

Agrin regulates acetylcholine receptors at the neuromuscular junction by locally stabilizing microtubules through the plus end tracking proteins CLASP2 and CLIP-170.
Abstract
Agrin is the major factor mediating the neuronal regulation of postsynaptic structures at the vertebrate neuromuscular junction, but the details of how it orchestrates this unique three-dimensional structure remain unknown. Here, we show that agrin induces the formation of the dense network of microtubules in the subsynaptic cytoplasm and that this, in turn, regulates acetylcholine receptor insertion into the postsynaptic membrane. Agrin acted in part by locally activating phosphatidylinositol 3-kinase and inactivating GSK3β, which led to the local capturing of dynamic microtubules at agrin-induced acetylcholine receptor (AChR) clusters, mediated to a large extent by the microtubule plus-end tracking proteins CLASP2 and CLIP-170. Indeed, in the absence of CLASP2, microtubule plus ends at the subsynaptic muscle membrane, the density of synaptic AChRs, the size of AChR clusters, and the numbers of subsynaptic muscle nuclei with their selective gene expression programs were all reduced. Thus, the cascade linking agrin to CLASP2-mediated microtubule capturing at the synaptic membrane is essential for the maintenance of a normal neuromuscular phenotype.
Introduction
The function of the dynamic cytoskeleton in synapse formation and maintenance is poorly understood. At the neuromuscular junction (NMJ) the array of synaptic proteins such as the acetylcholine receptors (AChRs) is determined by the specific set of genes induced by the nerve selectively in the muscle nuclei underlying the synapse. However, although the nerve-induced synaptic gene expression program can explain the set of proteins expressed in the synaptic region, it does not account for their focal insertion into the postsynaptic muscle membrane where the density of, e.g., AChRs declines sharply from ∼10,000/µm2 to <5/µm2 within a few micrometers of muscle fiber length. One possibility is a focal transport involving microtubules (MTs) oriented toward and captured at the subsynaptic muscle membrane. Indeed, the NMJ is associated with dense, subsynaptic networks of cortical actin filaments and microtubules (Jasmin et al., 1990; Ralston et al., 1999). Actin filaments are thought to be involved in anchoring AChRs along with other proteins that play a part in NMJ formation and maintenance (Dai et al., 2000; Borges and Ferns 2001), and they may be involved in MT capturing. However, the generation of the subsynaptic actin networks has not been investigated. Likewise, it is not known how MTs are organized, and whether and how they are involved in the formation of the postsynaptic membrane.
The behavior of MTs and their interactions with other intracellular components are largely regulated by MT plus-end tracking proteins (+TIPs), which specifically associate with the growing, or “plus” ends of the MTs (Galjart 2010). In motile fibroblasts, a paradigm for studying MT behavior in cultured cells (Akhmanova et al., 2001), a subset of MTs is oriented toward the leading edge and becomes selectively stabilized (Bulinski and Gundersen 1991). Posttranslational modifications on lattice-incorporated tubulin subunits, such as de-tyrosination or acetylation, accumulate on stable MTs, allowing their recognition with specific antibodies (Bulinski and Gundersen 1991). Specific signaling pathways that are activated at the leading edge mobilize downstream effectors to recruit stable leading edge–oriented MTs. Signaling molecules include lysophosphatidic acid (LPA), which triggers a pathway dependent on the Rho-GTPase and the formin mDia (Gundersen et al., 2004), and phosphatidylinositol 3-kinase (PI3-K) acting via glycogen synthase kinase-3 (GSK3; Akhmanova et al., 2001). Several +TIPs have been implicated in selective MT capturing and stabilization, including APC, ACF7, CLIP-170, and CLASP2 (Galjart 2010). Although CLASPs were discovered (and named) through their interaction with CLIP-115 and -170, the functional significance of this interaction is still unknown, and its in vivo relevance has not yet been described.
The major factor mediating the neural regulation of postsynaptic membrane assembly at the vertebrate NMJ is agrin, a heparansulfate proteoglycan secreted from motor nerve terminals and acting through its receptor/effector, LRP4/MuSK (muscle-specific kinase; Kim et al., 2008; Zhang et al., 2008; Wu et al., 2010), in the muscle fiber membrane. Agrin on its own is sufficient to induce differentiation of functional synaptic membranes in vivo in the absence of motor nerves (Jones et al., 1997). This major organizing function makes agrin-induced AChR clustering in cultured myotubes an ideal system in which to analyze how the subsynaptic MTs come to be organized and to elucidate their roles in synapse formation in vivo.
Here, we present evidence for a novel signaling pathway downstream of agrin that allows dynamic MTs probing the interior of the muscle fiber membrane to become immobilized and captured at the synaptic membrane. Specifically, in both adult muscle and cultured myotubes, agrin triggers the activation of PI3-K, leading to the phosphorylation of GSK3β at the subsynaptic muscle membrane. This serves to cause the local capture of the plus ends of dynamic MTs at the synaptic, but not at the extrasynaptic membrane via a process involving CLASP2 and CLIP-170. Agrin/CLASP2/CLIP-170–mediated MT capturing in turn directs the focal AChR insertion into the synaptic membrane. Our data reveal, for the first time, the in vivo relevance of a CLIP-170–CLASP2 interaction in this process.
Results
The nerve terminal and agrin induce a subsynaptic network of MTs oriented toward the subsynaptic membrane
The subsynaptic muscle membrane is associated with a dense network of MTs as visualized by immunofluorescent labeling of dynamic MTs (or Tyr-tubulin; Fig. 1 a) and total MTs (or β-tubulin; Fig. 1 b). Labeling of de-tyrosinated tubulin (or Glu-tubulin) reveals an even higher enrichment of a subpopulation of stable subsynaptic MTs (Fig. 1 b). These data suggest that MTs are enriched and specifically stabilized near the NMJ and suggest the involvement of specific +TIPs in this process.
Figure 1.

The subsynaptic network of MTs at the neuromuscular junction is induced by agrin. (a) NMJ in mouse sternomastoid muscle labeled for AChRs (red) and for Tyr-tubulin (green) reveals subsynaptic network of MTs. Bar, 10 µm. (b) MTs at the NMJ are posttranslationally modified by de-tyrosination. Labeling for β-tubulin, Glu-tubulin, and AChRs reveals synaptic and nonsynaptic MTs (red), selective de-tyrosination of subsynaptic MTs (green), and AChRs (blue). Note MTs are also present in presynaptic nerve terminal branches (arrowheads). Bar, 10 µm. (c) Network of MTs (tyr-tubulin, green) is present at nerve-free, ectopic postsynaptic membranes (marked by accumulation of AChRs [red]) induced by ectopic application of recombinant neural agrin to nonsynaptic region of adult muscle fiber. Note the accumulation of muscle nuclei, revealed by the surrounding MTs, at same site. Bar, 20 µm. (d) Tips of growing MTs are enriched at synaptic AChR clusters in epitrochleo-anconeus (ETA) muscle from GFP-Clip-170ki/ki mutant mouse. Top: en-face view of NMJ marked by AChRs (red). MT plus-ends are stained with anti-GFP antibody. Bar, 5 µm. Bottom: part of a synaptic AChR cluster (red) stained for CLIP-170 with anti-GFP antibody (green) and reconstructed in 3D from a stack of confocal images. Left, view from muscle side of AChR cluster; note the MT plus-ends approaching the synaptic AChR cluster from below the postsynaptic membrane. Right, view from presynaptic side of same AChR cluster; note absence of MT tips from primary synaptic gutter where nerve terminal was located (not stained).
Subsynaptic MT induction can be mediated by agrin in vivo. Specifically, agrin secreted from muscle fibers upon intracellular injection of expression plasmid into nonsynaptic regions of innervated muscle fibers (Jones et al., 1997) induces ectopic AChR clusters, which have a MT network associated with them similar to that observed in vivo (Fig. 1 c). This suggests that the nerve normally induces the subsynaptic MTs, at least in part, by the secretion of agrin.
To examine the orientation of subsynaptic MTs at the NMJ, muscle fibers of knock-in mice expressing GFP-tagged CLIP-170 (Akhmanova et al., 2005) at the plus ends of the MTs were fixed in a way that preserves dynamic MTs (see Materials and methods). Labeling with an antibody against GFP showed CLIP-170 enriched at synaptic AChR clusters (Fig. 1 d). 3D reconstruction of NMJs from confocal image stacks showed that the plus ends of the MTs were preferentially colocalized with the synaptic AChR cluster. In contrast, in many instances they were completely absent from the synaptic gutter containing the nerve terminal (Fig. 1 d). These findings indicate that the plus ends of MTs probing the interior of the cell cortex become preferentially associated with synaptic AChR clusters.
In cultured cells, MT stabilization is promoted by several +TIPs, including CLASP2, which are directly or indirectly linked to the actin network (Galjart 2010). Cortical actin is highly enriched at the NMJ (Dai et al., 2000; Bruneau et al., 2008), and is thought to be involved in the anchoring of the AChRs in the subsynaptic muscle membrane. Immunolabeling of CLASP2 suggested that it was organized in a pattern similar to that of CLIP-170, with an enrichment at the crests of the synaptic folds (i.e., between troughs marked by AChR-rich lines [Fig. 2 a], as well as along the edge of the AChR cluster [Fig. 2 c]). Interestingly, the AChR cluster edge is where new AChRs are inserted into the synaptic membrane of developing NMJs (Kummer et al., 2004). Imaging NMJs after 3 d of denervation, i.e., when nerve terminals were degenerated, produced similar results (not depicted). Thus, CLIP-170 and CLASP2 immunoreactivities were postsynaptic.
Figure 2.

Synaptic localization of CLASP2, Tyr-tubulin, and Glu-tubulin in wt and Clasp2−/− NMJs. (a) Staining of CLIP-170 and CLASP2 at NMJ of wild-type muscle. Like CLIP-170 (left, green), CLASP2-decorated MT plus-ends (right, green) are enriched at synaptic AChR clusters primarily at their edges and at the crests of synaptic folds (arrowheads; troughs of folds marked by the AChRs). Bar, 5 µm. Note that images in panel a are from the level of the synaptic folds, i.e., deeper in the synaptic gutter; in contrast, maximum intensity projections from optical slices taken at the level of the edge of the synaptic AChR cluster and thus of the synaptic gutter (such as in Fig. 1 d, top images) reveal pronounced CLIP-170 enrichment compared with adjacent perisynaptic membrane. This explains the apparent difference in CLIP-170 intensity along the edge of the clusters in the two images (Fig. 2 a vs. 1 d). (b) Double staining of CLIP-170 and CLASP2 at NMJ of wild-type muscle. Accumulation of both +TIPs at edge of synaptic AChR cluster (blue) and overlap of the two +TIPs (marked by asterisks) is consistent with cooperation between CLIP-170 and CLASP2 at MT plus-ends. Note that some of the CLIP-170 and CLASP2 staining does not overlap, which may reflect differences in antibody accessibility and staining, or, alternatively, independent functions of these proteins in the NMJ. For localization of CLIP-170 in Clasp2−/− NMJ, see Fig. 4. Bar, 2.5 µm. (c) Top: CLASP2 is enriched at edge of synaptic AChR cluster at wild-type NMJ. Bottom: absence of CLASP2 staining of NMJ from Clasp2−/− muscle shows specificity of antibody used. Bars, 5 µm. (d) Both Tyr-tubulin (top) and Glu-tubulin (bottom) are enriched at NMJs of Clasp2−/− muscle. Bars, 10 µm.
Double stainings of CLIP-170 and CLASP2 at the synapse revealed significant colocalization of these proteins at the NMJ (Fig. 2 b), indicating that subsynaptic MT ends contain both +TIPs. However, MT capture and stabilization can still occur in the absence of either of these proteins as indicated by the synaptic MT networks at Clasp2−/− (Fig. 2 d) and at Clip1−/−;Clip2−/− NMJs (not depicted) visualized by Tyr-tubulin and Glu-tubulin stainings. Although no impairment of stable synaptic MTs could be resolved by tubulin stainings in either of these mutants due to variable staining conditions, MT capture as revealed by CLIP-170 staining was reduced by the deletion of CLASP2 (see Fig. 4). Taken together, these findings suggest that agrin/MuSK triggers downstream signals that capture MTs at synaptic AChR clusters by a process that includes CLIP-170 and CLASP2, and that this, in turn, promotes the insertion of AChRs into the synaptic membrane.
Figure 4.

Absence of CLASP2 reduces the density of GFP-CLIP-170–decorated MTs at the synaptic membrane of the NMJ in vivo. (a) The localization of MT plus-ends (visualized by GFP labeling) at synaptic AChR clusters is reduced in Clasp2−/−;GFP-Clip170ki/ki myotubes. Bar, 5 µm. Graph shows percentage of synapses with GFP-CLIP-170 enriched at the edge of the AChR cluster in wild-type and Clasp2−/− synapses (means ± SEM, n = 13 wild-type and 18 CLASP2−/− synapses from 3 muscles each (*, P < 0.05; **, P < 0.01, Mann Whitney U-Test). A synapse was classified “blindly” by visual inspection as enriched in CLIP-170 (arrowheads), when edges of AChR clusters were decorated with GFP puncta along >80% of their lengths. Note that further information on, e.g., number and intensity of CLIP dots cannot be extracted from these confocal images (for details, see Materials and methods). (b) Analysis by structured illumination microscopy (SIMELRYA S.1; Carl Zeiss) of the localization of GFP-CLIP-170–decorated MTs at the synaptic membrane (marked by AChRs) in wild-type and Clasp2−/−;GFP-Clip-170ki/ki NMJs. NMJs stained for AChRs (red) and GFP (green). Bars, 1 µm, 3 µm, and 1.5 µm. Stacks of images of NMJs en face were taken through the entire depth of the synapse at 0.125-µm steps and processed. 10–20 µm of contour lines of AChRs (as the one illustrated by the white line in the 3D-reconstructed cluster in top panel) at each of three z-levels for each synapse were selected, and CLIP-170 puncta per length of contour line were counted (arrowheads). Bottom panels show sample AChR contour lines and CLIP-170 immunoreactivity, including enlarged insets. Graph shows combined data from three synapses each in wild-type and Clasp2−/− muscle (*, P < 0.05; **, P < 0.01, two-sided t test). Note the marked reduction in the density of CLIP-170 puncta on AChR contour lines in the absence of CLASP2.
NMJs are impaired in Clasp2−/− mice
To test this hypothesis, we investigated NMJs in mice in which CLASP2 had been genetically deleted. The phenotype of NMJs in adult Clasp2−/− mice was abnormal in several ways. Specifically, when compared with size-matched wild-type mice with similar muscle fiber diameters, the area of synaptic AChR clusters, the numbers of subsynaptic nuclei, and the density of synaptic AChRs were all reduced to ∼75% of control (Fig. 3, a and b), resulting in a decline of synaptic AChR number to <60% (0.75 × 0.75). Strikingly, the rate at which AChRs were replaced was also reduced in NMJs (Fig. 3 b), suggesting a role for CLASP2 in the accumulation of this important receptor, akin to its function in polarized vesicle trafficking in motile cells (Miller et al., 2009). Although the defects in the structure of NMJs in Clasp2−/− mice would be expected to lower the safety factor for neuromuscular impulse transmission, this effect was not great enough to precipitate impairment of impulse transmission as judged from the animal’s gross motor behavior.
Figure 3.
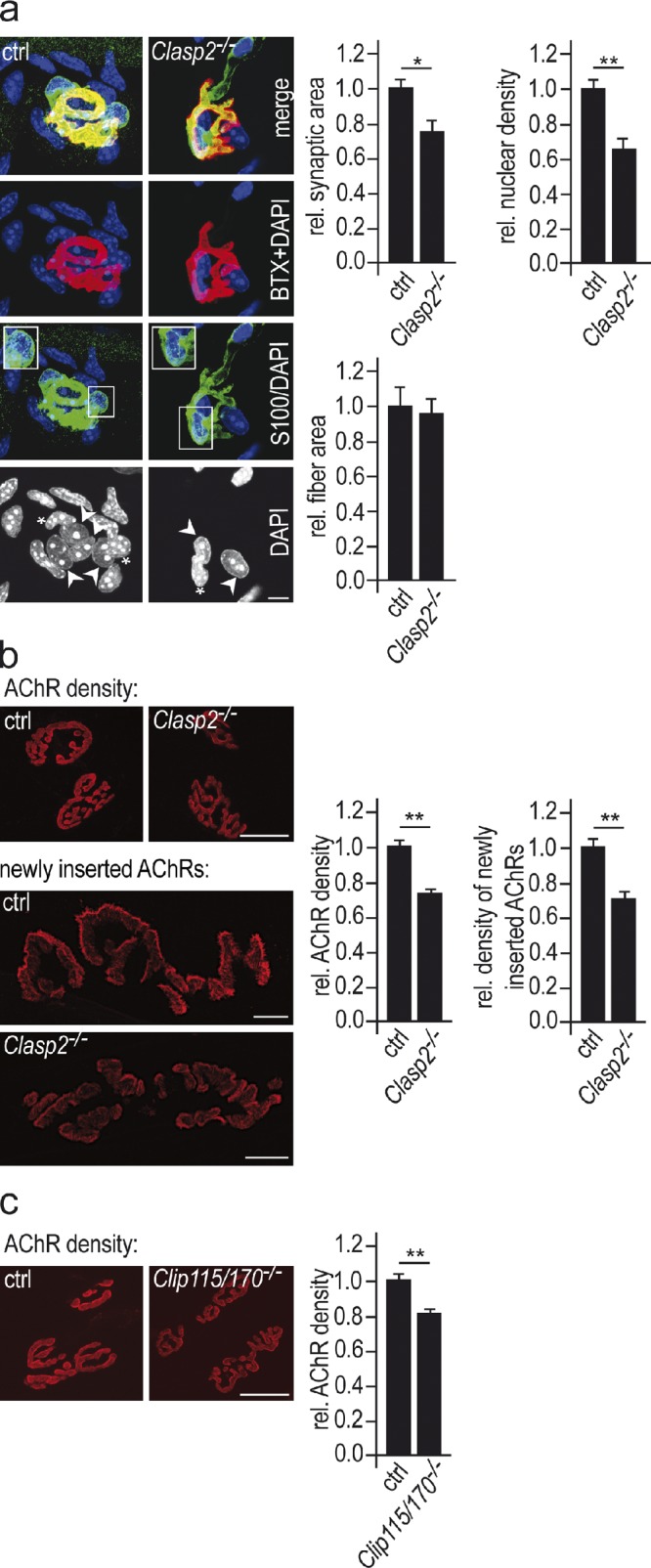
Genetic deletion of CLASP2 impairs NMJs in vivo. (a) The size of synaptic AChR clusters and the number of subsynaptic nuclei per synapse are reduced in Clasp2−/− muscle. NMJs in soleus muscles of wild-type and mutant animals of equal weights were stained for AChRs (red), the Schwann cell marker S-100 (green), and nuclei with DAPI (blue). Synaptic area was determined from AChR labeling of synapses lying en face (see Materials and methods). Nuclei surrounded by S-100 labeling were considered as terminal Schwann cells (see inset in panel labeled S100/DAPI). The number of subsynaptic muscle nuclei was estimated by subtracting the number of Schwann cell nuclei (marked by asterisks) from the number of nuclei underlying the AChR cluster (marked by arrowheads). Bar, 10 µm. Graphs show mean values of respective parameters ± SEM, n = 47 wild-type and 52 Clasp2−/− NMJs from 3 muscles of each genotype analyzed (*, P < 0.05; **, P < 0.01, two-sided t test). (b) AChR density and AChR insertion rates are reduced in Clasp2−/− muscle. To determine AChR densities sternomastoid muscles were saturated with α-BTX–Alexa 594. Bar, 25 µm. Means ± SEM, n = 63 and 72 NMJs from 3 wild-type and 3 Clasp2−/− muscles analyzed (*, P < 0.05; **, P < 0.01, two-sided t test). To estimate AChR insertion rates sternomastoid muscles were denervated to increase AChR turnover; 7 d later AChRs in superficial endplates were saturated in vivo with α-BTX–Alexa 488. After another 7 d (to allow AChR turnover), endplates were saturated with α-BTX–Alexa 594, and the average intensity of Alexa 488 fluorescence in Clasp2−/− NMJs was normalized to that in wild-type muscles. Bar, 25 µm. Means ± SEM, 28 wild-type and 25 Clasp2−/− endplates analyzed (*, P < 0.05; **, P < 0.01, two-sided t test). (c) AChR density is reduced in Clip1−/−;Clip2−/− muscle. Analysis similar as in b. Given are means ± SEM, n = 30 wild-type and 31 Clip1−/−;Clip2−/− (*, P < 0.05; **, P < 0.01, two-sided t test). Bar, 25 µm.
These data indicate that CLASP2 contributes to normal synaptic function in multiple ways: by increasing synaptic AChR density through delivery of AChRs to the synaptic membrane; by increasing synaptic area and, by inference, increased quantal content of the endplate potential; and through the recruitment of subsynaptic nuclei which, through their muscle activity–resistant expression of AChR subunit and other synaptic genes, contribute to the maintenance of the NMJ.
If both CLASP2 and CLIP-170 are involved in subsynaptic MT capturing, as is also suggested by their colocalization at the NMJ (Fig. 2 b), one would expect a similar NMJ phenotype in mice lacking CLIP-170 as in the CLASP2-deficient mice. We therefore measured synaptic AChR density in CLIP-115/170–deficient mice, and found a reduction to ∼80% of control (Fig. 3 c), similar to what we observed in the Clasp2 knockouts. These results suggest that the CLASP2–CLIP-170 interaction that was documented in vitro serves to regulate synaptic function in vivo.
CLASP2 promotes attachment of MT plus-ends to synaptic membranes of mature NMJs
To test for a role of CLASP2 in MT stabilization at the NMJ, we compared the localization of MT plus-ends as visualized by CLIP-170 labeling in wild-type and in Clasp2−/− mice. As described above, CLIP-170 was prominent at the edges of the synaptic AChR clusters at wild-type NMJs but was also present throughout the postsynaptic membrane in the primary synaptic fold. In Clasp2−/− mice the synaptic localization of CLIP-170 was reduced. This was most readily seen from the decrease of CLIP-170 immunoreactivity along the edges of the synaptic AChR clusters (Fig. 4 a).
Because the resolution of confocal microscopy is limited, in particular in the z-direction, some synapses were imaged by using structured illumination microscopy, which increases resolution both in x-y and in z-directions by a factor of 2 compared with conventional confocal microscopy (Gustafsson et al., 2008). In individual optical slices taken through synaptic AChR clusters oriented en face, AChR labeling appeared as contour lines following the synaptic membrane at that respective z-level. In wild-type animals, puncta of CLIP-170 immunoreactivity precisely followed these lines. This indicates that in wild-type mice the plus ends of the MTs were present exactly, within the limit of z-resolution, at the level of the synaptic membrane. In contrast, at Clasp2−/− synapses CLIP-170 puncta were largely absent from the AChR contour lines (Fig. 4 b). Thus, in the absence of CLASP2, the incidence of MT plus-ends at the level of the AChRs was markedly reduced. This observation implicates CLASP2 in the capturing of CLIP-170–labeled MT ends (and thus of MTs) at the agrin-induced AChR cluster.
Agrin phosphorylates GSK3β through PI3-K
Next we examined how agrin regulates CLASP2-mediated MT capturing. At the leading edge of motile fibroblasts CLASP2-mediated MT capturing is regulated by phosphatidylinositol 3-kinase (PI3-K) and glycogen synthase kinase-3 β (GSK3β; Akhmanova et al., 2001). In many instances, PI3-K activates AKT through its recruitment to inositol lipids and subsequent phosphorylation by PDK-1; AKT in turn phosphorylates GSK3β, which is active by default but is rendered inactive by phosphorylation on Serine 9 (Ser9). The inactivation of GSK3β triggered by PI3-K prevents phosphorylation of CLASP2 and, as a consequence, enhances its affinity to MT plus-ends (Akhmanova et al., 2005; Kumar et al., 2009; Watanabe et al., 2009). Blockade of PI3-K is known to inhibit agrin-induced AChR clustering in cultured myotubes (Nizhynska et al., 2007). We therefore tested the hypothesis that agrin, by activating PI3-K, induces phosphorylation of GSK3β at Ser9 (p-GSK3β).
To this end, C2C12 and primary wild-type myotubes were exposed to 5 nM of either agrin isoforms that are selectively expressed by motor neurons (neural agrin) or those expressed by nonneural tissues such as muscle. Of these, only neural agrin can induce synaptic membranes in muscle (Meier et al., 1998). Western blotting of lysates showed a pronounced but transient increase in both p-AKT and p-GSK3β within a few minutes in response selectively to neural, but not to nonneural agrin. Both AKT and GSK3β phosphorylation could be blocked by pretreating cells with PI3-K inhibitors (LY294002 or ZSTK474) or with AKT inhibitor (A6730; Fig. 5 a, left). Similar results were seen when myotubes were grown on a substrate impregnated with agrin, which more closely resembles physiological agrin presentation (Fig. 5 a, right; for full blots see Fig. S2). Thus agrin, by activating PI3-K and AKT, phosphorylates (i.e., inactivates) GSK3β in cultured myotubes.
Figure 5.

Agrin phosphorylates PI3-K, AKT, and GSK3β in muscle cells. (a) Neural, but not nonneural, agrin phosphorylates GSK3β via phosphorylation of PI3-K and AKT in cultured myotubes. Western blots of lysates from C2C12 myotubes to which soluble neural (5 nM) or nonneural (5 nM) agrin was added. Blots probed with antibodies as described in Materials and methods. Panels showing p-GSK3β and p-AKT are derived from same blot exposed for different times. Note that (1) AKT and GSK3β are not phosphorylated by nonneural agrin (top left panel, right lane); (2) their phosphorylation by neural agrin is inhibited by the PI3-K blocker LY294002 (50 µM), and the AKT inhibitor A6730 (0.5 µM, bottom left panel), respectively. Neural agrin also phosphorylates AKT and GSK3β when presented attached to culture substrate (top right panel, see Materials and methods). Full blots are shown in Fig. S1. Bar, 20 µm. (b) PI3-K is activated at the postsynaptic membrane of the NMJ. Soleus muscles of wild-type mice were electroporated with expression constructs for PH-BTK or PH-BTKmut, i.e., pleckstrin homology domain fragments of Brutton tyrosine kinase, which specifically bind (PH-BTK-GFP) or do not bind (PH-BTK_R28C-GFP) to PIP3, a read-out for PI3-K activity. Both constructs were tagged with eGFP (see Várnai et al., 1999; Bohnacker et al., 2009). Top: longitudinal confocal sections passing through NMJs of muscle fibers expressing elevated GFP (green) at synaptic AChR cluster (BTX594, red). Bars, 10 µm. Bottom: cross sections through fibers electroporated with the two constructs. Synaptic localization of PH-BTK-GFP is higher than that of PH-BTKmut-GFP, indicating synaptic PI3-K activity. Graph shows means ± SEM of fold change in GFP intensity at synaptic AChR clusters at 10–14 d after electroporation with PH-BTK or PH-BTKmut, respectively (n = 20 synapses on GFP-positive fibers per construct examined, *, P < 0.05; **, P < 0.01, two-sided t test). Bars, 7.5 µm and 3 µm. (c) P-GSK3β is enriched at the NMJ. Immunoreactivity is abolished by pretreatment of muscle with lambda-phosphatase. Bar, 10 µm. For specificity of antibody used, see Fig. S2. (d) High resolution confocal image of p-GSK3β immunoreactivity. GSK3β-(Ser9)P labeling was not distributed evenly. Rather, p-GSK3β labeling was preferentially located between the crests of the synaptic folds carrying the AChRs, as is the case for CLIP-170 and CLASP2 (Fig. 2 a). Enlarged inset and corresponding line profiles of AChR and GSK3β-P fluorescence are shown on the right. Bars, 3 µm and 0.5 µm.
We next examined whether activated PI3-K and p-GSK3β are present at the NMJ in vivo. PI3-K activity at NMJs was assessed by measuring relative synaptic GFP fluorescence levels in fibers electroporated with expression constructs encoding either a wild-type or a mutant fragment of Bruton’s tyrosine kinase (BTK). Both constructs comprised a GFP tag coupled to the respective wild-type and mutant BTK pleckstrin homology domains, of which the wild type, but not the mutant, binds to PtdIns(3,4,5)P3 formed by active PI3-K (Várnai et al., 1999). Fig. 5 b shows that the synaptic GFP fluorescence signal was greatly reduced when fibers expressed the mutant BTK-GFP construct, indicating that PI3-K is activated at the NMJ. Consistent with our hypothesis for the agrin-induced signaling cascade, we further observed by immunofluorescence that p-GSK3β was colocalized with the synaptic membrane of normal (Fig. 5 c; for specificity of the antibody used, see Fig. S2) and denervated NMJs (not depicted), excluding that the p-GSK3β immunoreactivity was presynaptic. p-GSK3β was not distributed evenly but appeared focally localized at the crests of the synaptic folds between the regions carrying the AChRs (Fig. 5 d), i.e., similar to CLIP-170 and CLASP2 (Fig. 2 a). When combined, these results indicate that agrin, by activating PI3-K, phosphorylates and thus inactivates GSK3β at the NMJ.
MTs are captured at agrin-induced AChR clusters by a process involving PI3-K and CLASP2
If agrin induces the trapping of plus ends of dynamic MTs at differentiating synaptic spots, it should be possible to visualize this in real time using GFP-tagged +TIPs. To mimic the stable presentation of agrin to the muscle at mature NMJs, we deposited neural agrin in small patches (20–60 µm in diameter) onto a laminin substrate. Myotubes that contact these agrin deposits form large AChR clusters that include stabilized AChRs and have associated with them a cluster of myonuclei that express the synapse-specific form of AChRs (Fig. 6 a; Jones et al., 1996). We therefore compared the dynamics of MTs in primary GFP-CLIP-170–expressing myotubes, inside and outside agrin-induced AChR clusters, using TIRF microscopy.
Figure 6.

Capturing of dynamic MTs at agrin-induced AChR clusters in cultured myotubes is abolished by pharmacological inhibition of PI3-K and by genetic elimination of Clasp2. (a) Scheme illustrating focal impregnation of culture substrate with neural agrin. Transfected COS1 cells secrete and locally deposit neural agrin on a laminin substrate, and the cells are then lysed. Stable AChR clusters form where GFP-Clip-170ki/ki myotubes contact agrin deposits. (b) MT plus-ends decorated with GFP-CLIP-170 as observed by TIRF microscopy at 1-s intervals for 160 s. Shown are AChR cluster, surface reflective interference contrast (SRIC) image of same myotube, first frame and maximum intensity projection of all 160 images of the stack. Note higher density and brighter GFP signal at AChR cluster (marked in red). Bars, 10 µm. (c) Representative examples of kymograms of type I and type II comet behavior. See text for discussion of comet behavior and criteria for comet classification. (d) Same as in b, except that myotubes had been incubated for 90 min in ZSTK474 (5 µM) before GFP analysis. Note that “cluster”-specific comet behavior is abolished. Bars, 10 µm. (e) Same as in b, except that myotubes derived from Clasp2−/−;GFP-Clip-170ki/ki mice were used. Note that “cluster”-specific comet behavior is abolished. Bars, 10 µm. (f) Quantification of comet behavior inside and outside agrin-induced AChR clusters. Comet immobilization by neural agrin is abolished by inhibition of PI3-K or the absence of CLASP2. Comets are grouped from kymograms into type I (mobile) and type II (stable) comets as described in the text, and the percentages of stable comets, both inside and outside agrin-induced AChR clusters, is given (means ± SEM, n = 4 myotubes for each condition; number of comets analyzed: wild type: 374; wild type, ZSTK474-treated: 342; Clasp2−/−: 323; *, P < 0.05; **, P < 0.01, two-sided t test).
Analysis of individual frames of time-lapse movies (see Video 1) of wild-type myotubes suggested a much higher density of GFP-CLIP-170–labeled MT plus-ends inside the AChR clusters than outside (Fig. 6 b, center). However, maximum intensity projections (MIPs) of all 160 frames of the time-lapse imaging experiment showed that also outside the cluster many MT ends do reach the membrane (Fig. 6 b, right). The fact that inside the cluster the difference in MT plus-end density between single frames and MIPs is less pronounced than outside suggests that MT ends reaching the cell membrane inside the cluster are more likely to be captured than they are outside the cluster.
Analysis of changes in GFP-CLIP-170 labeling and distribution in distance versus time plots (so-called kymographs) revealed two types of behavior (Fig. 6 c). One type (here called type I) consisted of largely continuous movement of GFP-CLIP-170 “comets” during the time they remained within the penetration range of the evanescent wave. These comets had an average speed of 0.15 ± 0.01 µm/s (SEM, n = 73 tracks) and represent the continuous advance of the plus ends of growing MTs. In the other type of behavior (here called type II), the GFP-CLIP-170 fluorescence appeared stable over a period of 160 s or longer, with fluorescence flashing up at discrete spots along the track for tens of seconds before fading. Type II behavior suggests reduced freedom of GFP-CLIP-170 movement, and the capture of GFP-CLIP-170–labeled MT ends. Conspicuously, GFP-CLIP-170 dynamics was different inside and outside agrin-induced AChR clusters. Inside clusters, ∼75% of GFP-CLIP-170 tracks observed were of type II, and the remaining 25% of type I. In contrast, this ratio was reversed outside receptor clusters, where only 25% were of type II (Fig. 6 f).
As suggested above for adult NMJs, MT stabilization at agrin-induced AChR clusters in cultured myotubes was mediated by PI3-K activation and CLASP2. Specifically, when MT dynamics were examined in primary GFP-CLIP-170 myotubes treated with the PI3-K inhibitor ZSTK474 (Fig. 6 d), or in myotubes derived from Clasp2−/−;GFP-Clip-170ki/ki (i.e., knockout/knock-in) mice (Fig. 6 e; see Video 2), type I and II comets were equally common in both cluster and noncluster regions (Fig. 6 f), suggesting nonspecific MT plus-end capturing both outside and inside the clusters. These experiments demonstrate that agrin enables the capture of CLIP-170–coated MT ends through a process requiring CLASP2, and that this process depends on PI3-K activation.
CLIP-170 is increased at plus ends of MTs in the proximity of agrin-induced AChR clusters
The GFP fluorescence of MT plus-ends imaged inside agrin-induced AChR clusters in control wild-type myotubes appeared brighter by eye than those in the same myotubes outside the AChR cluster. This difference could be due to MTs coming closer to the internal myotube membrane inside clusters than outside and/or to an increased load of CLIP-170 molecules on MT tips induced by agrin. To examine these possibilities, myotubes were labeled for CLIP-170 and EB3, a member of the EB1-family of “core” +TIPs that is expressed in myotubes (Fig. 7). Myotubes were then imaged by confocal microscopy, and the intensity of CLIP-170 fluorescence on MT plus-ends, identified by their EB3 immunoreactivity, was assayed inside and outside AChR clusters by line scans. At MT plus-ends inside AChR clusters in control myotubes the intensity of CLIP labeling was significantly higher than outside clusters, whereas EB3 was not changed (Fig. 7 a). This suggests that pathways activated by agrin increase the affinity of CLIP-170 for MT plus-ends.
Figure 7.
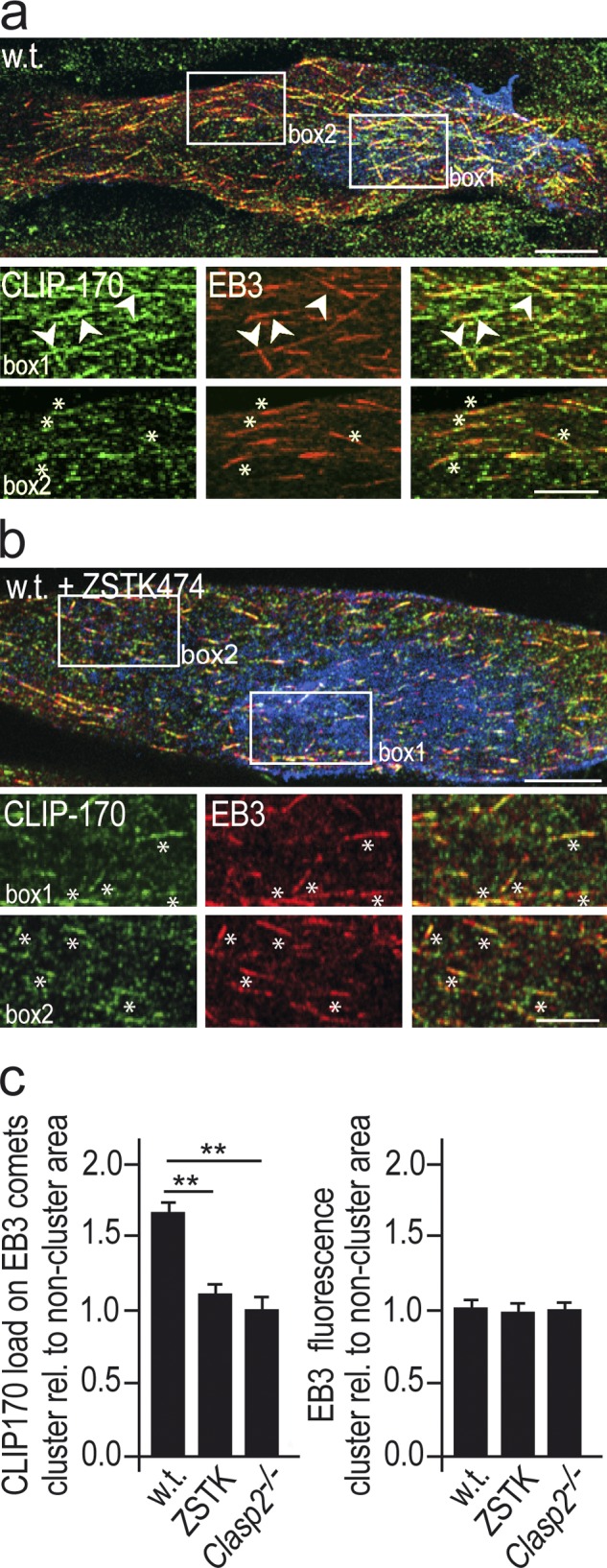
The CLIP-170 load relative to EB3 at MT plus-ends is greater inside than outside AChR clusters and is reduced by PI3-K inhibition and by deletion of Clasp2. (a) Comets in AChR clusters in GFP-Clip-170ki/ki myotubes revealed by immunolabeling of EB3 and of GFP, to visualize CLIP-170. Regions outlined by boxes within and outside AChR cluster are shown enlarged at bottom. Agrin-induced AChR clusters in blue, GFP-CLIP-170 in green, EB3 in red. Note increased load of CLIP-170 at comets in AChR cluster (arrowheads) compared with comets outside cluster lacking increased CLIP-170 (asterisks). Bars: (top) 10 µm; (bottom) 5 µm. (b) Same as panel a but after blockade of PI3K with ZSTK474. Note reduction in CLIP-170 load in PI3-K–blocked compared with nonblocked myotubes. Bars: (top) 10 µm; (bottom) 5 µm. Asterisks same as in panel a. (c) Quantification of CLIP-170 staining on EB3-stained comets inside relative to that of comets outside AChR clusters. The elevated CLIP-170 load at agrin-induced AChR clusters observed in wild-type myotubes is abolished by PI3-K inhibition and by genetic deletion of Clasp2. EB3 staining is similar inside and outside AChR clusters. Percentages are means ± SEM from 84 to 245 comets analyzed in wild-type, ZSTK474-blocked, and Clasp2−/− myotubes (4 cells each). *, P < 0.05; **, P < 0.01, two-sided t test.
Next, we examined whether the increase in CLIP-170 load was mediated through PI3K and CLASP2 by repeating the above experiment in wild-type myotubes treated with ZSTK474 (Fig. 7 b) and in Clasp2−/− mutant myotubes (not depicted). In both cases the enrichment of CLIP-170 was reduced to extrasynaptic levels (Fig. 7 c). Thus, in addition to CLASP2 deficiency, blockade of PI3-K inhibited CLIP-170 enrichment. These data strongly suggest that agrin leads to CLASP2-mediated MT capture and regulates the load of CLIP-170 at MT plus-ends, whereas EB3 remains unchanged; they provide further evidence for the in vivo relevance of a CLIP–CLASP interaction.
MTs are required for insertion of AChRs into agrin-induced AChR clusters in cultured myotubes in a manner dependent on PI3-K and CLASP2
Having found that CLASP2 deletion reduces the area and density of synaptic AChR clusters at NMJs in vivo, we examined the role of MT capturing in the maintenance of agrin-induced AChR clusters in cultured myotubes. In a first set of experiments, we asked whether MT integrity was essential for the maintenance of agrin-induced AChR clustering. Myotubes were treated with 10 µM nocodazole for 3 h, sufficient to depolymerize most MTs. At the end of the drug treatment, the AChR clusters were labeled with α-BTX, and their area measured. The clusters of AChR in nocodazole-treated myotubes were markedly (60%) smaller than those in untreated myotubes (Fig. 8 a). The density of AChRs was, however, unchanged. These data indicate that the number of AChRs in the clusters was decreased by nocodazole.
Figure 8.

Impairment of MT function by nocodazole, by inhibition of PI3-K and AKT, and by genetic deletion of CLASP2 impairs AChR insertion into agrin-induced AChR clusters in cultured myotubes. (a) Areas of agrin-induced AChR clusters in primary myotubes appear smaller when AChRs are stained after 3 h of treatment with the following drugs: nocodazole (10 µM); inhibitors of AKT (A6730, 0.5 µM); PI3-K (ZSTK474, 5 µM; only the effect of ZSTK474 is illustrated). However, the density of AChRs in clusters remains unchanged. Conversely, inhibiting GSK3β activity (CHIR99021, 0.1 µM) increases AChR cluster size. Bar, 30 µm. Means ± SEM. Total numbers of AChR clusters analyzed in two independent experiments: Control (wild type): 78; nocodazole: 67; A6730: 68; ZSTK474: 38; CHIR99021: 54 (*, P < 0.05; **, P < 0.01, two-sided t test). (b) In contrast, AChR clusters appear unchanged after 3 h of drug treatment when AChRs are stained at the time of drug addition; again, AChR densities in clusters remain unchanged. Bar, 60 µm. Means ± SEM. Total numbers of AChR clusters analyzed in two independent experiments: Control (wild type): 30; nocodazole: 46; ZSTK474: 27; CHIR99021: 21 (*, P < 0.05; **, P < 0.01, two-sided t test). (c) AChR clusters are smaller in Clasp2−/− than in wild-type muscle, and unlike in wild-type muscle (see panel a), cluster size is not affected by ZSTK474 (5 µM) or CHIR99021 (0.1 µM). AChRs were stained after drug treatment. Bar: 30 µm. Graphs give means ± SEM, 20–78 clusters analyzed from 2 cultures for each condition (*, P < 0.05; **, P < 0.01, two-sided t test). AChR clusters marked by arrowheads.
This decrease might occur through reduced AChR insertion into, or accelerated AChR removal from, the clusters. To distinguish between these possibilities the experiment was repeated, but with AChR clusters stained at the beginning of the drug incubation period. With this protocol, AChR cluster size was similar in control and in nocodazole-treated myotubes (Fig. 8 b). Thus, once AChRs had been inserted into the cluster, their removal was not affected by nocodazole. These experiments thus demonstrate that nocodazole reduces AChR cluster size by impairment of AChR insertion, and further, that in these experiments, measuring AChR cluster size can be used to differentiate between AChR insertion and removal.
Finally, we tested the effects of inhibitors of PI3-K, AKT, and GSK3β, on AChR insertion. Using the criteria just described, inhibition of PI3-K and AKT decreased and inhibition of GSK3β increased AChR insertion into AChR clusters (Fig. 8 a). Likewise, genetic deletion of Clasp2 (using Clasp2−/− myotubes) reduced AChR insertion (Fig. 8 c). In these Clasp2−/− myotubes, in contrast to wild-type myotubes, inhibition of PI3-K (ZSTK474) did not reduce AChR insertion any further (Fig. 8 c), nor did inhibition of GSK3β with CHIR 99021 increase receptor insertion, indicating that PI3-K, p-GSK3β, and CLASP2 are part of the same signaling cascade. Taken together, these experiments are consistent with our observations at mature NMJs (Fig. 3 b) and strongly support our model (see Fig. 9) that agrin, acting through PI3-K and p-GSK3β, promotes MT capturing at the synapse (Fig. 4 b and Fig. 6), and that this enhances AChR insertion into agrin-induced AChR clusters.
Figure 9.

Regulation and function of CLIP-170/CLASP2-dependent microtubule stabilization at the postsynaptic membrane of the neuromuscular junction. (a) Working model summarizing the role of neural agrin to capture MTs at the postsynaptic membrane of the NMJ. Agrin-induced MT capturing at AChR clusters is regulated through: (1) local GSK3β inactivation promoting the recruitment of unphosphorylated CLASP2 to MT plus-ends and lattice; (2) recruitment of CLIP-170 through CLIP/CLASP and CLIP/MT interactions; (3) local, PI3-K–dependent recruitment and immobilization of LL5β in the synaptic membrane. Molecules and their interactions inferred from work published in other systems rather than from present data in muscle cells are marked by broken symbols. (b) At developing NMJs, constitutively expressed synaptic AChRs are delivered to synaptic membrane by lateral diffusion in the plane of the cell membrane (arrows). At adult NMJs, MTs stabilized by mechanisms outlined in panel a ensure focal delivery of synaptic AChRs to the crests of synaptic folds. Cartoon in panel b is modified from Ruegg, 2005.
Discussion
The role of dynamic MTs in synaptic function is only beginning to be understood. It has only recently been reported that dynamic MTs enter dendritic spines transiently in a way that appears to be dependent on synaptic activity and correlated with LTP (Gu et al., 2008; Hu et al., 2008; Jaworski et al., 2009). However, neither the mechanisms that regulate MT entry into spines nor their role in modulating synaptic plasticity have been established.
Here, we have used the NMJ of a GFP-CLIP-170 knock-in mouse mutant as a model for studying the role of MTs at the synapse. This model has two major advantages. First, subsynaptic differentiation and maintenance of the NMJ, including the regulation of a stable subsynaptic MT network, are controlled by one major organizer, agrin. In our in vitro paradigm, agrin was presented in a locally stable fashion, similar to that in the muscle fiber’s synaptic basal lamina in vivo. Second, the use of myotubes derived from GFP-CLIP-170 knock-in mice ensures normal expression levels of this GFP-tagged +TIP, which functions in vivo like nontagged CLIP-170 (Akhmanova et al., 2005). This allows us to observe GFP-CLIP-170–decorated dynamic MTs at agrin-induced AChR clusters under completely physiological conditions.
The classical function of agrin is to cluster AChRs (and other synaptic proteins) in the subsynaptic muscle membrane. This involves the induction of a scaffold for anchoring these proteins at high density to the subsynaptic actin cytoskeleton. The present experiments, both at NMJs in vivo and in cultured myotubes, now reveal an additional aspect of agrin function in the clustering process, i.e., in the insertion of the AChRs into the synaptic receptor cluster. AChR insertion into the synaptic muscle membrane requires MTs captured at the synaptic membrane via a pathway downstream of agrin regulating, through PI3-K and GSK3β, the CLASP2-mediated MT capture at the subsynaptic membrane. Thus, synaptic MT capturing at the NMJ might be regulated similarly as at the leading edge of migrating cells where, by inactivation of GSK3β, CLASP2 is dephosphorylated, which increases its association with MTs (Kumar et al., 2009; Watanabe et al., 2009).
MTs can be linked through nonphosphorylated CLASP2 to cortical sites in two nonmutually exclusive ways: (1) through the PIP3 sensor LL5β (Lansbergen et al., 2006; Hotta et al., 2010), and (2) through direct binding of CLASP2 to actin (Tsvetkov et al., 2007). In addition, as suggested by the increase of the CLIP load on MTs at AChR clusters, increased (de-phosphorylated) CLASP2 at the clusters might increase binding of CLIP-170 via direct CLASP2–CLIP interactions. Whether increased CLIP also participates in MT capturing at the NMJ, e.g., through the Rac effector IQGAP1 to actin (Fukata et al., 2002; Watanabe et al., 2004) or through potentiating further CLASP2 binding, is not known, but would be consistent with the reduced AChR density at Clip115−/−;Clip170−/− NMJs (Fig. 3 c).
Of the potential CLASP2 interactors, LL5β is expressed in the endplate membrane and is localized at the crests between the AChR-rich regions at the mouth of the folds (Kishi et al., 2005), i.e., where p-GSK3β, CLIP-170, and CLASP2 are enriched (Figs. 1, 2, and 4). Further, its depletion inhibits AChR clustering (Kishi et al., 2005). LL5β is thus a strong candidate for mediating the GSK3-β/CLASP2–dependent MT capturing observed here. Our working model for CLASP2-mediated aspects of synaptic MT capturing is illustrated in Fig. 9.
MT capturing at the NMJ might also be mediated via the +TIPs APC (Zumbrunn et al., 2001) and ACF7 (Wu et al., 2011). Of these, APC is enriched at the NMJ, and overexpression of a dominant-negative APC truncation mutant inhibits agrin-dependent AChR clustering in cultured myotubes (Wang et al., 2003). Finally, ankyrin has been shown to be involved in the MT organization at the NMJ perhaps by acting as a membrane receptor for dynactin, with which it interacts (Ayalon et al., 2008), and which stabilizes MTs at adherens junctions (Shaw et al., 2007). Such additional mechanisms for synaptic MT capturing may account for the relatively mild phenotype at Clasp2−/− and at Clip1−/−;Clip2−/− NMJs.
The 30% reduction in the number of junctional nuclei at Clasp2−/− NMJs raises the alternative possibility that reduced synapse-specific AChR gene expression might account for the reduction in synaptic size and AChR density. Although we cannot exclude this for synaptic size, AChR density is unlikely to be affected by 30% reduced AChR expression, as AChR densities are similar at NMJs of AChRε+/+ and AChRε+/− synapses (Missias et al., 1997), and the amplitudes of miniature endplate currents do not change between the first and ninth postnatal weeks (Witzemann et al., 1996), when synaptic size and nuclear number increase dramatically.
The directed transport of synaptic components to the endplate membrane as proposed here for CLASP2-mediated synaptic MT capturing may be more important at mature NMJs than during early stages of their development. Early in NMJ development, muscle fibers express high levels of synaptic proteins, including AChRs, along their entire surface as part of their developmental program. This allows the recruitment of proteins to the developing synapse by lateral diffusion in the plane of the cell membrane (Anderson and Cohen 1977; Flanagan-Steet et al., 2005). In contrast, at later stages of NMJ maturation, when nerve-induced electrical muscle activity has down-regulated extrasynaptic AChR expression, the supply of synaptic molecules from nonsynaptic membrane is no longer available. This necessitates the transport of AChRs and other synaptic proteins from the TGN below the synaptic membrane where synapse-specific gene expression is maintained by the agrin-induced transcription from the fundamental muscle nuclei underlying the synapse (Brenner et al., 1990). The mechanisms described here can explain the highly focal nature of this transport to the synaptic muscle membrane through mechanisms similar to those controlling polarity and directed migration in motile cells.
Materials and methods
Animals
Generation of the Clasp2 single knockout strain and of the Clip1;Clip2 double knockout mouse strain will be described elsewhere. In brief, the genes encoding CLIP-170 (Clip1 gene) and CLIP-115 (Clip2 gene) were targeted by homologous recombination in embryonic stem (ES) cells. In the case of the Clip1 gene we inserted a GFP-loxP-Neo-loxP casette (where Neo indicates the neomycin resistance gene) into the exon containing the ATG translation initiation codon. This yielded the Clip1 knockout allele. Removal of the neomycin resistance gene in ES cells by Cre recombinase yielded the GFP-Clip170 knock-in (ki) allele (Akhmanova et al., 2005). In the case of the Clip2 gene we inserted a loxP-neo-loxP casette at the 5′ end of the gene and a loxP-Puro-loxP-LacZ casette (where Puro indicates the puromycin resistance gene, and LacZ the β-galactosidase gene) at the 3′ end of the gene. Cre-mediated recombination in ES cells yielded the Clip2 knockout allele (Hoogenraad et al., 2002). Single Clip1 and Clip2 knockout and GFP-Clip170ki mice were obtained by germline transmission of the modified alleles in chimeric mice, which were in turn obtained by injecting ES cells carrying the modified alleles into recipient blastocysts. The Clip1 and Clip2 single knockout mice were crossed to generate the double knockout line.
For electroporation and in vivo stainings, animals were anesthetized with ketamine (87 mg per kg body weight) and xylazine (13 mg per kg body weight). Postoperative analgesia was by 4 injections of buprenorphine at 12-h intervals. Mice were sacrificed with CO2. Animal handling was approved by the Cantonal Veterinary Office of Basel-Stadt.
Depending on experimental suitability, soleus (for electroporations), epitrochleo-anconeus (ETA; for optimal preservation of dynamic microtubules) or sternomastoideus (for estimates of AChR density from whole BTX-Alexa 488–stained NMJs) muscles were used.
Chemicals and antibodies
ZSTK474 (LC Laboratories) was used at 1 or at 5 µM on cultured myotubes analyzed biochemically or by imaging, respectively. All other reagents were applied in the following concentrations: nocodazole (10 µM; Sigma-Aldrich), LY294002 (50 µM; Sigma-Aldrich), A6730 (500 nM; Sigma-Aldrich), CHIR99021 (100 nM; Axon Medchem) λ-protein phosphatase (2,000 U; New England Biolabs, Inc.), and rapamycin (100 nM; LC Laboratories). AChRs were labeled with α-BTX–Alexa 488, 594, or 647 (Invitrogen). GFP-CLIP-170 (by staining for GFP), EB1, and CLASP2 were visualized with antibodies from Invitrogen, BD, and Absea, respectively. Polyclonal rabbit antibodies against human EB3 (image clone 714028) were custom made at Absea, using a bacterially expressed and purified GST-EB3 fusion protein as antigen, as described previously (Stepanova et al., 2003). Antibodies against AKT, p-AKT (S473), GSK3β, p-GSK3β (S9; no. 9336), and actin were purchased from Cell Signaling Technology. All other primary antibodies were obtained from Sigma-Aldrich (anti-tyrosinated α-tubulin, anti-neurofilament), Millipore (anti-detyrosinated α-tubulin), Santa Cruz Biotechnology, Inc. (anti-myc), and Dako (anti-S100; Z0311). Secondary antibodies were goat anti–rabbit, goat anti–mouse, or goat anti–rat antibodies conjugated to Alexa 488 (Invitrogen) as well as donkey anti–chicken Cy2 (Jackson ImmunoResearch Laboratories, Inc.). HRP-conjugated secondary antibodies were from Santa Cruz Biotechnology, Inc. Ringer’s solution was obtained from Braun. Phosphatase inhibitors PIC1 and 2 as well as Collagenase type 1A were purchased from Sigma-Aldrich, protease inhibitors and Fugene HD from Roche, and PDGF-BB from Peprotech. Rapamycin was a gift from M.N. Hall (Biozentrum, University of Basel, Basel, Switzerland).
Estimation of synaptic PI3-K activity
40 µg of pPH-BTK-GFP or pPH-BTK R28C-GFP (Várnai et al., 1999) in 10 µl 0.9% NaCl was injected with a Hamilton syringe into the soleus muscle of anesthetized C57BL76 mice (8–10 wk old). After suturing the skin, 8 pulses (20 ms, 1 Hz, 200 V/cm) were applied to the leg using an ECM 830 electroporation system. 10–14 d later, electroporated soleus muscles were dissected, AChRs stained with 1 µg/ml α-BTX-Alexa 594 for 1 h at RT, fixed in 4% paraformaldehyde (PFA) for 2 h, placed in 30% sucrose/PBS overnight, and frozen. Muscles were then embedded and sectioned at 12-µm thickness in a cryostat (CM 1950; Leica). Sections were mounted in Citifluor and GFP and α-BTX-Alexa 594 stainings imaged using an ACS APO 63x/1.3 NA objective on an SPE confocal microscope (DMI 4000B; Leica). For the measurement of synaptic GFP intensity, the α-BTX–stained synapse of GFP-positive fibers was defined as a region of interest (ROI). Using ImageJ (National Institutes of Health), green fluorescence in this synaptic ROI was measured, the ROI moved to the extrasynaptic region of the same fiber profile, and the extrasynaptic green fluorescence was measured. Because the level of GFP expression varies across fibers within an individual muscle, the ratio of green fluorescence at the synapse to that in nonsynaptic membrane was taken as an estimate of synaptic PH-BTK-GFP or PH-BTK R28C-GFP binding, respectively.
Confocal microscopy
Muscle fibers and myotubes were imaged with an SPE confocal scanning laser microscope (DMI 4000B; Leica) at a resolution of 1024 × 1024 pixels using an HCX PL APO 100x objective (NA 1.46) or an ACS APO 63x objective (NA 1.30). Image stacks were acquired with a step size of 300 nm. For images used in 3D reconstructions, step sizes of 100 nM were used, and image stacks were deconvolved using Huygens Essential software (Scientific Volume Imaging). For comparison of different samples the same laser settings were applied. Quantitative differences potentially due to changes in the light source or camera were excluded by imaging control and mutant muscles within the same session. Nevertheless, the quality of confocal images, taken at different NMJs, was too variable to allow quantitatively precise measurements of intensity and number of GFP-CLIP-170 or CLASP2 dots or of subsynaptic MT networks visualized by Tyr-tubulin or Glu-tubulin stainings for comparison between, e.g., wild-type and mutant NMJs. This was due to unequal fixation and/or unequal penetration of antibody in different fibers (for details, see Immunocytochemistry section). Preservation of CLIP-170– and CLASP2-positive MT plus-ends in adult muscle required the use of the thin epitrochlearis-anconeus (ETA) muscle, fixed in −80°C methanol; moreover, fibers were stained in bundles, such that not all endplates within a bundle were equally exposed to antibody; finally, images needed different contrasting to optimally resolve GFP-CLIP-170 or CLASP2 dots. As a consequence, only qualitative information such as the number of endplates with clear enrichment of GFP-CLIP-170 dots along edges of synaptic AChR clusters could be extracted.
3D reconstructions (surface renderings) were performed in Imaris x64 7.4.0 on stacks acquired with a sample distance of 0.05 × 0.05 × 0.1 µm.
Preparation of primary muscle cultures
Neonatal leg muscles from wild-type and mutant neonatal muscle were minced, dissociated with collagenase type IV and dispase type II, and cells were plated on a laminin substrate in DME containing 2 mM glutamine, 20% FCS, 5 ng/ml recombinant human basic FGF, and 1% antibiotic/antimycotic solution. After 2 d, they were resuspended in PBS by brief trypsinization, treated with rat monoclonal anti–mouse α7-integrin antibody, and purified using (magnetic) Dynabeads coated with sheep anti–rat IgG and a Dynal-MPC-L magnetic particle concentrator (Blanco-Bose et al., 2001; Escher et al., 2005). C2C12 or wild-type myotubes were cultured on laminin-coated dishes focally impregnated with agrin. For the preparation of the dishes, COS-1 cells, transfected with a plasmid coding for full-length chicken agrin (Jones et al., 1996), were seeded at a density of 7–20 x 103 cells per 30 mm laminin-coated culture dish. After 48 h cells were extracted for 1 h in 2% Triton X-100 in PBS, followed by intensive washing (6–8x 1 h PBS) and myoblast seeding (Schmidt et al., 2011). Subsequent differentiation was in DME, 5% horse serum, and 1% antibiotic/antimycotic solution. For biochemistry, 6-well dishes were coated with 10 µg/ml laminin (Invitrogen) followed by coating with agrin solution (0.5 µg/ml, 37°C, 2 h) before cell plating (Schmidt et al., 2011). It should be noted that throughout the present paper, agrin was applied attached to the culture substrate rather than in solution to mimic the in vivo situation.
Immunocytochemistry
All immunostainings of MTs and +TIPs were done on mice epitrochlearis-anconeus (ETA) muscle, which at its thinnest part contains only ∼6 fiber layers; this makes it optimally suited for snap fixation in cold methanol, which proved crucial for preservation of +TIP-decorated MT plus-ends. ETA muscles were pinned out after excision on Sylgard supports in a culture dish, allowing medium access from both sides. To allow recovery from potential preparative stress, muscles were bathed in DME gassed with 95% CO2/5% O2 at 37°C for 1 h and containing 1 µg/ml fluorescent α-BTX for AChR staining. After Collagenase type 1A treatment (0.5%, 37°C, 15 min), myotube culture dishes and recovered ETA muscles were rapidly transferred to −80°C methanol for 2 h, then transferred to a 1:1 mixture of methanol and 4% PFA at −20°C for 1 h, followed by 30-min periods at 4°C and at RT. Final fixation was in 4% PFA (RT, 10 min). Teased bundles of 4–10 muscle fibers were then incubated at 4°C for 15 min in 100 mM glycine and permeabilzed in 20% normal goat serum (NGS)/PBS/2% Triton X-100 at RT for 2 h. In the case of cultured myotubes, glycine treatment was followed by permeabilization in 20% NGS/PBS/0.5% Triton X-100 (RT, 1 h). Primary and secondary antibodies were diluted in 5% NGS/PBS/0.1% Triton X-100 and were applied at 4°C overnight (primaries) or at RT for 45 min (secondaries).
GSK3β phosphorylated at Ser9 was labeled in sternomastoid muscles treated with 1% Collagenase type 1A (37°C, 30 min). Muscles were fixed in 4% PFA (37°C, 20 min) and permeabilized. Teased fibers were then incubated in protein phosphatase buffer (50 mM Hepes, 100 mM NaCl, 2 mM DTT, 1 mM MnCl2, and 0.01% Brij-35, pH 7.5) with or without 2,000 U λ-protein phosphatase at RT for 1 h, washed 3x with PBS, and processed as described above.
Quantification of GFP-CLIP-170 load on EB3 comets
The mean fluorescence of GFP-CLIP-170 and EB3, visualized with the respective fluorescent antibodies, was determined in ImageJ (Plugin “Measure RGB”) in and outside of the AChR cluster area in nonsaturated, nonprocessed confocal images. The ratio of GFP-CLIP-170 and EB3 fluorescence served as a measure for the load of GFP-CLIP-170 on EB3 comets. For EB3 fluorescence quantitation, only the intensities of EB3 comets were taken into account.
Estimation of number of subsynaptic myonuclei
Soleus muscles from wild-type or clasp2−/− mice of similar weight were stained with 1 µg/ml of α-BTX–Alexa 594 in buffered L-15 medium (RT, 1 h), fixed with 4% PFA for 2 h, and individual fibers were teased and permeabilized in 20% NGS and 2% Triton X-100. Staining with S100 was performed overnight at 4°C. After incubation with secondary antibody, fibers were mounted in ProLong Gold containing DAPI. En-face synapses were imaged in 1-µm stacks using an ACS APO 63x/1.3 NA objective at the SPE confocal microscope (DMI 4000B; Leica). For synapse area measurements, synapses were outlined in the maximum intensity projection for each image manually and the area of the outlined region was calculated using ImageJ. Schwann cell nuclei marked by S100 staining were discounted from the total nuclear count as visualized by DAPI. For measurement of muscle fiber areas, muscles were treated and sectioned as described in Estimation of synaptic PI3-K activity. To delineate muscle fiber plasma membranes, cryosections were postfixed for 5 min in 2% PFA, washed, stained with Wheat Germ Agglutinin Oregon Green 488 overnight, and mounted. The analySIS software from Olympus was used to outline fiber ferrets and calculate cross-sectional area.
Structured illumination microscopy (SIM)
Fixed samples were imaged using the ELYRA S.1 structured illumination microscope (Carl Zeiss). Images were acquired using a 63x/1.40 oil Plan Apochromat objective and an EMCCD camera (iXon 885; Andor Technology). Stacks of images of NMJs en face were taken through the entire depth of the synapse at 0.125 µm. Image processing was performed using the Carl Zeiss Zen software to achieve a maximal resolution of 110 nm in x-y and 250 nm in z-directions. For colocalization experiments, precise pixel alignment between different acquisition channels was ensured by correcting potential pixel shifts via the channel alignment function within the Zen software (Car Zeiss) using simultaneously acquired images of multi-spec beads (200 nm) as a reference. Image analysis was performed using Imaris software (Bitplane). Contour lines of AChRs at three z-levels for each synapse were selected, and the number of CLIP-170 puncta per length of contour line was counted.
Total internal reflection microscopy (TIRF)
Dynamics of GFP-CLIP-170 comets were measured in myotubes derived from GFP-Clip-170ki/ki mice. Agrin-induced AChR clusters were stained with α-BTX–Alexa 594 (1 µg/ml, 30 min), and after washing with prewarmed DME cells were incubated at 37°C for 1 h before being mounted on a thermostat perfusion chamber. Cells were imaged at 37°C in Krebs Ringer's solution (140 mM NaCl; 5 mM KCl, 1 mM Mg2+, 2 mM Ca2+, 20 mM Hepes, 1 mM NaHPO4, and 5.5 mM glucose) at pH 7.4. Online fluorescence images were acquired using an inverted TIRF microscope (TE2000; Nikon) equipped with an oil immersion CFI Plan Apochromat 100× TIRF objective (1.49 NA) and an electron multiplier CCD camera (C9100-13, Hamamatsu Photonics; Treves et al., 2010). The focal plane corresponding to the coverglass/cell membrane contact was adjusted with a surface reflective interference contrast (SRIC) cube and maintained throughout the experiment with the help of a perfect focus system. GFP-CLIP-170 dynamics were imaged for 160 s with a rate of 1 frame per second (MetaMorph; Molecular Devices).
Quantitative analysis of GFP-CLIP-170 comet dynamics
Dynamics of GFP-CLIP-170 comets were analyzed inside and outside of agrin-induced AChR cluster with ImageJ (National Institutes of Health). The maxiprojection of all acquired 160 frames of the live imaging experiment was used to mark all the comet traces within the imaged area. Kymographs (time–space plots) of all the comets visualized in the TIRF plane were generated with the Kymograph plugin for ImageJ from J. Rietdorf (Friedrich Miescher Institute, Basel, Switzerland) and A. Seitz (EMBL Heidelberg, Heidelberg, Germany). To study the effect of PI3-K signaling as well as CLASP2 on GFP-CLIP-170 comet dynamics, comets from myotubes either treated with ZSTK474 (37°C, 60–80 min) or deficient in CLASP2 were used for the analysis.
Quantitative analysis of AChR cluster size
An effect of the drugs indicated or of CLASP2 deficiency on AChR cluster size was determined by incubating the cells for 3 h at 37°C in the absence or presence of pharmacological inhibitors. Cells were then fixed (4% PFA) and stained with α-BTX–Alexa 594 (1 µg/ml, 30 min) to label agrin-induced AChR clusters.
To elucidate AChR removal rates in culture, agrin-induced AChR clusters in primary cells were stained with α-BTX–Alexa 594 (1 µg/ml, 30 min) before the cells were incubated for 3 h at 37°C (to allow removal of fluorescent receptors) in the absence or presence of pharmacological inhibitors. Cells were then fixed (4% PFA) and imaged (HC Plan Apo 20x/0.70 objective; inverted microscope [DMI 6000B; Leica]; 1394 ORCA-ERA camera [Hamamatsu Photonics], Volocity 6.0.1 [PerkinElmer]). Cluster sizes after 3 h were determined by measuring the area of the remaining AChR fluorescence in ImageJ (National Institutes of Health).
Quantification of AChR density and insertion in vivo
To determine the density of AChRs in CLASP2-deficient and age-matched control littermates, the sternomastoid muscles of anaesthetized mice were exposed and bathed for 90 min in Ringer's solution containing 5 µg/ml α-BTX–Alexa 594 (a dose that has previously been demonstrated to be sufficient to saturate all receptors; Bruneau et al., 2005). Mice were sacrificed, and muscles were washed (Ringer’s solution) and fixed in 4% PFA for 20 min. Dissection of the muscles was followed by mounting and imaging of superficial synapses only using confocal microscopy (63x objectve, oil immersion). AChR fluorescence was measured in nonsaturated, nonprocessed images (ImageJ).
Staining of newly inserted AChRs was performed in denervated sternomastoid muscles of anaesthetized mice. Muscle denervation was followed immediately by labeling of AChRs in the synaptic as well as perisynaptic muscle membrane with a saturating dose of α-BTX–Alexa 488 (5 µg/ml, 90 min). After the removal of excess α-BTX–Alexa 488 with Ringer's solution, wounds were sutured and allowed to heal for 1 wk. Mice were re-anaesthetized, sternomastoid muscles were exposed, and newly inserted AChRs were stained with α-BTX–Alexa 594 (5 µg/ml, 90 min). Mice were then sacrificed. Fixation, mounting, image acquisition, and quantitation was done as described above.
Immunoblotting
Myotubes were lysed in 3x SDS-lysis buffer (150 mM Tris, pH 6.8, 300 mM DTT, 6% SDS, 0.2% bromophenol blue, and 30% glycerol) supplemented with phosphatase inhibitors (1:100), protease inhibitors (1/10 tablet per ml lysis buffer), and 100 mM DTT. Lysates were denatured at 95°C for 5 min and loaded on a 10% SDS-PAGE. Gels were transferred onto PVDF membranes and developed with ECL after incubation with primary and secondary antibodies.
Statistical analyses
Data are given as mean ± SEM. The Shapiro-Wilk test was used to test if datasets belong to a normally distributed population. If so, quantitative comparisons of numerical datasets were tested for statistical significance by means of the parametric, nonpaired, two-sided Student's t test. Otherwise the nonparametric, two-sided U test (Mann & Whitney) was applied. One asterisk marks P values below 5% (P < 0.05), whereas two asterisks correspond to P values below 1% (P < 0.01).
Online supplemental material
Fig. S1 shows the full blots from which Fig. 5 a was extracted. They show the effect of neural vs. nonneural agrin on phosphorylation of GSK3β and AKT and the effects of AKT, mTOR, and PI3-K inhibitors. Fig. S2 shows the tests used to demonstrate the specificity of the anti-phospho-GSK3β antibody (no. 9336; Cell Signaling Technology), using myc-tagged GSK3β wt and S9A mutant transfected into COS cells. Video 1 shows capturing of GFP-CLIP-170 comets at agrin-induced AChR clusters in primary of GFP-CLIP-170ki/ki myotubes, using TIRF microscopy. Video 2 shows inhibition of capturing of GFP-CLIP-170 comets at agrin-induced AChR clusters in Clasp2−/−;GFP-Clip-170ki/ki primary myotubes, using TIRF microscopy. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201111130/DC1.
Supplementary Material
Acknowledgments
We thank Dr. Matthias Wymann for suggesting the expression of PH-BTK fragments to test for PI3K activity in vivo, Michele Courtet for technical assistance, and Dr. Clarke Slater for critical reading of the manuscript.
This work was supported by the Swiss National Science Foundation, The Swiss Foundation for Research on Muscle Diseases, The Neuromuscular Research Association Basel (NeRAB), the Netherlands Organization for Health Research and Development (ZonMW), and the Dutch Cancer Genomics Center (CGC).
Footnotes
Abbreviations used in this paper:
- AChR
- acetylcholine receptor
- GSK3
- glycogen synthase kinase-3
- MT
- microtubule
- NMJ
- neuromuscular junction
- PI3-K
- phosphatidylinositol 3-kinase
- +TIP
- plus-end tracking protein
References
- Akhmanova A., Hoogenraad C.C., Drabek K., Stepanova T., Dortland B., Verkerk T., Vermeulen W., Burgering B.M., De Zeeuw C.I., Grosveld F., Galjart N. 2001. Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell. 104:923–935 10.1016/S0092-8674(01)00288-4 [DOI] [PubMed] [Google Scholar]
- Akhmanova A., Mausset-Bonnefont A.L., van Cappellen W., Keijzer N., Hoogenraad C.C., Stepanova T., Drabek K., van der Wees J., Mommaas M., Onderwater J., et al. 2005. The microtubule plus-end-tracking protein CLIP-170 associates with the spermatid manchette and is essential for spermatogenesis. Genes Dev. 19:2501–2515 10.1101/gad.344505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson M.J., Cohen M.W. 1977. Nerve-induced and spontaneous redistribution of acetylcholine receptors on cultured muscle cells. J. Physiol. 268:757–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayalon G., Davis J.Q., Scotland P.B., Bennett V. 2008. An ankyrin-based mechanism for functional organization of dystrophin and dystroglycan. Cell. 135:1189–1200 10.1016/j.cell.2008.10.018 [DOI] [PubMed] [Google Scholar]
- Blanco-Bose W.E., Yao C.C., Kramer R.H., Blau H.M. 2001. Purification of mouse primary myoblasts based on alpha 7 integrin expression. Exp. Cell Res. 265:212–220 10.1006/excr.2001.5191 [DOI] [PubMed] [Google Scholar]
- Bohnacker T., Marone R., Collmann E., Calvez R., Hirsch E., Wymann M.P. 2009. PI3Kgamma adaptor subunits define coupling to degranulation and cell motility by distinct PtdIns(3,4,5)P3 pools in mast cells. Sci. Signal. 2:ra27 10.1126/scisignal.2000259 [DOI] [PubMed] [Google Scholar]
- Borges L.S., Ferns M. 2001. Agrin-induced phosphorylation of the acetylcholine receptor regulates cytoskeletal anchoring and clustering. J. Cell Biol. 153:1–12 10.1083/jcb.153.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner H.R., Witzemann V., Sakmann B. 1990. Imprinting of acetylcholine receptor messenger RNA accumulation in mammalian neuromuscular synapses. Nature. 344:544–547 10.1038/344544a0 [DOI] [PubMed] [Google Scholar]
- Bruneau E., Sutter D., Hume R.I., Akaaboune M. 2005. Identification of nicotinic acetylcholine receptor recycling and its role in maintaining receptor density at the neuromuscular junction in vivo. J. Neurosci. 25:9949–9959 10.1523/JNEUROSCI.3169-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau E.G., Brenner D.S., Kuwada J.Y., Akaaboune M. 2008. Acetylcholine receptor clustering is required for the accumulation and maintenance of scaffolding proteins. Curr. Biol. 18:109–115 10.1016/j.cub.2007.12.029 [DOI] [PubMed] [Google Scholar]
- Bulinski J.C., Gundersen G.G. 1991. Stabilization of post-translational modification of microtubules during cellular morphogenesis. Bioessays. 13:285–293 10.1002/bies.950130605 [DOI] [PubMed] [Google Scholar]
- Dai Z., Luo X., Xie H., Peng H.B. 2000. The actin-driven movement and formation of acetylcholine receptor clusters. J. Cell Biol. 150:1321–1334 10.1083/jcb.150.6.1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escher P., Lacazette E., Courtet M., Blindenbacher A., Landmann L., Bezakova G., Lloyd K.C., Mueller U., Brenner H.R. 2005. Synapses form in skeletal muscles lacking neuregulin receptors. Science. 308:1920–1923 10.1126/science.1108258 [DOI] [PubMed] [Google Scholar]
- Flanagan-Steet H., Fox M.A., Meyer D., Sanes J.R. 2005. Neuromuscular synapses can form in vivo by incorporation of initially aneural postsynaptic specializations. Development. 132:4471–4481 10.1242/dev.02044 [DOI] [PubMed] [Google Scholar]
- Fukata M., Watanabe T., Noritake J., Nakagawa M., Yamaga M., Kuroda S., Matsuura Y., Iwamatsu A., Perez F., Kaibuchi K. 2002. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell. 109:873–885 10.1016/S0092-8674(02)00800-0 [DOI] [PubMed] [Google Scholar]
- Galjart N. 2010. Plus-end-tracking proteins and their interactions at microtubule ends. Curr. Biol. 20:R528–R537 10.1016/j.cub.2010.05.022 [DOI] [PubMed] [Google Scholar]
- Gu J., Firestein B.L., Zheng J.Q. 2008. Microtubules in dendritic spine development. J. Neurosci. 28:12120–12124 10.1523/JNEUROSCI.2509-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen G.G., Gomes E.R., Wen Y. 2004. Cortical control of microtubule stability and polarization. Curr. Opin. Cell Biol. 16:106–112 10.1016/j.ceb.2003.11.010 [DOI] [PubMed] [Google Scholar]
- Gustafsson M.G., Shao L., Carlton P.M., Wang C.J., Golubovskaya I.N., Cande W.Z., Agard D.A., Sedat J.W. 2008. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 94:4957–4970 10.1529/biophysj.107.120345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad C.C., Koekkoek B., Akhmanova A., Krugers H., Dortland B., Miedema M., van Alphen A., Kistler W.M., Jaegle M., Koutsourakis M., et al. 2002. Targeted mutation of Cyln2 in the Williams syndrome critical region links CLIP-115 haploinsufficiency to neurodevelopmental abnormalities in mice. Nat. Genet. 32:116–127 10.1038/ng954 [DOI] [PubMed] [Google Scholar]
- Hotta A., Kawakatsu T., Nakatani T., Sato T., Matsui C., Sukezane T., Akagi T., Hamaji T., Grigoriev I., Akhmanova A., et al. 2010. Laminin-based cell adhesion anchors microtubule plus ends to the epithelial cell basal cortex through LL5alpha/beta. J. Cell Biol. 189:901–917 10.1083/jcb.200910095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Viesselmann C., Nam S., Merriam E., Dent E.W. 2008. Activity-dependent dynamic microtubule invasion of dendritic spines. J. Neurosci. 28:13094–13105 10.1523/JNEUROSCI.3074-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin B.J., Changeux J.P., Cartaud J. 1990. Compartmentalization of cold-stable and acetylated microtubules in the subsynaptic domain of chick skeletal muscle fibre. Nature. 344:673–675 10.1038/344673a0 [DOI] [PubMed] [Google Scholar]
- Jaworski J., Kapitein L.C., Gouveia S.M., Dortland B.R., Wulf P.S., Grigoriev I., Camera P., Spangler S.A., Di Stefano P., Demmers J., et al. 2009. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron. 61:85–100 10.1016/j.neuron.2008.11.013 [DOI] [PubMed] [Google Scholar]
- Jones G., Herczeg A., Ruegg M.A., Lichtsteiner M., Kröger S., Brenner H.R. 1996. Substrate-bound agrin induces expression of acetylcholine receptor epsilon-subunit gene in cultured mammalian muscle cells. Proc. Natl. Acad. Sci. USA. 93:5985–5990 10.1073/pnas.93.12.5985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G., Meier T., Lichtsteiner M., Witzemann V., Sakmann B., Brenner H.R. 1997. Induction by agrin of ectopic and functional postsynaptic-like membrane in innervated muscle. Proc. Natl. Acad. Sci. USA. 94:2654–2659 10.1073/pnas.94.6.2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N., Stiegler A.L., Cameron T.O., Hallock P.T., Gomez A.M., Huang J.H., Hubbard S.R., Dustin M.L., Burden S.J. 2008. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 135:334–342 10.1016/j.cell.2008.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi M., Kummer T.T., Eglen S.J., Sanes J.R. 2005. LL5beta: a regulator of postsynaptic differentiation identified in a screen for synaptically enriched transcripts at the neuromuscular junction. J. Cell Biol. 169:355–366 10.1083/jcb.200411012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P., Lyle K.S., Gierke S., Matov A., Danuser G., Wittmann T. 2009. GSK3beta phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J. Cell Biol. 184:895–908 10.1083/jcb.200901042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer T.T., Misgeld T., Lichtman J.W., Sanes J.R. 2004. Nerve-independent formation of a topologically complex postsynaptic apparatus. J. Cell Biol. 164:1077–1087 10.1083/jcb.200401115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansbergen G., Grigoriev I., Mimori-Kiyosue Y., Ohtsuka T., Higa S., Kitajima I., Demmers J., Galjart N., Houtsmuller A.B., Grosveld F., Akhmanova A. 2006. CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5beta. Dev. Cell. 11:21–32 10.1016/j.devcel.2006.05.012 [DOI] [PubMed] [Google Scholar]
- Meier T., Marangi P.A., Moll J., Hauser D.M., Brenner H.R., Ruegg M.A. 1998. A minigene of neural agrin encoding the laminin-binding and acetylcholine receptor-aggregating domains is sufficient to induce postsynaptic differentiation in muscle fibres. Eur. J. Neurosci. 10:3141–3152 10.1046/j.1460-9568.1998.00320.x [DOI] [PubMed] [Google Scholar]
- Miller P.M., Folkmann A.W., Maia A.R., Efimova N., Efimov A., Kaverina I. 2009. Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat. Cell Biol. 11:1069–1080 10.1038/ncb1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missias A.C., Mudd J., Cunningham J.M., Steinbach J.H., Merlie J.P., Sanes J.R. 1997. Deficient development and maintenance of postsynaptic specializations in mutant mice lacking an ‘adult’ acetylcholine receptor subunit. Development. 124:5075–5086 [DOI] [PubMed] [Google Scholar]
- Nizhynska V., Neumueller R., Herbst R. 2007. Phosphoinositide 3-kinase acts through RAC and Cdc42 during agrin-induced acetylcholine receptor clustering. Dev. Neurobiol. 67:1047–1058 10.1002/dneu.20371 [DOI] [PubMed] [Google Scholar]
- Ralston E., Lu Z., Ploug T. 1999. The organization of the Golgi complex and microtubules in skeletal muscle is fiber type-dependent. J. Neurosci. 19:10694–10705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegg M.A. 2005. Organization of synaptic myonuclei by Syne proteins and their role during the formation of the nerve-muscle synapse. Proc. Natl. Acad. Sci. USA. 102:5643–5644 10.1073/pnas.0501516102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt N., Akaaboune M., Gajendran N., Martinez-Pena y Valenzuela I., Wakefield S., Thurnheer R., Brenner H.R. 2011. Neuregulin/ErbB regulate neuromuscular junction development by phosphorylation of α-dystrobrevin. J. Cell Biol. 195:1171–1184 10.1083/jcb.201107083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw R.M., Fay A.J., Puthenveedu M.A., von Zastrow M., Jan Y.N., Jan L.Y. 2007. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 128:547–560 10.1016/j.cell.2006.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova T., Slemmer J., Hoogenraad C.C., Lansbergen G., Dortland B., De Zeeuw C.I., Grosveld F., van Cappellen G., Akhmanova A., Galjart N. 2003. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein). J. Neurosci. 23:2655–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treves S., Vukcevic M., Griesser J., Armstrong C.F., Zhu M.X., Zorzato F. 2010. Agonist-activated Ca2+ influx occurs at stable plasma membrane and endoplasmic reticulum junctions. J. Cell Sci. 123:4170–4181 10.1242/jcs.068387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov A.S., Samsonov A., Akhmanova A., Galjart N., Popov S.V. 2007. Microtubule-binding proteins CLASP1 and CLASP2 interact with actin filaments. Cell Motil. Cytoskeleton. 64:519–530 10.1002/cm.20201 [DOI] [PubMed] [Google Scholar]
- Várnai P., Rother K.I., Balla T. 1999. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton’s tyrosine kinase pleckstrin homology domain visualized in single living cells. J. Biol. Chem. 274:10983–10989 10.1074/jbc.274.16.10983 [DOI] [PubMed] [Google Scholar]
- Wang J., Jing Z., Zhang L., Zhou G., Braun J., Yao Y., Wang Z.Z. 2003. Regulation of acetylcholine receptor clustering by the tumor suppressor APC. Nat. Neurosci. 6:1017–1018 10.1038/nn1128 [DOI] [PubMed] [Google Scholar]
- Watanabe T., Wang S., Noritake J., Sato K., Fukata M., Takefuji M., Nakagawa M., Izumi N., Akiyama T., Kaibuchi K. 2004. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev. Cell. 7:871–883 10.1016/j.devcel.2004.10.017 [DOI] [PubMed] [Google Scholar]
- Watanabe T., Noritake J., Kakeno M., Matsui T., Harada T., Wang S., Itoh N., Sato K., Matsuzawa K., Iwamatsu A., et al. 2009. Phosphorylation of CLASP2 by GSK-3beta regulates its interaction with IQGAP1, EB1 and microtubules. J. Cell Sci. 122:2969–2979 10.1242/jcs.046649 [DOI] [PubMed] [Google Scholar]
- Witzemann V., Schwarz H., Koenen M., Berberich C., Villarroel A., Wernig A., Brenner H.R., Sakmann B. 1996. Acetylcholine receptor epsilon-subunit deletion causes muscle weakness and atrophy in juvenile and adult mice. Proc. Natl. Acad. Sci. USA. 93:13286–13291 10.1073/pnas.93.23.13286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Xiong W.C., Mei L. 2010. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 137:1017–1033 10.1242/dev.038711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Shen Q.T., Oristian D.S., Lu C.P., Zheng Q., Wang H.W., Fuchs E. 2011. Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3β. Cell. 144:341–352 10.1016/j.cell.2010.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Luo S., Wang Q., Suzuki T., Xiong W.C., Mei L. 2008. LRP4 serves as a coreceptor of agrin. Neuron. 60:285–297 10.1016/j.neuron.2008.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumbrunn J., Kinoshita K., Hyman A.A., Näthke I.S. 2001. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr. Biol. 11:44–49 10.1016/S0960-9822(01)00002-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.