

Abstract
The objective of this study was to analyze genome-wide differential methylation patterns in maternal leukocyte DNA in early pregnant and non-pregnant states. This is an age and body mass index matched case-control study comparing the methylation patterns of 27,578 cytosine-guanine (CpG) sites in 14,495 genes in maternal leukocyte DNA in early pregnancy (n = 14), in the same women postpartum (n = 14), and in nulligravid women (n = 14) on a BeadChip platform. Transient widespread hypomethylation was found in early pregnancy as compared with the non-pregnant states. Methylation of nine genes was significantly different in early pregnancy compared with both postpartum and nulligravid states (< 10% False Discovery Rate). Early pregnancy may be characterized by widespread hypomethylation compared with non-pregnant states; there is no apparent permanent methylation imprint after a normal term gestation. Nine potential candidate genes were identified as differentially methylated in early pregnancy and may play a role in the maternal adaptation to pregnancy.
Keywords: DNA Methylation, epigenetics, immune, leukocyte, maternal, postpartum, pregnancy
Introduction
Epigenetics is the study of an altered phenotype resulting from changes in a chromosome without alterations in its DNA sequence.1,2 These epigenetic changes can be mediated through post-translational modification of histone tails by methylation, acetylation, ubiquitination and phosphorylation, altered transcription factors, interactions with non-protein coding RNAs that regulate transcription and, perhaps the most studied epigenetic modification, DNA methylation.3
DNA methylation is the addition of a methyl group to the DNA molecule at cytosine-guanine (CpG) sites. Many of these CpG sites are clustered in areas called CpG islands (CPI). There are more than 30,000 islands currently identified in the human genome. Many CPIs are positioned at the 5′ end of a gene in the vicinity of the promoter or the first exon/transcriptional start site. Methylation at the 5′ promoter end of a gene may result in altered chromatin structure, affecting access to the machinery that results in gene transcription.4
DNA methylation is an epigenetic mechanism that modulates gene expression in oncogenesis, in non-cancer pathologies (such as neuro-developmental, psychiatric, cognitive and autoimmune diseases) and in normal mammalian development.4,5 The progression of the human female from the nulligravid state, through pregnancy, to the postpartum state may involve changes in methylation patterns in multiple tissues, including maternal leukocytes, which may regulate implantation, trophoblastic invasion, vasculogenesis and maternal immunotolerance. Genome-wide changes in the methylation profiles in different maternal tissues at different time points during pregnancy have not been previously described.
In this report, we aimed to compare the methylation status of 27,578 CpG sites in 14,495 genes in maternal leukocyte DNA in normal early pregnancy, postpartum and in the nulligravid state. We postulated that the methylation profile in early pregnancy is different from that in the non-pregnant state, the latter being defined either as postpartum in the same individual, or as the non-pregnant state of nulligravid, unrelated women. In addition, we aimed to use differential methylation patterns in maternal leukocyte DNA to identify possible novel candidate genes that may be involved in maternal adaptation to pregnancy.
Results
Characteristics of sample groups
Nulligravid women (n = 14) were matched to women in their first pregnancy (n = 14) by age (nulligravid mean age 25.4; pregnant mean age 24.9) and BMI (nulligravid mean BMI 26.1 kg/m2; pregnant mean BMI 26.9kg/m2). The pregnant women had samples obtained at 9 gestational weeks on average (range 7–15 weeks), and 6–8 weeks postpartum, for a composite total of 42 samples. All women were of Caucasian/White race.
Quality control
Of the 42 samples, 5 were randomly chosen to be run in duplicate on different or the same chips (each chip can hold 12 samples) as technical replicates. The 5 randomly selected technical replicates were from 5 different subjects: 2 at the early pregnancy time point, 2 from the nulligravid controls and 1 from the postpartum time point. They were all randomly allocated to one of the 8 BeadChips—no two technical replicates randomized to the same BeadChip—and all chips were processed at the same time. The 5 technical replicates were highly reproducible, as estimated by a Pearson correlation coefficient (r2 > 0.994). In other words, 99.4% of concordance of the β values can be accounted for between the two replicates. Lastly, whole genome amplification (WGA) DNA was used as a negative external control, and was also run in duplicate and performed as expected.
Review of the 8 types of embedded control probes: staining, hybridization, target removal, extension, bisulfite conversion, G/T mismatch, negative control and non-polymorphic controls demonstrated that overall assay performance was as expected. Overall sample quality was determined by the total number of detected CpGs at detection p value < 0.05, the average detection p-value across all CpG sites, and the distribution of average β for all CpGs. CpGs that were not reliably detected in 95% samples based on detection p values (n = 475), or samples that did not have > 95% call for each CpG site based on the average detection p-value (n = 0), were excluded. All 42 patient samples and 98% or 27,103 CpG sites met quality control standards and were included for analysis.
All samples were randomly allocated to 8 BeadChips and run concurrently, thus eliminating issues of “batch effect.” However, differences related to chip effect (first chip vs. last chip) or placement on the chip (left vs. right, for example) can exist. The initial principal components analysis (PCA) of our entire cohort did not detect any significant chip effect, except with one chip that tended to separate from the others. This minor chip effect was not seen in other chip effect assessment tools, such as unsupervised clustering. As normalization/batch correction made a negligible difference in the results, the original data were presented in this paper.6
Differentially methylated CpG sites genome-wide
Differential methylation in the pregnant vs. the postpartum state
In the longitudinal comparison of the genome-wide methylation profiles of women early in their pregnancies with those at 6 weeks postpartum, 1524 of 27,103 CpG sites displayed differential methylation. Of these, 1384 were less methylated and 140 were more methylated in the first half of pregnancy than at six weeks postpartum. Using the false discovery rate (FDR) method of correction for multiple comparisons, 574 met the 10% FDR cutoff and 275 sites met the 5% cutoff. Of note, these significantly differentially methylated genes were located on all 22 autosomes and the X chromosome as well. (Figs. 1, 2 and Table S1)
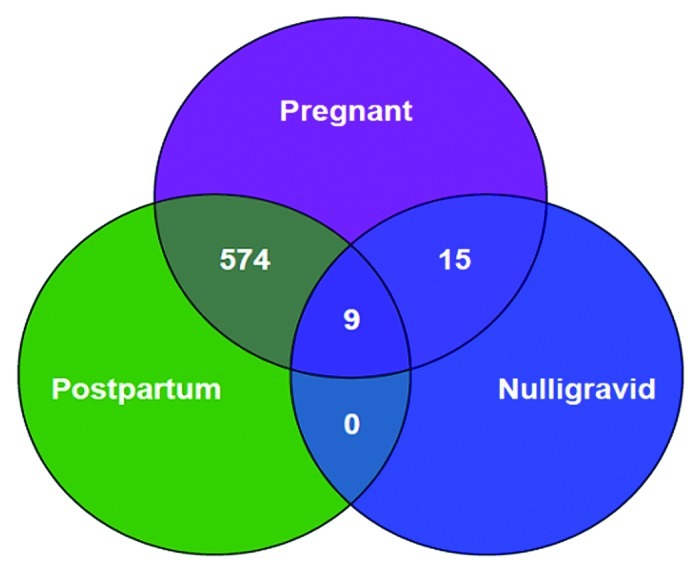
Figure 1. Venn diagram showing the number of CpG sites that demonstrate differential methylation when comparing groups. Differential methylation is defined as a mean absolute methylation level difference > 2% and significance was tested by using a two-group Students t-test (for nulligravid vs. either early pregnant or postpartum), or paired t-test (early pregnant vs. postpartum), after correction for multiple comparisons with FDR < 0.1. The power of the pair-wise comparison is demonstrated by the large number of significant CpG sites in the pregnant vs. postpartum comparison, as contrasted with the pregnant vs. nulligravid comparison. There were 9 genes differentially methylated in the early pregnant vs. postpartum state, which were also differentially methylated in the early pregnant vs. nulligravid state

Figure 2. A volcano plot illustrating the global methylation differences between pregnant and postpartum samples. Each dot represents a comparison of mean methylation at an individual CpG site. The x-axis is the methylation mean difference (pregnant - postpartum). The two dashed lines delineate minus 2% and plus 2% methylation mean difference. The y axis is the negative Log10 of the p value; dots in red have an FDR q value < 0.1, those in green have a p value < 0.05, and black sites have non-significant differences. Note the large number of highly significant differentially hypomethylated sites in pregnancy (arrow).
Differential methylation in the pregnant vs. the nulligravid state
In the comparison of the genome-wide methylation profiles of women early in their pregnancies with the methylation profiles in nulligravid women, 1407 of 27,103 CpG sites had differential methylation. Of these, 1031 CpG sites were less methylated and 376 were more methylated in the pregnant women compared with the nulligravid women. After correction for multiple comparisons, 15 met the 10% FDR cutoff. (Fig. 1)
Differential methylation between two non-pregnant states: nulligravid vs. postpartum
When postpartum samples were compared with nulligravid samples, 699 CpG sites were differentially methylated. Of these, 620 sites were more methylated postpartum and 79 were less methylated postpartum, compared with the nulligravid samples. No CpG site met the FDR cutoff of 10% (Fig. 1).
Identification of differentially methylated CpG sites in candidate genes
To identify differentially methylated novel candidate genes associated with early pregnancy, two separate comparisons were made between early pregnant and the non-pregnant states: nulligravid and postpartum. Genes that were statistically significantly differentially methylated and found to be common between the two comparisons are potential candidate genes involved in maternal adaptation to pregnancy. This analysis revealed that nine genes were differentially hypomethylated in pregnancy, at a q value of < 0.1 in both comparisons. (Fig. 1 and Table 1)
Table 1. Genes with at least one CpG site differentially methylated in early pregnancy compared with both non–pregnancy groups (nulligravid and postpartum) that were significant at the 10% FDR level (q < 0.10).
| Gene Symbol |
Mean methylation Nulligravid |
q value (Nulligravid vs. Pregnant) |
Mean Methylation Pregnant |
q value (Postpartum vs. Pregnant) |
Mean Methylation Postpartum |
|---|---|---|---|---|---|
|
TREML1 |
0.213 |
0.086 |
0.166 |
0.011 |
0.213 |
|
NDUFS2 |
0.338 |
0.086 |
0.267 |
0.025 |
0.333 |
|
IL1R2 |
0.251 |
0.086 |
0.185 |
0.044 |
0.250 |
|
PTPRJ |
0.396 |
0.086 |
0.323 |
0.053 |
0.380 |
|
SPAG4 |
0.551 |
0.086 |
0.464 |
0.011 |
0.557 |
|
HPR |
0.459 |
0.086 |
0.387 |
0.032 |
0.458 |
|
CCIN |
0.401 |
0.089 |
0.285 |
0.044 |
0.389 |
|
PC |
0.584 |
0.097 |
0.514 |
0.092 |
0.578 |
| CTGLF1 | 0.824 | 0.098 | 0.788 | 0.025 | 0.824 |
Discussion
Our study indicates that altered methylation, favoring hypomethylation, can be found in maternal leukocyte DNA in early pregnancy compared with non-pregnant states. As differentially methylated genes are present on every chromosome, our results further suggest that this hypomethylation is widespread across the genome. In addition, the non-pregnant states (whether nulligravid or postpartum) have demonstrated similar methylation patterns. Taken together, our findings suggest that altered methylation in leukocyte DNA may be a transient epigenetic mechanism by which maternal immunotolerance and adaptation to pregnancy occurs.
In addition to describing global patterns of methylation, the Illumina Human Methylation-27 Assay can identify possible candidate genes involved in maternal adaptation to pregnancy via altered methylation profiles. Among nine genes that had statistically significant differences in the early pregnant compared with both non-pregnant states, two genes have been previously recognized as important in pregnancy. IL1R2 (Interleukin 1 Receptor Type 2) is a cytokine receptor decoy, which binds interleukin 1 polypeptides released from macrophages, thus antagonizing their proinflammatory actions.7 Our results showed a 6.5% decrease in methylation of a CpG site in the IL1R2 gene (change in β value from 0.250 to 0.185). We did not measure gene transcription or protein levels; however, decreased methylation could correlate with an increase in the availability of the IL1R2 receptor decoy, if it leads to an increase in gene expression. Although there are no studies quantifying a change in the protein level of IL1R2 in maternal sera associated with the pregnant state, it is well accepted that the IL1 family as a whole plays an important role in the establishment and progression of normal pregnancy. For example, interleukin 1 β (IL1 b) levels are known to increase at the maternal–fetal interface at the time of implantation and early placentation,8,9and also are increased in high sensitivity assays of normal pregnant sera at 14–16 weeks compared with known values in healthy non-pregnant controls.10,11 It is interesting to note that several other genes in the IL1 family are differentially methylated in early pregnancy at a q value of < 0.05 in our study, also suggesting an important role of this cytokine family in maternal adaptation to pregnancy (Table S1).
HPR (haptoglobin-related protein) is a secreted plasma protein associated with high-density lipoprotein (HDL) particles, part of the innate immune response.7 We saw that a decrease in the β value from 0.458 to 0.387 at a CpG site in the HPR gene was associated with early pregnancy vs. the non-pregnant states. Others have noted increased HPR expression in pregnancy,12-14 and although we did not measure either mRNA or protein levels, the altered DNA methylation we have shown may be one mechanism by which this occurs.
Other genes have not been specifically studied within the context of pregnancy, but are involved in cellular processes known to be critical to reproduction, such as the immune response (TREML1, triggering receptor expressed on myeloid cells-like 1), gametogenesis (CCIN, calicin; SPAG4, sperm associated antigen 4), and cell cycle control and cell differentiation (PTPRJ, protein tyrosine phosphatase, receptor type, J). Other differentially methylated genes include PC (pyruvate carboxylase), a housekeeping gene involved in gluconeogenesis, lipogenesis and insulin secretion; NDUFS2 (NADH dehydrogenase/ubiquinone Fe-S protein 2), a core subunit of the mitochondrial membrane respiratory chain; and AGAP4/CTGLF1 whose function is unknown.7
Limited work has addressed DNA methylation changes in pregnancy. Most have focused on specific genes,15-19 rather than a genome-wide approach,20,21 and have concentrated on disorders of pregnancy, such as preeclampsia,16,19-22 intrauterine growth restriction,17 in vitro fertilization23 and gestational diabetes,24 rather than normal pregnancy.15,25 The changes in methylation have been described primarily from fetal-derived tissues, such as placenta15,19-21,23-25 or free fetal DNA in maternal plasma,16,19 and only rarely in maternal DNA.25 Our data reflect the methylation changes that occur in the DNA of white blood cells, which represent a major component of the buffy coats that were used in our experiments.
Leukocytes play a major role in the mechanisms of proliferation, invasion, inflammation, and immune tolerance, which are all critical for maternal adaptation to pregnancy and normal placental and fetal development.26 Normal pregnancy is a state of enhanced innate immune response. Natural killer cells and macrophages are key components in regulating trophoblastic invasion and angiogenesis, while neutrophils increase in both number, as well as cytotoxic activity in the systemic circulation. Normal pregnancy is also a state of altered adaptive immunity, with a shift from a Th1/Th2 balance toward the helper T cell 2 state. Depressed Th1 cell mediated immunity and a relative anti-inflammatory state in mid-gestation may act synergistically in preventing rejection of the semi-allogenic fetus.27
Epigenetic mechanisms, such as DNA methylation, may play a role in this maternal immune adaptation. As the presence of a methyl group at key CpG sites in CpG islands located in or near promoters can lead to structural changes in the DNA molecule that block gene transcription, the widespread hypomethylation we observed in maternal leukocyte DNA in pregnancy may indicate that many of these genes are either turned on or upregulated to facilitate maternal adaptation to trophoblast invasion and gestational tolerance and support. These changes do not appear to persist after normal pregnancy ends.
The major limitation of our study is the relatively small number of women comprising each group. There are hundreds of other genes that were also significantly differentially methylated (even after correction for multiple comparisons) in the early pregnant to postpartum comparison; many of these same genes were also significant, with p values < 0.001 in the nulligravid to pregnant comparison, but did not remain statistically significant after the conservative multiple comparisons adjustment. We hope to replicate the present findings in a larger, independent sample, and to identify other differentially methylated genes that are crucial to human reproduction.
Many factors can influence the establishment and maintenance of DNA methylation over time. We controlled for the putative factors that may influence global DNA methylation4 by matching the two groups of women for age (within 5 y), tobacco status (all non-smokers), and BMI (within 5 kg/m2). All women were free of major medical co-morbidities. It is important to emphasize that when defining “significantly different,” it is unclear what level of differential methylation is clinically meaningful, as this area of epigenetics is currently in its infancy. We chose an absolute change of 2% as the lower limit for this initial exploratory analysis, as our goal was not to exclude any statistically significant genes, accepting that some of these changes might not be clinically significant. This represented a relative change in methylation at any given CpG site, ranging from 2% to > 100%. For example, a specific CpG site in the TREML1 gene is methylated 21.3% of the time in non-pregnant subjects (postpartum and nulligravid), but only 16.6% in pregnancy, representing a 4.7% absolute difference, but a 22% relative change in methylation. Although the precise relationship between altered methylation and resultant gene expression is complex and poorly understood, we believe it is very plausible that these differences are not only statistically, but clinically significant as well.
Other limitations include the possibility of confounding by single nucleotide polymorphisms (SNPs) in the CpG sites interrogated in the methylation assay, which may falsely implicate or obscure differential methylation patterns in individual genes when comparisons are made between different groups of individuals, such as our pregnant cases and nulligravid controls. However, this should not impact our paired comparison of pregnant and postpartum samples in the same individual, as the primary sequence should be the same. None of our nine potential candidate genes have known SNPs at the interrogated CpG sites. In future studies, we plan to use the latest version of the Illumina chip, the Infinium HumanMethylation450 BeadChip, which takes SNPs into account when analyzing output data.
Lastly, the identification of nine potential candidate genes involved in maternal adaptation to normal early pregnancy requires replication before a true association can be confirmed. As we have learned with genome-wide association studies (GWAS) for SNPs the nature of genome-wide platform analysis can lead to false positive associations, despite appropriate correction for multiple comparisons.28 Traditionally, this is done through exact replication in an independent data set, although the question of what should be the “gold standard” for confirming an association29 and the applicability to epigenome-wide association studies is still in question.30 In future work, we plan to not only repeat genome-wide analysis on a larger independent sample, but we will utilize pyrosequencing to confirm altered methylation in individual genes, and explore downstream transcriptional and translational implications via gene expression arrays and protein product quantification.
Despite these limitations, a major strength of our study is the paired longitudinal sampling in pregnancy and, then again, during postpartum, such that women serve as their own non-pregnant controls. The statistical power of this approach is evident, as 574 CpG sites were significant even after correction for multiple comparisons.
Our data provide initial evidence that early pregnancy is associated with altered maternal leukocyte DNA methylation, primarily hypomethylation, in genes across all chromosomes, compared with non-pregnancy. We identified nine genes with altered DNA methylation and postulate that such epigenetic modification may be partially responsible for maternal adaptation to pregnancy. We also found no permanent methylation imprint after a normal term gestation. Whether pathologic pregnancy states, such as preeclampsia, may have a distinct methylation profile compared with normotensive pregnancy will be the subject of our future research. The analysis of genome-wide differential methylation patterns in both normal pregnancy and in pregnancy-associated disease states is a promising new and powerful tool to identify novel genes involved in these processes.
Materials and Methods
This study was approved by the Mayo Clinic Institutional Review Board and informed consent was obtained prior to enrollment. The methylation profiles from 14 primigravid patients at < 16 gestational weeks (GW) were compared with their respective profiles 6 weeks postpartum (the longitudinal arm of the study). In addition, the < 16 GW methylation profiles were compared with those of non-pregnant, nulligravid women who were age and body mass index (BMI) matched (the cross-sectional arm of the study). This paper focuses on 42 samples plus technical replicates selected from a larger study (total samples n = 96) of normotensive and preeclamptic women sampled at different time points in gestation. Both the source of the technical replicates and their positions on the BeadChips were determined randomly.
Source of pregnant/postpartum DNA
Plasma and buffy coat samples from maternal blood were collected longitudinally from 14 primigravid pregnant women at < 16 GW and at the 6 week postpartum visit; the samples were stored at -80°C until assayed.
Source of nulligravid DNA
For cross-sectional comparison, we utilized the Mayo Clinic Biobank to obtain DNA from 14 normotensive nulligravid women (defined as no prior pregnancy; no history of infertility), who were age and BMI matched to the primigravid pregnant women. Nulligravid women were selected due to the possibility of persistent methylation changes in women who previously had been pregnant.
Clinical Data
All medical records were abstracted for data regarding gravidity and parity, ethnicity, age, BMI, and confirmed absence of medical or obstetrical co-morbidities, including prior malignancy, chronic hypertension, diabetes mellitus, chronic kidney disease, tobacco use, and preeclampsia.
DNA extraction and processing
Genomic DNA was extracted using the QIAgen Flexigene Reagent kit and AutoGenFlex DNA purification kit from the frozen buffy coats, which were collected in a 10 ml EDTA tube, quantified with a NanoDrop spectrophotometer, normalized with standard PicoGreen methodology, and plated in 1000 ng aliquots. Bisulfite modification was performed using the EZ DNA Methylation Kit (Zymo Research).
Methylation assay
The Illumina Human Methylation-27 Assay (Illumina) was used to evaluate the methylation status of 27,578 CpG sites in 14,495 genes. A total of 96 samples were randomly allocated to 8 BeadChips and run at the same time, and this paper describes a subset.
Data analysis
The Illumina GenomeStudio (version 2010.2) with methylation module (version 1.7) was used to process the raw data from BeadArray Reader. This generated a mean β value for each CpG site between 0 and 1, representing the average ratio of methylated cytosine residues to the total number of cytosine residues at that site for each sample. A detection p value measuring the reliability of the mean β was also calculated for each CpG.
A series of quality control metrics were applied to ensure that the data were of high quality. Overall sample performance was determined by the total number of detected CpGs, the average detection p-value across all CpG sites, and the distribution of average β value for all CpGs. Call rates for each CpG site (across samples) and sample (across all markers) were determined. Methylation sites and samples were excluded if the unreliable call rate (detection p value > 0.05) was greater than 5%.
Additional quality assessment of the array was conducted using the “Control Dashboard” in the software package which includes a graphical inspection of the 8 types of embedded control probes: staining, hybridization, target removal, extension, bisulfite conversion, G/T mismatch, negative control and non-polymorphic controls.
All samples were modified, plated, and run concurrently, so no batch effect should occur. However, even when processed at the same time, BeadChips can have variation in assay integrity leading to the “chip” effect. Data were examined for the chip effects using various approaches, including principal components analysis (PCA) and unsupervised hierarchical clustering.31,32
Data were analyzed as quantitative variables. A two-group Student’s t-test was used for cross-sectional comparison of the mean methylation level at each CpG site between the nulligravid vs. either early pregnant or postpartum samples. When the mean methylation value at each CpG site was compared longitudinally in the same patient at the early pregnant (< 16 GW) and postpartum time points, the paired Student’s t test was used. To characterize global differences in methylation between groups, differential methylation at any CpG site was defined as significant when there was > 2% absolute mean methylation difference and p < 0.05.
To identify novel candidate genes that may be involved in maternal adaptation to pregnancy, a more stringent correction for multiple comparisons was performed on all the CpG sites found to be differentially methylated in the longitudinal early pregnancy and postpartum comparison using the false discovery rate (FDR) method. This method, commonly used in genomics when large scale multiple testing is performed, produces a q value, which is the expected proportion of false discoveries among all results deemed significant.33
This candidate gene identification process was repeated with a second independent cross-sectional comparison between the nulligravid and early pregnancy samples. The CpG sites that were differentially methylated (defined as q < 0.10) and were common to both comparisons were deemed potential novel candidate genes.
The GEO submission ID for our dataset is GSE37722.
Supplementary Material
Acknowledgments
We thank the Mayo Clinic Center for Individualized Medicine for access to the Biobank samples. This study was approved by the Mayo Clinic Institutional Review Board and informed consent was obtained prior to enrollment.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was funded by the Mayo Clinic Foundation: Mary Kathryn and Michael B. Panitch Career Development Award and K08HD051714 (Vesna D. Garovic) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health and Human Development or the National Institutes of Health.
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/20388
References
- 1.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–8. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 2.Richards EJ. Inherited epigenetic variation--revisiting soft inheritance. Nat Rev Genet. 2006;7:395–401. doi: 10.1038/nrg1834. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 4.Tost J. DNA methylation: an introduction to the biology and the disease-associated changes of a promising biomarker. Mol Biotechnol. 2010;44:71–81. doi: 10.1007/s12033-009-9216-2. [DOI] [PubMed] [Google Scholar]
- 5.Feinberg AP. Genome-scale approaches to the epigenetics of common human disease. Virchows Arch. 2010;456:13–21. doi: 10.1007/s00428-009-0847-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun Z, Chai HS, Wu Y, White WM, Donkena KV, Klein CJ, et al. Batch effect correction for genome-wide methylation data with Illumina Infinium platform. BMC Med Genomics. 2011;4:84. doi: 10.1186/1755-8794-4-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.GeneCards (Internet). Rehovet: Weizmann Institute of Science; 1996 2012. www.genecards.org.
- 8.Librach CL, Feigenbaum SL, Bass KE, Cui TY, Verastas N, Sadovsky Y, et al. Interleukin-1 beta regulates human cytotrophoblast metalloproteinase activity and invasion in vitro. J Biol Chem. 1994;269:17125–31. [PubMed] [Google Scholar]
- 9.Herrmann-Lavoie C, Rao CV, Akoum A. Chorionic gonadotropin down-regulates the expression of the decoy inhibitory interleukin 1 receptor type II in human endometrial epithelial cells. Endocrinology. 2007;148:5377–84. doi: 10.1210/en.2007-0368. [DOI] [PubMed] [Google Scholar]
- 10.Chow SS, Craig ME, Jones CA, Hall B, Catteau J, Lloyd AR, et al. Differences in amniotic fluid and maternal serum cytokine levels in early midtrimester women without evidence of infection. Cytokine. 2008;44:78–84. doi: 10.1016/j.cyto.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Product Information and Manual: Human IL-1B High Sensitivity ELISA BMS224HS. Vienna: eBioscience; 2010. [Google Scholar]
- 12.Kuhajda FP, Piantadosi S, Pasternack GR. Haptoglobin-related protein (Hpr) epitopes in breast cancer as a predictor of recurrence of the disease. N Engl J Med. 1989;321:636–41. doi: 10.1056/NEJM198909073211003. [DOI] [PubMed] [Google Scholar]
- 13.Kuhajda FP, Katumuluwa AI, Pasternack GR. Expression of haptoglobin-related protein and its potential role as a tumor antigen. Proc Natl Acad Sci U S A. 1989;86:1188–92. doi: 10.1073/pnas.86.4.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffman LH, Winfrey VP, Blaeuer GL, Olson GE. A haptoglobin-like glycoprotein is produced by implantation-stage rabbit endometrium. Biol Reprod. 1996;55:176–84. doi: 10.1095/biolreprod55.1.176. [DOI] [PubMed] [Google Scholar]
- 15.Avila L, Yuen RK, Diego-Alvarez D, Peñaherrera MS, Jiang R, Robinson WP. Evaluating DNA methylation and gene expression variability in the human term placenta. Placenta. 2010;31:1070–7. doi: 10.1016/j.placenta.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 16.Bellido ML, Radpour R, Lapaire O, De Bie I, Hösli I, Bitzer J, et al. MALDI-TOF mass array analysis of RASSF1A and SERPINB5 methylation patterns in human placenta and plasma. Biol Reprod. 2010;82:745–50. doi: 10.1095/biolreprod.109.082271. [DOI] [PubMed] [Google Scholar]
- 17.Bourque DK, Avila L, Peñaherrera M, von Dadelszen P, Robinson WP. Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta. 2010;31:197–202. doi: 10.1016/j.placenta.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Chiu RW, Chim SS, Wong IH, Wong CS, Lee WS, To KF, et al. Hypermethylation of RASSF1A in human and rhesus placentas. Am J Pathol. 2007;170:941–50. doi: 10.2353/ajpath.2007.060641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao A, Cheng Y, Li X, Li Q, Wang L, Xu J, et al. Promoter hypomethylation of COMT in human placenta is not associated with the development of pre-eclampsia. Mol Hum Reprod. 2011;17:199–206. doi: 10.1093/molehr/gaq092. [DOI] [PubMed] [Google Scholar]
- 20.Yuen RK, Avila L, Peñaherrera MS, von Dadelszen P, Lefebvre L, Kobor MS, et al. Human placental-specific epipolymorphism and its association with adverse pregnancy outcomes. PLoS One. 2009;4:e7389. doi: 10.1371/journal.pone.0007389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuen RK, Peñaherrera MS, von Dadelszen P, McFadden DE, Robinson WP. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet. 2010;18:1006–12. doi: 10.1038/ejhg.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kulkarni A, Chavan-Gautam P, Mehendale S, Yadav H, Joshi S. Global DNA methylation patterns in placenta and its association with maternal hypertension in pre-eclampsia. DNA Cell Biol. 2011;30:79–84. doi: 10.1089/dna.2010.1084. [DOI] [PubMed] [Google Scholar]
- 23.Katari S, Turan N, Bibikova M, Erinle O, Chalian R, Foster M, et al. DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum Mol Genet. 2009;18:3769–78. doi: 10.1093/hmg/ddp319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouchard L, Thibault S, Guay SP, Santure M, Monpetit A, St-Pierre J, et al. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care. 2010;33:2436–41. doi: 10.2337/dc10-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Müller HM, Ivarsson L, Schröcksnadel H, Fiegl H, Widschwendter A, Goebel G, et al. DNA methylation changes in sera of women in early pregnancy are similar to those in advanced breast cancer patients. Clin Chem. 2004;50:1065–8. doi: 10.1373/clinchem.2003.030387. [DOI] [PubMed] [Google Scholar]
- 26.Veenstra van Nieuwenhoven AL, Heineman MJ, Faas MM. The immunology of successful pregnancy. Hum Reprod Update. 2003;9:347–57. doi: 10.1093/humupd/dmg026. [DOI] [PubMed] [Google Scholar]
- 27.Mor G, Cardenas I. The immune system in pregnancy: a unique complexity. Am J Reprod Immunol. 2010;63:425–33. doi: 10.1111/j.1600-0897.2010.00836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ioannidis JP, Thomas G, Daly MJ. Validating, augmenting and refining genome-wide association signals. Nat Rev Genet. 2009;10:318–29. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu YJ, Papasian CJ, Liu JF, Hamilton J, Deng HW. Is replication the gold standard for validating genome-wide association findings? PLoS One. 2008;3:e4037. doi: 10.1371/journal.pone.0004037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12:529–41. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–6. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics. 2010;3:33. doi: 10.1186/1755-8794-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storey JD, Tibshirani R. Statistical methods for identifying differentially expressed genes in DNA microarrays. Methods Mol Biol. 2003;224:149–57. doi: 10.1385/1-59259-364-X:149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.