction and accelerates SOCS3 degradation. J Biol Chem. 2003;278:31972–9. doi: 10.1074/jbc.M303170200. [DOI] [PubMed] [Google Scholar]
- He B, You L, Uematsu K, Matsangou M, Xu Z, He M, McCormick F, Jablons DM. Cloning and characterization of a functional promoter of the human SOCS-3 gene. Biochem Biophys Res Commun. 2003;301:386–91. doi: 10.1016/s0006-291x(02)03071-1. [DOI] [PubMed] [Google Scholar]
- Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D, Nicola NA. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A. 1998;95:114–9. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortner M, Nielsch U, Mayr LM, Heinrich PC, Haan S. A new high affinity binding site for suppressor of cytokine signaling-3 on the erythropoietin receptor. Eur J Biochem. 2002a;269:2516–26. doi: 10.1046/j.1432-1033.2002.02916.x. [DOI] [PubMed] [Google Scholar]
- Hortner M, Nielsch U, Mayr LM, Johnston JA, Heinrich PC, Haan S. Suppressor of cytokine signaling-3 is recruited to the activated granulocyte-colony stimulating factor receptor and modulates its signal transduction. J Immunol. 2002b;169:1219–27. doi: 10.4049/jimmunol.169.3.1219. [DOI] [PubMed] [Google Scholar]
- Irandoust MI, Aarts LH, Roovers O, Gits J, Erkeland SJ, Touw IP. Suppressor of cytokine signaling 3 controls lysosomal routing of G-CSF receptor. Embo J. 2007 doi: 10.1038/sj.emboj.7601640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegalian AG, Wu H. Regulation of Socs gene expression by the proto-oncoprotein GFI-1B: two routes for STAT5 target gene induction by erythropoietin. J Biol Chem. 2002;277:2345–52. doi: 10.1074/jbc.M105575200. [DOI] [PubMed] [Google Scholar]
- Karlsen AE, Ronn SG, Lindberg K, Johannesen J, Galsgaard ED, Pociot F, Nielsen JH, Mandrup-Poulsen T, Nerup J, Billestrup N. Suppressor of cytokine signaling 3 (SOCS-3) protects beta -cells against interleukin-1beta - and interferon-gamma -mediated toxicity. Proc Natl Acad Sci U S A. 2001;98:12191–6. doi: 10.1073/pnas.211445998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–50. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- Lejeune D, Demoulin JB, Renauld JC. Interleukin 9 induces expression of three cytokine signal inhibitors: cytokine-inducible SH2-containing protein, suppressor of cytokine signalling (SOCS)-2 and SOCS-3, but only SOCS-3 overexpression suppresses interleukin 9 signalling. Biochem J. 2001;353:109–116. [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Gong Y, Patel V, Karner CM, Fischer E, Hiesberger T, Carroll TJ, Pontoglio M, Igarashi P. Mutations of HNF-1beta inhibit epithelial morphogenesis through dysregulation of SOCS-3. Proc Natl Acad Sci U S A. 2007;104:20386–91. doi: 10.1073/pnas.0705957104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf D, Greenhalgh CJ, Viney E, Willson TA, Starr R, Nicola NA, Hilton DJ, Alexander WS. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature. 2000;405:1069–73. doi: 10.1038/35016611. [DOI] [PubMed] [Google Scholar]
- Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–9. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- Naka T, Matsumoto T, Narazaki M, Fujimoto M, Morita Y, Ohsawa Y, Saito H, Nagasawa T, Uchiyama Y, Kishimoto T. Accelerated apoptosis of lymphocytes by augmented induction of Bax in SSI-1 (STAT-induced STAT inhibitor-1) deficient mice. Proc Natl Acad Sci U S A. 1998;95:15577–82. doi: 10.1073/pnas.95.26.15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, Akira S, Kishimoto T. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–9. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- Nicholson SE, De Souza D, Fabri LJ, Corbin J, Willson TA, Zhang JG, Silva A, Asimakis M, Farley A, Nash AD, Metcalf D, Hilton DJ, Nicola NA, Baca M. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc Natl Acad Sci U S A. 2000;97:6493–8. doi: 10.1073/pnas.100135197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piessevaux J, Lavens D, Montoye T, Wauman J, Catteeuw D, Vandekerckhove J, Belsham D, Peelman F, Tavernier J. Functional cross-modulation between SOCS proteins can stimulate cytokine signaling. J Biol Chem. 2006;281:32953–66. doi: 10.1074/jbc.M600776200. [DOI] [PubMed] [Google Scholar]
- Piessevaux J, Lavens D, Peelman F, Tavernier J. The many faces of the SOCS box. Cytokine Growth Factor Rev. 2008;19:371–81. doi: 10.1016/j.cytogfr.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Qin H, Roberts KL, Niyongere SA, Cong Y, Elson CO, Benveniste EN. Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007;179:5966–76. doi: 10.4049/jimmunol.179.9.5966. [DOI] [PubMed] [Google Scholar]
- Robb L, Boyle K, Rakar S, Hartley L, Lochland J, Roberts AW, Alexander WS, Metcalf D. Genetic reduction of embryonic leukemia-inhibitory factor production rescues placentation in SOCS3-null embryos but does not prevent inflammatory disease. Proc Natl Acad Sci U S A. 2005;102:16333–8. doi: 10.1073/pnas.0508023102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Robb L, Rakar S, Hartley L, Cluse L, Nicola NA, Metcalf D, Hilton DJ, Alexander WS. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc Natl Acad Sci U S A. 2001;98:9324–9. doi: 10.1073/pnas.161271798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, Inagaki-Ohara K, Yoshida T, Yamanaka A, Sasaki M, Yasukawa H, Koromilas AE, Yoshimura A. The N-terminal truncated isoform of SOCS3 translated from an alternative initiation AUG codon under stress conditions is stable due to the lack of a major ubiquitination site, Lys-6. J Biol Chem. 2003;278:2432–6. doi: 10.1074/jbc.C200608200. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Yasukawa H, Suzuki A, Kamizono S, Syoda T, Kinjyo I, Sasaki M, Johnston JA, Yoshimura A. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells. 1999;4:339–51. doi: 10.1046/j.1365-2443.1999.00263.x. [DOI] [PubMed] [Google Scholar]
- Song MM, Shuai K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J Biol Chem. 1998;273:35056–62. doi: 10.1074/jbc.273.52.35056. [DOI] [PubMed] [Google Scholar]
- Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, Hilton DJ, Alexander WS. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci U S A. 1998;95:14395– 9. doi: 10.1073/pnas.95.24.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–21. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- Tannahill GM, Elliott J, Barry AC, Hibbert L, Cacalano NA, Johnston JA. SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol Cell Biol. 2005;25:9115–26. doi: 10.1128/MCB.25.20.9115-9126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woldman I, Varinou L, Ramsauer K, Rapp B, Decker T. The Stat1 binding motif of the interferon-gamma receptor is sufficient to mediate Stat5 activation and its repression by SOCS3. J Biol Chem. 2001;276:45722–8. doi: 10.1074/jbc.M105320200. [DOI] [PubMed] [Google Scholar]
- Wong PK, Egan PJ, Croker BA, O’Donnell K, Sims NA, Drake S, Kiu H, McManus EJ, Alexander WS, Roberts AW, Wicks IP. SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J Clin Invest. 2006;116:1571–81. doi: 10.1172/JCI25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, Ohtsuka S, Imaizumi T, Matsuda T, Ihle JN, Yoshimura A. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. Embo J. 1999;18:1309–20. doi: 10.1093/emboj/18.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, Hirano T, Chien KR, Yoshimura A. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551–6. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, Kile BJ, Kent SB, Alexander WS, Metcalf D, Hilton DJ, Nicola NA, Baca M. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci U S A. 1999;96:2071–6. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.