

Abstract
Persistent inflammation is associated with a shift in spinal GABAA signaling from inhibition to excitation such that GABAA-receptor activation contributes to inflammatory hyperalgesia. We tested the hypothesis that the primary afferent is the site of the persistent inflammation-induced shift in GABAA signaling which is due to a Na+-K+-Cl−-co-transporter (NKCC1)-dependent depolarization of the GABAA current equilibrium potential (EGABA). Acutely dissociated retrogradely labeled cutaneous dorsal root ganglion (DRG) neurons from naïve and inflamed (3 days after a subcutaneous injection of complete Freund’s adjuvant) adult male rats were studied with Ca2+ imaging, western blot and gramicidin perforated patch recording. GABA evoked a Ca2+ transient in a subpopulation of small- to medium-diameter capsaicin-sensitive cutaneous neurons. Inflammation was associated with a significant increase in the magnitude of GABA-induced depolarization as well as the percentage of neurons in which GABA evoked a Ca2+ transient. There was no detectable change in NKCC1 protein or phosphoprotein at the whole ganglia level. Furthermore, the increase in excitatory response was comparable in both HEPES- and HCO3−-buffered solutions, but was only associated with a depolarization of EGABA in HCO3−-based solution. In contrast, under both recording conditions, the excitatory response was associated with an increase in GABAA current density, a decrease in low threshold K+ current density, and resting membrane potential depolarization. Our results suggest that increasing K+ conductance in afferents innervating a site of persistent inflammation may have greater efficacy in the inhibition of inflammatory hyperalgesia than attempting to drive a hyperpolarizing shift in EGABA.
Keywords: Inflammatory pain, nociceptor sensitization, neuroplasticity
Introduction
In the absence of tissue injury in adult animals, spinal ionotropic GABAA receptor signaling is inhibitory and plays a critical role in establishing nociceptive threshold. Consequently, exogenous administration of GABAA receptor agonists is antinociceptive while that of antagonists results in a dramatic decrease in nociceptive threshold (Yamamoto and Yaksh, 1991, Anseloni and Gold, 2008). In the presence of injury, however, there appears to be a shift in the spinal GABAA receptor signaling, such that low concentrations of the GABAA agonist, muscimol facilitates inflammatory hyperalgesia and the GABAA antagonist, gabazine is antinociceptive (Anseloni and Gold, 2008, Lagraize et al., 2010).
While GABAA receptors are present on both afferent terminals and dorsal horn neurons (Todd, 2002, Takazawa and MacDermott, 2010), and GABAergic interneurons in the spinal cord dorsal horn appear to be the primary source of GABA on afferent terminals (Takazawa and MacDermott, 2010) evidence from acute injury and inflammation models suggest that the primary afferent may be the site of the inflammation-induced shift in spinal GABAA receptor signaling. The acute inflammation-induced increase in antidromic activity initiated at the central terminals of nociceptive afferents, referred to as the dorsal root reflex (DRR), is blocked by GABAA receptor antagonist bicuculline (Rees et al., 1995, Lin et al., 1999). Bicuculline also blocks the spinal release of excitatory amino acids from primary afferents in an acute inflammation model (Sluka et al., 1994). Furthermore, while a number of mechanisms could underlie this shift in spinal GABAA signaling, a prevailing hypothesis is that an upregulation of Na+-K+-Cl− -cotransporter 1 (NKCC1), which facilitates the accumulation of intracellular Cl−, drives a depolarization of the GABAA receptor equilibrium potential (EGABA) (Price et al., 2009). In contrast to neurons in the central nervous system, where expression of NKCC1 is developmentally downregulated, expression of this co-transporter persists in adult sensory neurons (Plotkin et al., 1997a, Plotkin et al., 1997b). Consequently, the persistent expression of NKCC1 in primary afferents is not only thought to be the primary mechanism underlying primary afferent depolarization (Alvarez-Leefmans et al., 1988, Sung et al., 2000), a phenomenon dependent on Cl− efflux via GABAA receptor activation, but the substrate underlying the acute injury/inflammation-induced shift in spinal GABAA signaling (Price et al., 2009).
Available evidence in support of a role for NKCC1 in acute injury/inflammation-induced shift in the spinal GABAA signaling is compelling. Disruption of NKCC1 function either by NKCC1 blocker bumetanide or with genetic deletion leads to a hyperpolarizing shift of EGABA in sensory neurons (Alvarez-Leefmans et al., 1988, Sung et al., 2000, Laird et al., 2004) and an increase in nociceptive threshold (Sung et al., 2000, Pitcher et al., 2007). NKCC1 function is rapidly upregulated via phosphorylation in response to peripheral capsaicin injection (Galan and Cervero, 2005) and inflammatory mediators (Funk et al., 2008), as well as more slowly upregulated via membrane translocation in response to capsaicin (Galan and Cervero, 2005). Furthermore, mechanical allodynia and the sensitization of spinal nociceptive neurons associated with peripheral capsaicin administration is blocked in NKCC1 knockout mice (Laird et al., 2004), and attenuated with spinal bumetanide administration (Granados-Soto et al., 2005, Pitcher et al., 2007, Pitcher and Cervero, 2010).
Based on the available data, an upregulation of NKCC1 in primary afferents is a reasonable hypothesis for the shift in GABAA signaling observed in the presence of persistent inflammation. However, there is no direct evidence indicating that the persistent inflammation-induced shift in GABAA signaling is due to an NKCC1 dependent shift in EGABA in primary afferent neurons as the upregulation in NKCC1 and changes associated with spinal bumetanide and the NKCC1 knock out are not necessarily specific to primary afferents. Moreover, the DRR can also be attributed to non-GABAergic mechanisms that are indirectly triggered by spinal GABAA signaling. Therefore, the purpose of the present study was to test the hypothesis that up-regulation of NKCC1 in primary afferents contributes to the persistent inflammation-induced switch in spinal GABAA signaling from inhibition to excitation.
To test this hypothesis, western blot analysis was performed on total protein harvested from L4/L5 DRG, and Ca2+ imaging and whole cell patch clamp techniques were used to study retrogradely labeled cutaneous DRG neurons from naïve rats and from rats 3 days after the induction of inflammation with complete Freund’s adjuvant (CFA).
Experimental Procedures
2.1 Animals
Adult male Sprague Dawley rats (Harlan-Sprague Dawley, Indianapolis, IN) weighing between 250 and 350 g were used for all the experiments. Rats were housed two per cage in the University of Pittsburgh AAALAC approved animal facility on a 12:12 light: dark schedule with food and water available ad libitum. All procedures involving animals were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.2 Labeling and inflammation
DRG neurons that innervate the glabrous skin of rat hind paw were retrogradely labeled with 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI, 17 mg/ml in DMSO and saline), which was injected (3–5 sites at 2–3 μl/site) with a 30g needle, 14–17 days prior to tissue harvest and analysis. CFA (Sigma-Aldrich, St Louis MO; mixed 1:1 with saline), was injected (100 μl) into the site previously injected with DiI. Inflamed DRG neurons were studied 72 hours after CFA injection. Both DiI and CFA were injected under isofluorane-induced anesthesia.
2.3 Preparation of isolated DRG neurons
Prior to tissue harvest, rats were deeply anesthetized with a subcutaneous injection (1 ml/kg) of a cocktail containing ketamine (55 mg/ml), xylaxine (20 mg/ml) and acepromazine (5.5 mg/ml). L4 and L5 DRG were harvested, cleaned of connective tissue, enzymatically treated and mechanically dispersed as previously described (Lu et al., 2010). Isolated neurons were plated on poly-L-lysine coated coverslips and electrophysiology experiments were done 2–8 h after plating.
2.4 Ca2+ imaging
DRG neurons were loaded with 10 μM Ca2+ indicator fura-2 AM ester (TEF Labs, Austin TX) dispersed with 0.025% Pluronic F-127 for 20 min at room temperature. Following fura-2 loading, DRG neurons were placed in a chamber on an inverted microscope (Nikon Eclipse TE2000-U, Japan) and were continuously superfused with bath solution. Excitation of fura-2 (340/380 nm) was controlled by a lambda DG-4 filter changer (Sutter Instrument Co, Novato CA). Fluorescence data were acquired with Metafluor software (Molecular Devices, Sunnyvale, CA, USA) via a charge coupled-device (CCD) camera (Quant-EM 512sc, from Photometrics, Tucson, AZ) at 1 Hz during drug application. In a subset of experiments, DRG neurons were incubated with FITC-conjugated isolectin B4 (IB4, Sigma-Aldrich) for 10 minutes after incubation with fura-2 AM, and IB4 binding was examined under epifluorescence before the calcium imaging experiment.
One of two bath solutions was used in these experiments. HCO3− bath: NaCl 130 mM, KCl 3 mM, NaHCO3 26 mM, NaH2PO4 1.25 mM, CaCl2 2.5 mM, MgCl2 0.6 mM and Glucose 10 mM, osmolality was adjusted to 325 mOsm with sucrose, bubbled with 5% CO2 and 95% O2; HEPES bath: NaCl 130 mM, KCl 3 mM, CaCl2 2.5 mM, MgCl2 0.6 mM, HEPES 10 mM, Glucose 10 mM; pH was adjusted to 7.4 with Tris-Base and osmolality was adjusted to 325 mOsm with sucrose. All salts were obtained from Sigma-Aldrich.
A HEPES buffered bath solution was used for initial experiments performed on cutaneous neurons. This solution was used for two major reasons. First, although the GABAA receptor coupled anion channel is also permeable to HCO3− (Kaila et al., 1993), due to its higher permeability to Cl− and the high concentration of Cl− in the bath solution, the theoretical impact of [HCO3−] on EGABA is negligible. Second, since nearly all the previous experiments on EGABA in sensory neurons were done with HEPES buffered bath solution (Alvarez-Leefmans et al., 1988, Sung et al., 2000, Gilbert et al., 2007, Rocha-Gonzalez et al., 2008) using a HEPES bath buffered solution would enable us to compare our data with previous results.
GABA or other test compounds were applied with a piezo driven perfusion system (SF-77B Perfusion Fast-Step; Warner instruments LLC, Hamden, CT), that enabled drug on- and off-rates of under 10 ms. To minimize the potential impact of GABAB receptor activation, all GABA applications were performed in the presence of the GABAB receptor antagonist CGP 55845 (1 μM, Tocris Bioscience, Minneapolis, MN).
2.5 Electrophysiology
Voltage clamp recordings were performed using an Axopatch 200B amplifier (Molecular Devices) controlled with pClamp software (Version 10.2, Molecular Devices) via a Digidata 1320A A/D converter. Data were low-pass filtered at 10 kHz and digitally sampled at 2 kHz. For voltage-clamp protocols, capacity transients were canceled via amplifier circuitry. Borosilicate glass (WPI, Sarasota Springs, FL) patch electrodes were pulled on a Sutter P-2000 horizontal puller (Sutter Instruments) and were ~1.5 – 2.5 MΩ in resistance when filled with electrode solution of the following composition: K-Methansulphonate 140 mM, MgCl2.6H2O 2 mM, CaCl2 1 mM, EGTA 11 mM, HEPES 10 mM and NaCl 5 mM; pH was adjusted to 7.2 with Tris Base, and osmolality was adjusted to 317 mOsm with sucrose.
Gramicidin (Sigma-Aldrich) perforated patch was used for all voltage and current clamp recordings. Gramicidin was used in order to obtain electrical access with a minimal impact on resting intracellular Cl− concentration (Akaike, 1996). A stock solution of gramicidin (1.5 mg/100 μl) was prepared in DMSO. This was diluted with electrode solution in a 1:300 ratio (1 to 300 μl) to give a final concentration of 50 μg/ml. The gramicidin containing electrode solution was vortexed for 15s. No filtering was applied. The tip of the electrode was loaded with a small volume of gramicidin free electrode solution in order to avoid interference of the antibiotic with seal formation. Gramicidin containing electrode solution was back loaded. The progress of perforation was monitored with the capacitative transient to a 5 mV step. Experiments were not started until access resistance was less than 7 MΩ.
Voltage-clamp recording: Series resistance compensation (> 70%) was employed for all voltage-clamp recording. A ramp protocol was used to determine EGABA. Neurons were held at −70 mV. The membrane potential was depolarized to −30 for 2 s to inactivate transient outward and inward currents and then hyperpolarized from −30 to −80 mV at a rate 0.125 mV/ms. The membrane potential was then returned to rest at −70 mV. The same protocol was run twice in the presence and absence of 60 μM GABA. EGABA was determined from the difference between the two current traces at the point on the voltage ramp where the corresponding GABA current was 0 pA. The linear phase of the GABA current was extrapolated to 0 pA to determine EGABA when it was more depolarized than −30 mV. To ensure the accuracy of extrapolated EGABA measurements, we used a low concentration of GABA (60 μM) applied at least 100 ms prior to the start of the ramp to ensure the stability, and therefore linearity of the GABA current during the ramp.
To assess changes in low threshold currents active between resting membrane potential (Vrest) and action potential threshold (~30 mV), current was evoked with 100 ms 10 mV voltage steps from −70 to −30 mV. In order to detect inflammation-induced changes in both leak currents and voltage-activated currents, no leak subtraction protocol was employed.
For current clamp recordings, the holding current was set to 0 pA so as to determine Vrest, as well as the magnitude of GABA evoked changes in membrane potential, from Vrest. Input resistance was assessed with direct current injection, based on the relationship between the current required to produce a linear change in membrane potential around Vrest.
2.6 Western blot
L4 and L5 DRG were homogenized with Teflon tube and mortar for less than 10 strokes in ice cold RIPA buffer supplied with protease inhibitors (aprotinin, leupeptin, pepstatin, E-64, trypsin inhibitor, and PMSF, all at a final concentration of 2 ng/ml except PMSF, which was used at a final concentration of 1 mM). All protease inhibitors were obtained from Sigma-Aldrich. Lysates were collected in 0.5 ml tubes. Teflon tubes were rinsed with RIPA buffer and the solutions were combined with the lysates previously collected. Lysates were centrifuged for 5 minutes at 10,000 rpm at 4 °C. Protein concentration was determined via BCA protein assay using a BCA assay kit (Thermo scientific, Rockford, IL). Lysates were then mixed with Laemmli buffer (2×, 400 μl + 100μL β-mercaptoethanol) and boiled for 5 minutes before loading. Protein (30 μg) from one animal was then loaded per lane and separated on a 7 % SDS-PAGE gel and transferred to nitrocellulose membrane. Membranes were blocked with 5% milk for 1 hour at room temperature and then incubated with primary antibody at 4°C overnight (1:1000 for NKCC1 (Millipore, AB3560P), 1:5000 for anti-phospho-NKCC1 R5 antibody (a gift from Dr. Biff Forbush of Yale University School of Medicine (Flemmer et al., 2002)), diluted with 5% milk/(Tris buffered saline with Tween 20 (TBST, Sigma-Aldrich). The blots were washed and then incubated with peroxidase conjugated secondary antibody (1:2000 in 5% milk/TBST, Jackson ImmunoResearch Laboratories Inc. West Grove, PA) for an hour at room temperature. An ECL kit (Amersham Biosciences, Piscataway, NJ) was used for detection of immunoreactivity, and luminescence data were collected on an LAS3000 imager (Fujifilm Inc, Japan).
2.7 Test compounds
Capsaicin, γ-aminobutyric acid (GABA), and gramicidin were from Sigma-Aldrich. CGP 55845 was from Tocris Bioscience (Minneapolis, MN). GABA was dissolved in water, CGP 55845 was dissolved in DMSO and capsaicin was dissolved in ethanol to make stock solutions. Stock solutions were then kept in a −20°C freezer and diluted with bath solution in 1:1000 ratios to their working concentration before use. The high K+ solution (high K) used to assess changes in the impact of GABA on the excitability of cutaneous neurons contained 30 mM K+ in a solution made by an isoionic substitution of KCl for NaCl in our normal HEPES buffered bath solution.
2.8 Statistics
All pooled data are presented as mean ± standard error of the mean. Chi-square Fisher Exact tests were used to compare the proportion neurons responsive to GABA in Ca2+ imaging experiments. Student’s t-test was used for two-group comparisons (i.e., naive versus inflamed), one-way ANOVA was used for multiple group comparisons (i.e., EGABA in neurons in which GABA evoked an excitatory response in from naïve and inflamed rats), and mixed design two-way ANOVA was used to assess the interaction between membrane potential and treatment group on outward currents evoked in cutaneous neurons. The Tukey test was used for post-hoc pair wise multiple comparisons. p < 0.05 was considered statistically significant.
Results
3.1 Inflammation increases the excitatory actions of GABA in cutaneous DRG neurons
Two initial sets of experiments were performed to determine whether inflammation can increase GABA mediated excitation of cutaneous DRG neurons. In both, changes in the concentration of intracellular Ca2+ were used as an indirect measure of excitation. In the first experiment, GABA was co-applied with bath solution containing 30 mM K+ (high K). Preliminary experiments indicated that it was necessary to use a brief (250 ms) high K application to detect increases or decreases in the high K evoked Ca2+ transient. We have previously demonstrated that the high K evoked Ca2+ transient in DRG neurons is entirely dependent on a membrane depolarization-induced activation of voltage-gated Ca2+ channels (Lu et al., 2006, Lu and Gold, 2008). Thus, an indirect measure of excitability, the ability of GABA to influence the high K-evoked Ca2+ transient should be a reflection of the change in membrane potential and therefore the number of Ca2+ channels activated by high K. Ca2+ transients induced by the combination of GABA and high K were compared to those induced by high K alone. A neuron was considered a “responder” in this experiment, if the co-application of GABA and high K resulted in a change in the Ca2+ transient greater than 2 standard deviations from the mean of the baseline transient evoked with high K alone: GABA was considered inhibitory when the high K evoked transient was decreased in the presence of GABA and excitatory when the high K-evoked transient was increased in the presence of GABA. In naïve rats, 12/39 cutaneous neurons demonstrated a GABA-induced inhibition (Figure 1A). However, this inhibition was detected in a significantly smaller proportion of cutaneous neurons from inflamed rats (2/40, p < 0.01). Conversely, none of the neurons tested from naïve rats demonstrated an excitatory response in the presence of GABA, whereas excitation was detected in 12/40 neurons test from inflamed rats (Figure 1A). This difference is also statistically significant (p < 0.01). Both excitation and inhibition were concentration dependent as illustrated by pooled data from neurons in which GABA produced inhibitory (naïve) and excitatory (inflamed) effects (Figure 1B).
Figure 1.
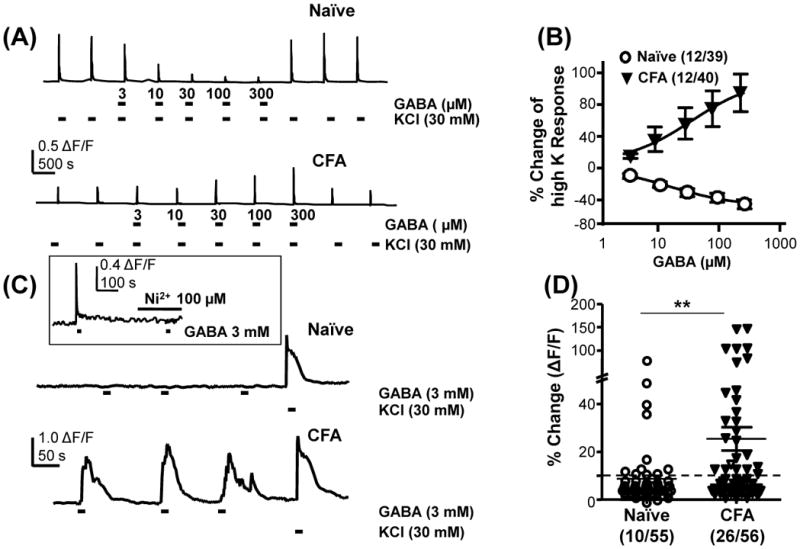
Inflammation-induced increase in the excitatory actions of GABA in cutaneous DRG neurons. A) High K (30 mM KCl) was used to evoke Ca2+ transients in neurons from naïve and inflamed (CFA) rats. Co-application of GABA with high K results in a concentration dependent suppression of the Ca2+ transient in a subpopulation of neurons from naïve rats, but an increase in the transient in a subpopulation of neurons from inflamed rats. B) Pooled data from neurons in which GABA increased (CFA) or decreased (Naïve) high K evoked transients. C) Direct application of GABA to cutaneous neurons from naïve rats resulted in little change in the intracellular Ca2+ concentration in the majority of neurons tested. However, in close to half of the neurons from inflamed rats, direct application of GABA resulted in a transient increase in intracellular Ca2+. High K was used to confirm neurons could respond to a depolarizing stimulus. Inset, GABA evoked Ca2+ transients were completely blocked by 100 μM Ni2+. D) Pooled data tested with direct activation of GABA. The proportion of cutaneous neurons from inflamed rats directly activated by GABA was significantly greater than that from naive rats. ** p < 0.01, χ2 test.
In the second experiment, we sought to determine whether application of GABA alone was sufficient to evoke a Ca2+ transient. Three mM GABA was used based on the results of the first experiment, which indicated that saturation of the GABA concentration-response curve at a GABA concentration greater than 1 mM. A neuron was considered a “responder”, when the application of GABA was associated with a Ca2+ transient greater than 10% above baseline (Figure 1C). This threshold was chosen as it was greater than 3 standard deviations above the average baseline fluctuation. The transient was completely blocked in presence of the low voltage activated (LVA) Ca2+ channel blocker Ni2+ (100 μM) in the bath solution (n = 4, inset to Figure 1C) and in the presence of nominally Ca2+ free bath solution (in which Ca2+ was omitted from the HEPES buffered bath solution, data not shown) confirming that the GABA-evoked Ca2+ transient was due to Ca2+ influx via voltage-gated Ca2+ channels. These results suggest that the GABA evoked Ca2+ transient was largely due to a depolarization-induced activation of LVA Ca2+ channels. The proportion of responders in cutaneous neurons from inflamed rats (26/56) was significantly (p < 0.01) greater than that in neurons from naïve rats (10/55; Figure 1D). Results from these of experiments are consistent with the hypothesis that there is an inflammation-induced increase in the excitatory effect of GABA in the primary afferents.
The majority of the GABA responders had a small (<30 μm, 47/60) or medium (between 30 and 40 μm, 12/60) diameter cell body and all of them responded to capsaicin (500 nM) applied at the end of each experiment. There was no significant difference in IB4 binding between GABA responders and non-responders: the proportion of IB4+ cutaneous non-responders from naïve rats, and non-responders and responders from inflamed rats were 21/31, 13/20 and 5/9, respectively.
3.2 GABA induced membrane depolarization is increased in a subpopulation of neurons from inflamed animals
While the results from the Ca2+ imaging experiments are consistent with an increase in the excitatory actions of GABA in the presence of inflammation, Ca2+ imaging is an indirect measure of excitability. We therefore performed current clamp recordings on cutaneous neurons from naïve and inflamed animals. Three mM GABA application was associated with membrane depolarization in every neuron tested in both naive and inflamed rats (Figure 2A). GABA depolarized neurons from inflamed animals to a potential (−35.7 ± 2.2 mV, n = 21) significantly (p < 0.01) more depolarized than that observed in neurons from naïve rats (−46.4 ± 2.6 mV, n = 9). However, action potentials were evoked in none of the neurons from naïve animals (n = 9) and in only 1/21 neurons tested from inflamed animals (not shown). Thus, the GABA-evoked increase in intracellular Ca2+ observed with Ca2+ imaging must have been due to the activation of voltage-gated Ca2+ channels rather than action potential firing. In support of the suggestion that the impact of inflammation was restricted to a subpopulation of cutaneous neurons, the difference in the GABA-evoked depolarization between groups appeared to be largely due to the greater depolarization observed in neurons “responsive” to GABA as determined with Ca2+-imaging experiments performed prior to electrophysiological analysis (Figure 2B): GABA depolarized “responders” from inflamed rats to a potential significantly greater than that observed in “non-responders” from inflamed rats (p < 0.05) or neurons from naïve rats (p < 0.01). Thus, these current clamp results are consistent with the Ca2+ imaging data and suggest that an “excitatory” response to GABA is only detectable when the GABA-induced depolarization activates a sufficient number of LVA Ca2+ channels to produce a detectable increase in intracellular Ca2+.
Figure 2.
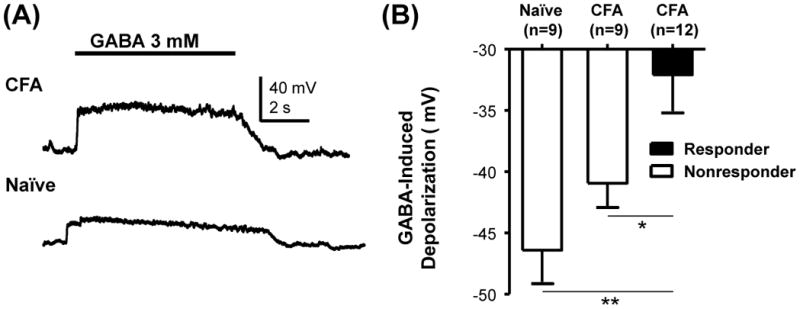
Inflammation-induced increase in GABA induced membrane depolarization. A) Three mM GABA induced a larger membrane depolarization in a cutaneous neurons from an inflamed rat that responded to GABA with a Ca2+ transient (upper trace) than in a nonresponder from naive rat (lower trace). The cell body diameter of both neurons was 30 μm, and both were responsive to capsaicin. Pooled GABA-induced depolarization data obtained in HEPES buffered bath: The membrane potential was significantly more depolarized in responders from inflamed rats than in non-responders from inflamed rats or naïve rats. * is p < 0.05, ** is p < 0.01.
3.3 Persistent inflammation has no influence on NKCC1 or phospho-NKCC1 protein levels in intact DRG
To explore the potential contribution of NKCC1 to the increase in GABA-induced depolarization in cutaneous neurons we assessed protein expression levels of NKCC1 and phospho-NKCC1 in intact ganglia from naïve and inflamed rats with Western blot. Results from an analysis of tissue from 4 animals in each group indicate that there is no detectable (p > 0.05) change in either the total NKCC1 protein level or the relative protein level of phosphor-NKCC1 associated with persistent inflammation (Fig 3).
Figure 3.
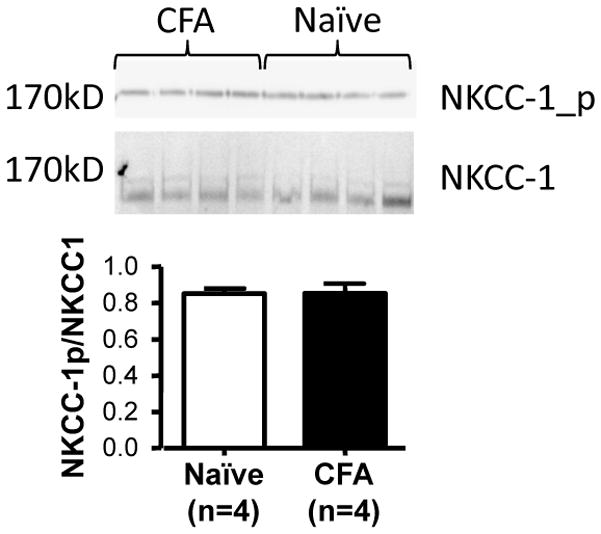
No detectable influence of inflammation on total or phosphorylated NKCC1 protein in DRG. Top panel is the blot used to assess changes in protein levels. Each lane was loaded with tissue from one rat that was either inflamed (CFA, 3 days post injection) or naïve. Bottom panel, pooled densitometric analysis reveals no difference in the relative band intensity in DRG harvested from naïve or inflamed rats (p>0.05).
3.4 Persistent inflammation has no influence on EGABA in HEPES buffered bath
Although no increase in NKCC1 protein or phosphorylation was detected with western blot analysis of whole ganglia, it was possible that an increase in NKCC1 protein and/or phosphorylation was masked by the majority of the neurons in the ganglia that was not inflamed or not influenced by the presence of inflammation. We therefore used gramicidin-perforated patch to directly measure the EGABA in cutaneous neurons from naïve and inflamed rats. Because a Ca2+ transient was observed in less than half of the cutaneous neurons from inflamed rats and the GABA-induced depolarization was significantly larger in GABA “responders” as defined by the presence of Ca2+ transient, Ca2+ imaging was used to prescreen the cutaneous neurons before subsequent electrophysiological analysis. All subsequent analyses were performed on 3 groups: non-responders from naïve rats, non-responders from inflamed rats and responders from inflamed rats.
EGABA was measured with a ramp protocol (Fig 4A, B) as described in Methods. Surprisingly, EGABA in cutaneous neurons from inflamed rats that responded to GABA with a Ca2+ transient (−19.0 ± 3.3 mV, n = 14) was comparable to that in non-responders from inflamed rats (−22.9 ± 2.2 mV, n = 8) as well as non-responders from naïve rats (−21.3 ± 3.4 mV, n = 15; p > 0.05, Figure 4C).
Figure 4.
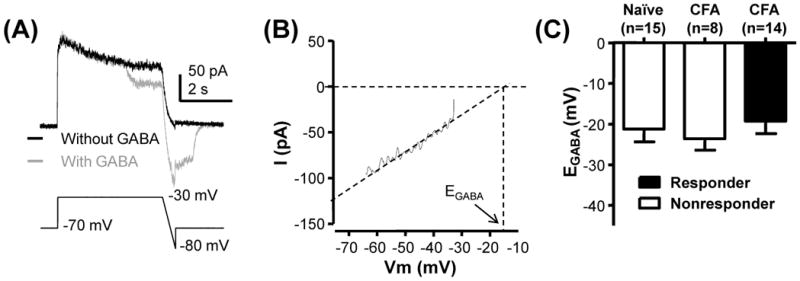
No detectable influence of inflammation on EGABA. A) A ramp protocol from −30 to −80 mV run with (gray trace) and without (black trace) GABA (60 μM) application was used to determine EGABA. B) The linear phase of the difference currents shown in A was extrapolated to 0 pA to estimate EGABA. C) Pooled data from cutaneous neurons from naïve and inflamed rats (CFA) in which direct application of GABA evoked a Ca2+ transient (responders) or failed to do so (nonresponders) are plotted. There is no statistically significant difference between groups (p>0.05).
3.5 EGABA is more depolarized in responders from inflamed rats in HCO3− buffered bath
Our results from the western blot analysis and EGABA measurements in HEPES buffered bath solution argued against a role for an increase in NKCC1 activity in the emergence of excitatory GABAA signaling with persistent inflammation. However, there is evidence that Na+-independent mechanisms contribute to the regulation of intracellular Cl− concentration (Rocha-Gonzalez et al., 2008), and that a HCO3−-dependent mechanism can also regulate the concentration of intracellular Cl− (Irie et al., 1998, Planelles, 2004). As the potential contribution of the latter would be minimized in a HEPES buffered bath solution, we assessed the impact of inflammation on EGABA with a HCO3− buffered bath solution. As with the experiments performed in HEPES buffered bath solution, neurons were first assayed with Ca2+ imaging to distinguish GABA responsive from non-responsive neurons. Results of this experiment were comparable to those obtained in HEPES buffered bath solution with a significantly (p < 0.01) greater proportion of cutaneous neurons from inflamed rats (19/42) responsive to GABA than from naïve rats (4/45, Figure 5A). As with the results obtained in the HEPES buffered bath solution, GABA depolarized responders (n = 10) from inflamed animals to a potential significantly greater than that observed in non-responders from inflamed animals (p < 0.05, n = 7) and non-responders from naïve animals (p < 0.01, n = 17, Figure 5B).
Figure 5.
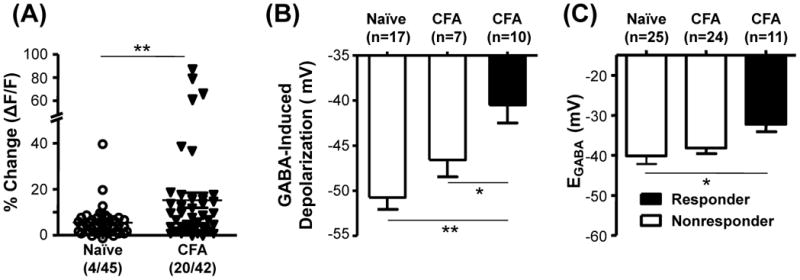
Impact of inflammation on the GABA response and EGABA in HCO3− buffered bath solution. A) Inflammation (CFA) resulted in a significant increase in the proportion of neurons that responded to GABA with a Ca2+ transient (responders). B) The GABA-induced depolarization was assessed as in Figure 2 and data from cutaneous neurons from naïve and inflamed rats were pooled according to whether they were GABA responders or non-responders. C) EGABA was determined as in Figure 4 in HCO3− buffered bath solution. In contrast to the results obtained in HEPES buffered bath solution, EGABA was significantly more depolarized in responders than in non-responders from naïve rats. The difference between EGABA in responders and non-responders from inflamed rats was not significant. * is p < 0.05, and ** is p < 0.01.
However, there were two striking differences between HCO3− and HEPES buffered bath with respect to EGABA measurements. First, EGABA was significantly (p<0.01, two-way ANOVA) more hyperpolarized in HCO3− than in HEPES (Figure 5C vs Figure 4C). Second, EGABA was significantly more depolarized in GABA-responsive neurons from inflamed rats (−32.2 ± 1.9 mV; n = 11) than non-responsive neurons from naïve rats (−40.1 ± 2.1 mV, n = 25; p < 0.05). EGABA in non-responsive neurons from inflamed rats (−38.2 ± 1.43, n = 24), did not differ significantly, however, from that in responsive neurons from naïve rats (Figure 4B). These results are consistent with an inflammation-induced increase in NKCC1 that contributes to the increase in the “excitatory” actions of GABA, but also raise the possibility that inflammation is associated with a decrease in the activity of the mechanism responsible for the hyperpolarizing shift in EGABA in HCO3− buffered bath solution.
3.6 Resting membrane potential is depolarized in GABA-responsive neurons from inflamed rats
While the depolarization of EGABA observed in responders from inflamed rats in HCO3− buffered bath solution is consistent with an inflammation-induced increase in NKCC1 activity, the absence of a comparable change in HEPES buffered bath suggests that other changes contribute to the inflammation-induced increase in GABA responsive neurons. Therefore, we first assessed the impact of inflammation on passive electrophysiological properties. These included resting membrane potential (Vrest), input resistance (Rin), and membrane capacitance. Mean values for each of these parameters measured in HEPES and HCO3− buffered solution are listed in Table 1. Of these three parameters, the only one consistently different under both recording conditions is Vrest, which was significantly (p < 0.01) more depolarized in responsive neurons from inflamed rats than that in either non-responsive neurons from inflamed rats or non-responsive neurons from naïve rats.
Table 1.
Passive properties of cutaneous DRG neurons
| Bath Solution | Group (N) | Capacitance (MΩ) | Vrest (mV) | Rin (GΩ) |
|---|---|---|---|---|
| HEPES | Responder Inflamed (13) | 46.1 ± 4.6 | −59.0 ± 1.69 * | 0.80 ±0.1** |
| Non-responder Inflamed (7) | 52.2 ± 2.0 ** | −64.5 ± 1.47 | 0.87 ±0.11** | |
| Non-responder Naïve (12) | 37.2 ± 4.0 | −68.2 ± 2.17 | 1.42 ± 0.14 | |
| HCO3− | Responder Inflamed (13) | 34.2 ± 2.4 | −62.8 ± 1.45* | 1.12 ± 0.22 |
| Non-responder Inflamed (15) | 38.8 ± 2.0 | −68.4 ± 1.48 | 1.15 ± 0.19 | |
| Non-responder Naïve (21) | 33.9 ± 2.2 | −69.4 ± 1.63 | 1.03 ± 0.11 |
(N) is the number of neurons in each group. Vrest is resting membrane potential. Rin is input resistance
is p < 0.05 and
is p < 0.01.
3.7 Increase in GABAA current density in neurons from inflamed rats
The inflammation-induced depolarization of Vrest is likely to contribute to the increase in the number of GABA responsive neurons in inflamed rats. However, the observation that the magnitude of the GABA-induced depolarization in responders from inflamed rats in both HEPES (24.7 ± 2.5 mV) and HCO3− (24.2 ± 2.1 mV) buffered bath solution was significantly (p < 0.05) larger than that in neurons from naïve rats (15.9 ± 2.9 mV and 18.2 ± 1.6 mV, in HEPES and HCO3− buffered bath solution, respectively), suggests that other mechanisms are also likely to contribute to the inflammation-induced changes in GABA signaling. Several mechanisms that could contribute include an increase in the magnitude of GABA evoked current, an increase in a low threshold voltage-activated current that facilitates GABA-induced depolarization, or a decrease in an outward current that normally attenuates the magnitude of the GABA-induced depolarization. With respect to the first possibility, the response to 3 mM GABA was assessed in cutaneous neurons from naïve and inflamed rats in voltage clamp from a holding potential of −60 mV (Figure 6A). A significant increase in the magnitude of the GABA current density was detected in cutaneous neurons from inflamed rats (p<0.05, Figure 6B).
Figure 6.
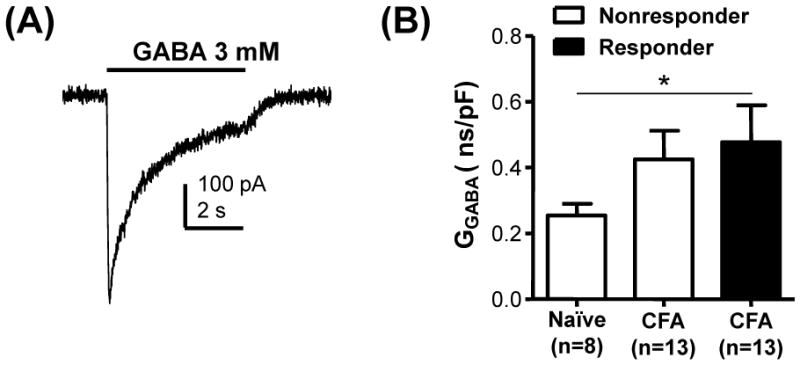
Inflammation-induced increase in GABA current density. A) The magnitude of the GABA current evoked in voltage clamp from −60 mV was used to determine current density. The current shown is typical of that evoked in small diameter neurons from inflamed rats in which GABA was able to evoke a Ca2+ transient prior to electrophysiological analysis. B) Pooled conductance density was plotted and analyzed with a one way ANOVA which revealed a significant influence of inflammation (p < 0.01).
3.8 Low threshold outward current is decreased in responsive neurons from inflamed rats
With respect to the second possibility, voltage-clamp analysis indicated that in cutaneous neurons, no low threshold inward current was detected with depolarizing voltage steps between −55 and −40 mV from a holding potential of −60 mV. Furthermore, previous analysis of inflammation-induced changes in voltage-gated Ca2+ currents in cutaneous neurons indicates that there is no inflammation-induced increase in LVA Ca2+ current (Lu et al., 2010). Rather than an inward current, outward currents dominated the low threshold voltage-activated currents in cutaneous neurons (Figure 7A, B). There is a significant decrease in both peak (Figure 7C, p<0.01, two-way repeated measure ANOVA) and sustained (not shown) outward current density in GABA responders from inflamed rats. While the differences between groups were significant, heterogeneity in low threshold outward current may account for the subpopulations of neurons from naïve animals in which GABA was able to evoke a Ca2+ transient. The reversal potential for the low threshold outward current observed in cutaneous neurons was ~ −68 mV, as estimated from tail currents. This is reversal potential depolarized relative to that predicted for a K+ current reversal potential by the Nernst equation under the recording conditions employed, but comparable to that for K+ currents previously described in cutaneous DRG neurons (Gold et al., 1996b).
Figure 7.
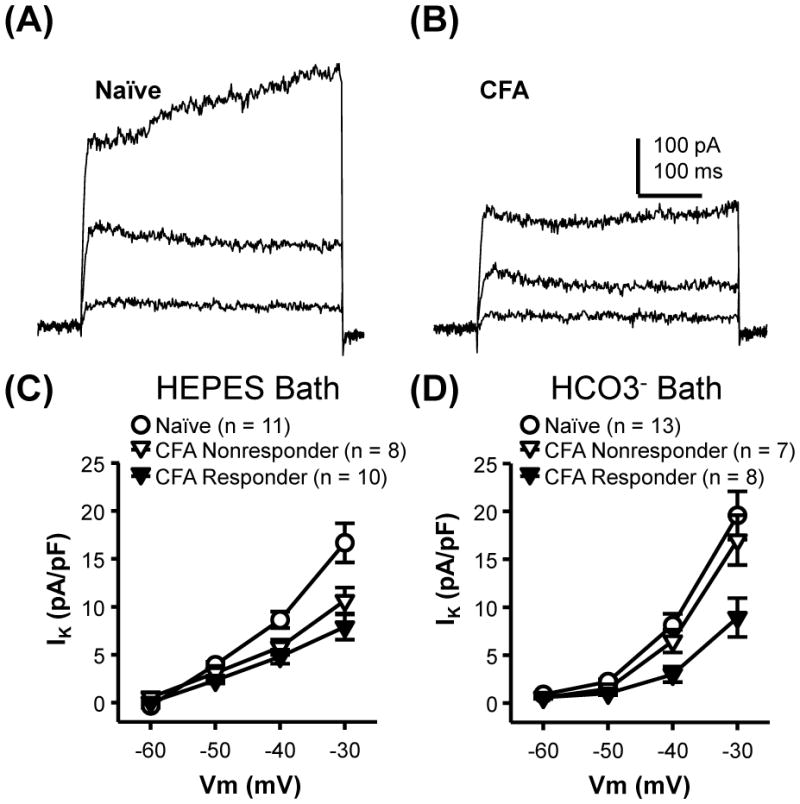
Inflammation-induced decrease in outward current in cutaneous neurons. Typical non- or weakly-inactivating outward current evoked with voltage steps to −50, −40 and −30 mV from −60 mV in cutaneous neurons from naïve (A) and inflamed (B) rats. The currents showed in B, were from a neuron that responded to GABA with a Ca2+ transient. Note the absence of low threshold inward current in either in A or B which was typical of cutaneous neurons studied from naïve and inflamed rats. C) Pooled data from naïve rats and responders and nonresponders from inflamed rats collected in HEPES buffered bath solution. D) Pooled data from the same 3 groups of neurons collected in HCO3- buffered bath solution. For data plotted in both C and D, two-way mixed design ANOVA revealed a significant influence of voltage (p < 0.01) and group (p < 0.01), as well as a significant interaction between the two. Post-hoc analysis indicated that current density in responders from inflamed rats evoked at −40 and −30 mV was significantly smaller that than in neurons from naïve rats.
Discussion
The hypothesis tested in the current study was that there is a switch of GABAA signaling in primary afferent neurons following persistent inflammation due to an NKCC1-dependent depolarizing shift in EGABA. Our data provided partial support for both components of this hypothesis. That is, an increase in the excitatory actions of GABA were detected in a subpopulation of cutaneous neurons from inflamed rats that appeared to be due to a depolarization-induced activation of LVA Ca2+ currents. However, despite a detectable membrane depolarization in all groups, GABA failed to drive the membrane potential above the action potential threshold in isolated neurons. Similarly, inflammation was associated with a significant depolarization of EGABA in a subpopulation of cutaneous neurons consistent with an increase in NKCC1 activity. However, this shift was observed when EGABA was assessed in HCO3− buffered but not in HEPES buffered bath solution. Furthermore, we detected no change in total or phospho-NKCC1 protein in DRG from inflamed rats. The additional changes in cutaneous neurons that were associated with a shift in GABA signaling included a decrease in Vrest, an increase in GABA current density, and a decrease in outward current density around the action potential threshold (i.e., −30 mV).
While our results are consistent with the prediction that the primary afferent is a site of the shift in spinal GABAA signaling observed in the presence of persistent inflammation (Anseloni and Gold, 2008), the absence of GABA-evoked action potentials in isolated cutaneous neurons suggests one of at least three possibilities. First, the inflammation-induced increase in DRR activity observed with acute noxious or inflammatory stimuli (Rees et al., 1994) does not persist in the presence of ongoing inflammation. This would suggest that the shift in GABAA signaling observed in the presence of persistent inflammation reflects changes in the modulatory activity of GABA rather than one of a direct excitatory nature. This possibility would be consistent with the presence of hypersensitivity three days after CFA injection rather than ongoing pain behavior (Boegel et al., 2011). Arguing against this possibility, however, is evidence of DRR activity in a model of arthritis (Rees et al., 1996). Second, DRR activity is present, but not manifest in the isolated cell body due to the particular biophysical constraints of the cell body which include a relatively small surface to volume ratio and a particularly high density of high threshold Na+ currents (England et al., 1996). GABA-induced spike initiation may be possible in the processes. Third, the emergence of DRR activity is not only due to persistent changes in Vrest, GABA current, and K+ current, but the ongoing activity of inflammatory mediators that are released in the periphery and dorsal horn (McMahon et al., 2005). For example, inflammatory mediators such as PGE2 increases Na+ currents (Gold et al., 1996a) and may further decrease K+ currents (Nicol et al., 1997). Nevertheless, the GABA-evoked increase in Ca2+ is still considered excitatory, given the profound impact of Ca2+ on signal transduction, transmitter release and gene transcription (Eshete and Fields, 2001, Medvedeva et al., 2008, Kawano et al., 2009).
Our results suggest that a depolarizing shift in EGABA is not sufficient to account for the increase in GABA-induced excitation in cutaneous neurons. However, this does exclude a role for NKCC1 in the excitatory actions of GABA, as this co-transporter contributes to the depolarized EGABA, even in the absence of inflammation. Furthermore, acute inflammatory mediator-induced modulation of NKCC1 (Funk et al., 2008) may further contribute to the shift in EGABA in vivo, as further depolarization will create a greater driving force on GABA currents resulting in greater of EGABA membrane depolarization.
The observation that EGABA was more hyperpolarized in HCO3− buffered bath was surprising, given that EHCO3- is generally considered to be relatively depolarized (Kaila et al., 1993). Furthermore, the relatively limited HCO3− permeability of the GABAA channel suggests that this ion should have little influence on EGABA. The −20 mV shift in EGABA in the presence of HCO3− bath suggests that there is a robust HCO3− dependent Cl− extrusion mechanism in cutaneous neurons. HCO3−/Cl− exchangers have not been considered a major player in the regulation of Cl− equilibrium in primary afferent neurons, as nearly all previous EGABA measurements in sensory neurons have been made with HEPES bath solution. Consequently, estimates of EGABA and intracellular Cl− concentrations have been comparable to those we obtained in HEPES buffered solution (Alvarez-Leefmans et al., 1988, Gilbert et al., 2007, Rocha-Gonzalez et al., 2008). EGABA estimates obtained in 3 mM HCO3− buffered bath solution in chick sensory neurons (Kenyon, 2000) were comparable to those obtained in the present study, suggesting the HCO3−/Cl− exchange is not only robust, but conserved across species.
Identification of the HCO3−/Cl− exchanger(s) underlying Cl− extrusion in cutaneous neurons is beyond the scope of the present study. However, members of the SLC4, HCO3− exchanger family, in particular SLC4A8, are promising candidates. Not only does SLC4A8 facilitate HCO3− loading (resulting in a decrease in intracellular Cl−), it is expressed in neuronal tissues, and is electroneutral (Chen et al., 2008, Sinning et al., 2011). These properties are consistent with the fact that there is little difference between resting membrane potential of cutaneous neurons tested in HEPES and HCO3− buffered bath solutions. Given the decrease in intracellular pH associated with neural activity (Sinning et al., 2011), inflammation-induced changes in the expression and/or activity of a HCO3− exchanger may result in alterations in the dynamic regulation of GABA signaling in primary afferents.
The observation that GABAA receptor activation was associated with a greater membrane depolarization in neurons responsive to 3 mM GABA begs the question as to whether this depolarization will lead to facilitation or inhibition of action potential generation and/or propagation. It has been suggested that one of the primary mechanisms underlying GABAA receptor-mediated presynaptic inhibition is the depolarization-induced inactivation of voltage-gated Na+ channels (Graham and Redman, 1994). Such a mechanism, however, relies on the biophysical properties of the Na+ channels in the neuron of interest. The vast majority of Na+ channels in the CNS as well as non-nociceptive afferents are subject to steady-state inactivation over a voltage-range that spans typical resting membrane potentials (i.e., −80 to −50 mV). However, there appears to be a relatively high density of tetrodotoxin-(TTX) resistant Na+ channel in both the peripheral (Khasar et al., 1998, Zimmermann et al., 2007) and central (Jeftinija, 1994, Gu and MacDermott, 1997) terminals of nociceptive afferents and these channels are relatively resistant to steady-state inactivation (Kostyuk et al., 1981, Elliott and Elliott, 1993, Gold et al., 1996a. Thus, in nociceptive afferents, GABA-induced depolarization should not inactivate the majority of Na+ channels and therefore should not block action potential propagation via this mechanism.
The other mechanism thought to underlie GABAA receptor-mediated presynaptic inhibition is shunting. The increase in membrane conductance associated with the opening of Cl− channels is typically thought to mediate this effect (Segev, 1990, Cattaert and El Manira, 1999). However, in sensory neurons, where GABAA receptor activation does not simply result in an increase in membrane conductance, but also membrane depolarization, this increase in conductance can be far more excitatory than inhibitory as we have recently demonstrated in dural afferents (Vaughn and Gold, 2010). Rather, the high density of low-threshold persistent K+ current activated with GABA-mediated depolarization may be a mechanism underlying the depolarization-induced inhibitory actions of GABA in cutaneous neurons. Thus, the inflammation-induced suppression of these currents appears to have a significant influence on the valence of the actions of GABA, where depolarization-induced inactivation of K+ (Gold et al., 1996b) as well as the acute inflammatory mediator-induced suppression of these currents (Nicol et al., 1997) should further facilitate the pronociceptive actions of GABA.
Highlights.
There is a persistent inflammation-induced shift in spinal GABAA signaling
This is thought to be due to an NKCC1-dependent depolarization of EGABA in nociceptive afferents
Our results suggest other mechanisms contribute to the shift
These include a depolarization of Vrest, an increase in IGABA and a decrease in low threshold IK
A HCO3−-dependent exchanger also plays an important role in the determination of EGABA
Acknowledgments
We thank Dr. Biff Forbush for the generous gift of R5 antibody and Nicole Scheff, and Drs. Ronald Dubner, Ke Ren, Man-Kyo Chung and Steven Prescott for helpful discussions during the preparation of this manuscript. This work was supported in part by a Grant from the National Institutes of Health, NS063010 (MSG).
Footnotes
Author Contributions:
YZ was involved in all aspects of this study including experimental design, data acquisition and analysis and the generation of the manuscript. SGL was involved in the Ca2+ imaging experiments. MSG was involved in the experimental design and data analysis and the generation of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akaike N. Gramicidin perforated patch recording and intracellular chloride activity in excitable cells. Prog Biophys Mol Biol. 1996;65:251–264. doi: 10.1016/s0079-6107(96)00013-2. [DOI] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ, Gamino SM, Giraldez F, Nogueron I. Intracellular chloride regulation in amphibian dorsal root ganglion neurones studied with ion-selective microelectrodes. J Physiol. 1988;406:225–246. doi: 10.1113/jphysiol.1988.sp017378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anseloni VC, Gold MS. Inflammation-induced shift in the valence of spinal GABA-A receptor-mediated modulation of nociception in the adult rat. J Pain. 2008;9:732–738. doi: 10.1016/j.jpain.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boegel K, Gyulai FE, Moore KK, Gold MS. Deleterious impact of a gamma-aminobutyric acid type A receptor preferring general anesthetic when used in the presence of persistent inflammation. Anesthesiology. 2011;115:782–790. doi: 10.1097/ALN.0b013e318215e1cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaert D, El Manira A. Shunting versus inactivation: analysis of presynaptic inhibitory mechanisms in primary afferents of the crayfish. J Neurosci. 1999;19:6079–6089. doi: 10.1523/JNEUROSCI.19-14-06079.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LM, Kelly ML, Parker MD, Bouyer P, Gill HS, Felie JM, Davis BA, Boron WF. Expression and localization of Na-driven Cl-HCO(3)(−) exchanger (SLC4A8) in rodent CNS. Neuroscience. 2008;153:162–174. doi: 10.1016/j.neuroscience.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott AA, Elliott JR. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J Physiol (Lond) 1993;463:39–56. doi: 10.1113/jphysiol.1993.sp019583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonaatal rat dorsal root ganglion neurons via the cyclic AMP-protein kinase A cascade. J Physiol (London) 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshete F, Fields RD. Spike frequency decoding and autonomous activation of Ca2+-calmodulin-dependent protein kinase II in dorsal root ganglion neurons. J Neurosci. 2001;21:6694–6705. doi: 10.1523/JNEUROSCI.21-17-06694.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemmer AW, Gimenez I, Dowd BF, Darman RB, Forbush B. Activation of the Na-K-Cl otransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem. 2002;277:37551–37558. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- Funk K, Woitecki A, Franjic-Wurtz C, Gensch T, Mohrlen F, Frings S. Modulation of chloride homeostasis by inflammatory mediators in dorsal root ganglion neurons. Mol Pain. 2008;4:32. doi: 10.1186/1744-8069-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan A, Cervero F. Painful stimuli induce in vivo phosphorylation and membrane mobilization of mouse spinal cord NKCC1 co-transporter. Neuroscience. 2005;133:245–252. doi: 10.1016/j.neuroscience.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Gilbert D, Franjic-Wurtz C, Funk K, Gensch T, Frings S, Mohrlen F. Differential maturation of chloride homeostasis in primary afferent neurons of the somatosensory system. Int J Dev Neurosci. 2007;25:479–489. doi: 10.1016/j.ijdevneu.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci U S A. 1996a;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Shuster MJ, Levine JD. Characterization of six voltage-gated K+ currents in adult rat sensory neurons. J Neurophysiol. 1996b;75:2629–2646. doi: 10.1152/jn.1996.75.6.2629. [DOI] [PubMed] [Google Scholar]
- Graham B, Redman S. A simulation of action potentials in synaptic boutons during presynaptic inhibition. J Neurophysiol. 1994;71:538–549. doi: 10.1152/jn.1994.71.2.538. [DOI] [PubMed] [Google Scholar]
- Granados-Soto V, Arguelles CF, Alvarez-Leefmans FJ. Peripheral and central antinociceptive action of Na+-K+-2Cl− cotransporter blockers on formalin-induced nociception in rats. Pain. 2005;114:231–238. doi: 10.1016/j.pain.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Gu JG, MacDermott AB. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature. 1997;389:749–753. doi: 10.1038/39639. [DOI] [PubMed] [Google Scholar]
- Irie T, Hara M, Yasukura T, Minamino M, Omori K, Matsuda H, Inoue K, Inagaki C. Chloride concentration in cultured hippocampal neurons increases during long-term exposure to ammonia through enhanced expression of an anion exchanger. Brain Res. 1998;806:246–256. doi: 10.1016/s0006-8993(98)00700-8. [DOI] [PubMed] [Google Scholar]
- Jeftinija S. The role of tetrodotoxin-resistant sodium channels of small primary afferent fibers. Brain Res. 1994;639:125–134. doi: 10.1016/0006-8993(94)91772-8. [DOI] [PubMed] [Google Scholar]
- Kaila K, Voipio J, Paalasmaa P, Pasternack M, Deisz RA. The role of bicarbonate in GABAA receptor-mediated IPSPs of rat neocortical neurones. J Physiol. 1993;464:273–289. doi: 10.1113/jphysiol.1993.sp019634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T, Zoga V, Gemes G, McCallum JB, Wu HE, Pravdic D, Liang MY, Kwok WM, Hogan Q, Sarantopoulos C. Suppressed Ca2+/CaM/CaMKII-dependent K(ATP) channel activity in primary afferent neurons mediates hyperalgesia after axotomy. Proc Natl Acad Sci U S A. 2009;106:8725–8730. doi: 10.1073/pnas.0901815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon JL. The reversal potential of Ca(2+)-activated Cl(−) currents indicates that chick sensory neurons accumulate intracellular Cl(−) Neurosci Lett. 2000;296:9–12. doi: 10.1016/s0304-3940(00)01610-4. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Gold MS, Levine JD. A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat. Neurosci Lett. 1998;256:17–20. doi: 10.1016/s0304-3940(98)00738-1. [DOI] [PubMed] [Google Scholar]
- Kostyuk PG, Veselovsky NS, Fedulova SA, Tsyndrenko AY. Ionic currents in the somatic membrane of rat dorsal root ganglion neurons - I. Sodium currents. Neuroscience. 1981;6:2424–2430. doi: 10.1016/0306-4522(81)90090-7. [DOI] [PubMed] [Google Scholar]
- Lagraize SC, Guo W, Yang K, Wei F, Ren K, Dubner R. Spinal cord mechanisms mediating behavioral hyperalgesia induced by neurokinin-1 tachykinin receptor activation in the rostral ventromedial medulla. Neuroscience. 2010;171:1341–1356. doi: 10.1016/j.neuroscience.2010.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird JM, Garcia-Nicas E, Delpire EJ, Cervero F. Presynaptic inhibition and spinal pain processing in mice: a possible role of the NKCC1 cation-chloride co-transporter in hyperalgesia. Neurosci Lett. 2004;361:200–203. doi: 10.1016/j.neulet.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Lin Q, Wu J, Willis WD. Dorsal root reflexes and cutaneous neurogenic inflammation after intradermal injection of capsaicin in rats. J Neurophysiol. 1999;82:2602–2611. doi: 10.1152/jn.1999.82.5.2602. [DOI] [PubMed] [Google Scholar]
- Lu SG, Gold MS. Inflammation-induced increase in evoked calcium transients in subpopulations of rat dorsal root ganglion neurons. Neuroscience. 2008;153:279–288. doi: 10.1016/j.neuroscience.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol. 2006;577:169–190. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SG, Zhang XL, Luo ZD, Gold MS. Persistent inflammation alters the density and distribution of voltage-activated calcium channels in subpopulations of rat cutaneous DRG neurons. Pain. 2010;151:633–643. doi: 10.1016/j.pain.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Cafferty WB, Marchand F. Immune and glial cell factors as pain mediators and modulators. Exp Neurol. 2005;192:444–462. doi: 10.1016/j.expneurol.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Medvedeva YV, Kim MS, Usachev YM. Mechanisms of prolonged presynaptic Ca2+ signaling and glutamate release induced by TRPV1 activation in rat sensory neurons. J Neurosci. 2008;28:5295–5311. doi: 10.1523/JNEUROSCI.4810-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol GD, Vasko MR, Evans AR. Prostaglandins suppress an outward potassium current in embryonic rat sensory neurons. J Neurophysiol. 1997;77:167–176. doi: 10.1152/jn.1997.77.1.167. [DOI] [PubMed] [Google Scholar]
- Pitcher MH, Cervero F. Role of the NKCC1 co-transporter in sensitization of spinal nociceptive neurons. Pain. 2010;151:756–762. doi: 10.1016/j.pain.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Pitcher MH, Price TJ, Entrena JM, Cervero F. Spinal NKCC1 blockade inhibits TRPV1-dependent referred allodynia. Mol Pain. 2007;3:17. doi: 10.1186/1744-8069-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planelles G. Chloride transport in the renal proximal tubule. Pflugers Arch. 2004;448:561–570. doi: 10.1007/s00424-004-1309-y. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Kaplan MR, Peterson LN, Gullans SR, Hebert SC, Delpire E. Expression of the Na(+)-K(+)-2Cl− cotransporter BSC2 in the nervous system. Am J Physiol. 1997a;272:C173–183. doi: 10.1152/ajpcell.1997.272.1.C173. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA’s excitatory role in immature brain. J Neurobiol. 1997b;33:781–795. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Price TJ, Cervero F, Gold MS, Hammond DL, Prescott SA. Chloride regulation in the pain pathway. Brain Res Rev. 2009;60:149–170. doi: 10.1016/j.brainresrev.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees H, Sluka KA, Lu Y, Westlund KN, Willis WD. Dorsal root reflexes in articular afferents occur bilaterally in a chronic model of arthritis in rats. J Neurophysiol. 1996;76:4190–4193. doi: 10.1152/jn.1996.76.6.4190. [DOI] [PubMed] [Google Scholar]
- Rees H, Sluka KA, Westlund KN, Willis WD. Do dorsal root reflexes augment peripheral inflammation? Neuroreport. 1994;5:821–824. doi: 10.1097/00001756-199403000-00021. [DOI] [PubMed] [Google Scholar]
- Rees H, Sluka KA, Westlund KN, Willis WD. The role of glutamate and GABA receptors in the generation of dorsal root reflexes by acute arthritis in the anaesthetized rat. J Physiol. 1995;484 (Pt 2):437–445. doi: 10.1113/jphysiol.1995.sp020676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha-Gonzalez HI, Mao S, Alvarez-Leefmans FJ. Na+, K+, 2Cl− Cotransport and Intracellular Chloride Regulation in Rat Primary Sensory Neurons: Thermodynamic and Kinetic Aspects. J Neurophysiol. 2008;100:169–184. doi: 10.1152/jn.01007.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segev I. Computer study of presynaptic inhibition controlling the spread of action potentials into axonal terminals. J Neurophysiol. 1990;63:987–998. doi: 10.1152/jn.1990.63.5.987. [DOI] [PubMed] [Google Scholar]
- Sinning A, Liebmann L, Kougioumtzes A, Westermann M, Bruehl C, Hubner CA. Synaptic glutamate release is modulated by the Na+-driven Cl-/HCO exchanger Slc4a8. J Neurosci. 2011;31:7300–7311. doi: 10.1523/JNEUROSCI.0269-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluka KA, Willis WD, Westlund KN. Inflammation-induced release of excitatory amino acids is prevented by spinal administration of a GABAA but not by a GABAB receptor antagonist in rats. J Pharmacol Exp Ther. 1994;271:76–82. [PubMed] [Google Scholar]
- Sung KW, Kirby M, McDonald MP, Lovinger DM, Delpire E. Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. J Neurosci. 2000;20:7531–7538. doi: 10.1523/JNEUROSCI.20-20-07531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takazawa T, MacDermott AB. Synaptic pathways and inhibitory gates in the spinal cord dorsal horn. Ann N Y Acad Sci. 2010;1198:153–158. doi: 10.1111/j.1749-6632.2010.05501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ. Anatomy of primary afferents and projection neurones in the rat spinal dorsal horn with particular emphasis on substance P and the neurokinin 1 receptor. Exp Physiol. 2002;87:245–249. doi: 10.1113/eph8702351. [DOI] [PubMed] [Google Scholar]
- Vaughn AH, Gold MS. Ionic mechanisms underlying inflammatory mediator-induced sensitization of dural afferents. J Neurosci. 2010;30:7878–7888. doi: 10.1523/JNEUROSCI.6053-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Yaksh TL. Spinal pharmacology of thermal hyperesthesia induced by incomplete ligation of sciatic nerve. I. Opioid and nonopioid receptors. Anesthesiology. 1991;75:817–826. doi: 10.1097/00000542-199111000-00014. [DOI] [PubMed] [Google Scholar]
- Zimmermann K, Leffler A, Babes A, Cendan CM, Carr RW, Kobayashi J, Nau C, Wood JN, Reeh PW. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature. 2007;447:855–858. doi: 10.1038/nature05880. [DOI] [PubMed] [Google Scholar]