

Abstract
Regulating endothelial cell behavior is a key step in understanding and controlling neovascularization for both pro-angiogenic and anti-angiogenic therapeutic strategies. Here, we characterized the effects of a covalently immobilized peptide mimic of vascular endothelial growth factor, herein referred to as VEGF receptor-binding peptide (VR-BP), on human umbilical vein endothelial cell (HUVEC) behavior. Self assembled monolayer arrays presenting varied densities of covalently immobilized VR-BP and varied densities of the fibronectin-derived cell adhesion peptide Gly-Arg-Gly-Asp-Ser-Pro (GRGDSP) were used to probe for changes in HUVEC attachment, proliferation and tubulogenesis. In a soluble form, VR-BP exhibited pro-angiogenic effects in agreement with previous studies, indicated by increases in HUVEC proliferation. However, when presented to cells in an insoluble context, covalently immobilized VR-BP inhibited several pro-angiogenic HUVEC behaviors, including attachment and proliferation, and also inhibited HUVEC response to soluble recombinant VEGF protein. Furthermore, substrates with covalently immobilized VR-BP also modulated HUVEC tubulogenesis when a matrigel overlay assay was used to provide cells with a pseudo-three dimensional environment. Taken together, these results demonstrate that the context in which ligands are presented to cell surface receptors strongly influences their effects, and that the same ligand can be an agonist or an antagonist depending on the manner of presentation to the cell.
Keywords: self-assembled monolayers, RGD, VEGF, umbilical vein endothelial cells, array
Introduction
The vascular endothelial growth factor (VEGF) family of glycoproteins represents a key class of soluble cytokines that regulate neovascularization in physiological and pathophysiological processes. VEGF binding to receptor tyrosine kinases expressed by endothelial cells can result in activation of intracellular signaling cascades that direct cellular behavior. In humans, the VEGF family consists of four VEGF-A isoforms (121, 165, 189, 206 amino acids), as well as VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF), which bind to VEGF receptors including VEGFR-1 (also known as Flt-1), VEGFR-2 (also known as Flk-1 or KDR), and VEGFR-3.[1, 2] In the adult vascular endothelium VEGFR-2 and VEGF-A have been identified as major regulators of endothelial cell proliferation, survival, migration and vascular permeability. VEGF-A is a homodimeric protein with symmetrical receptor-binding sites that promote receptor dimerization, phosphorylation, and subsequent signal transduction. As a result, development of pro- and anti-angiogenic therapies have focused on regulating soluble VEGF-A concentrations as well as methods to interfere with VEGFR-2 dimerization or activation.[3, 4]
Peptide analogues of VEGF have been investigated as modulators of VEGF signaling, and peptide fragments derived from a single VEGFR binding motif of VEGF-A have been used to antagonize VEGF activity.[5] For example, amino acid sequence 116–159 of VEGF-A165 has been shown to bind to VEGFR without promoting receptor dimerization and activation, resulting in VEGF inhibition.[5] Similarly, VEGF-A mutants in which one wild type monomer forms a heterodimer with a mutant monomer results in a molecule that can bind VEGFR-2 but cannot promote receptor dimerization and activation.[6–8] However, this effect is not universal, as some receptor-binding peptide fragments have been shown to stimulate VEGF signaling and, in some cases, have been reported to synergize with VEGF-A signaling to create more robust biological responses in vitro and in vivo.[9, 10] Therefore, previous studies suggest that VEGF receptor-binding ligands can be used to inhibit or to stimulate VEGF signaling, and there is a need to more clearly understand the mechanisms by which VEGFR-binding molecules can differentially influence VEGF signaling.
One particularly well-studied VEGFR-binding peptide, designed to mimic the VEGFR binding site on VEGF-A (KLTWQELYQLKYKGI, here referred to as VR-BP), has been shown to stimulate VEGF signaling and also synergize with soluble VEGF-A to promote increased endothelial cell proliferation and tubule formation.[9–11] Furthermore, when tested in normotensive wistar kyoto rats, soluble VR-BP promoted increased angiogenesis in an ischemic hindlimb model, increased wound healing in dorsal full thickness skin wounds, and increased capillary infiltration into subcutaneously implanted matrigel plugs.[10] However, in addition to delivery of this VR-BP via injection, some strategies have attempted to locally regulate neovascularization by delivering it from biomaterials. For example, recently modular versions of VR-BP have been engineered to bind to either collagen or hydroxyapatite biomaterials. Lee and coworkers demonstrated that a modular peptide combining VR-BP and a hydroxyapatite-binding domain bound to hydroxyapatite materials and slowly released over time.[12] When seeded with C166 mouse endothelial cells, patterned coatings of the modular peptide exhibited pro-angiogenic properties, including locally increased endothelial cell proliferation and increased in vitro wound healing response. Similarly, Chan and coworkers demonstrated that a modular peptide combining VR-BP with a collagen-binding domain could generate collagen biomaterials that locally increased HUVEC tubulogenesis and increased endothelial sprouting.[13] Covalent chemistries have also been used to tether VR-BP within hydrogels. Recently, Leslie-Barbick and coworkers demonstrated that when VR-BP was covalently immobilized within poly(ethylene glycol) (PEG) hydrogels using a 3400 Da PEG linker, the peptide increased in vitro HUVEC migration and tubulogenesis and in vivo angiogenesis in a mouse corneal pocket.[14] Taken together, these previous results suggest that VR-BP exhibits pro-angiogenic properties when delivered as a soluble molecule, when reversibly associated with a biomaterial, and when conjugated within a biomaterial via a flexible polymer linkage.
In this current work, we sought to precisely control the manner of VR-BP presentation (soluble versus insoluble), and thereby study the influence of the context on VR-BP effects. To clearly characterize the effects of insoluble VR-BP on endothelial cell behavior we used chemically-defined alkanethiolate self-assembled monolayer (SAM) arrays formed via a stencil-based approach (Figure 1, Supplement Figure 1). SAMs bearing oligo(ethylene-glycol) functionalities prevent non-specific protein interactions, and we hypothesized that SAMs would thus allow us to clearly attribute changes in endothelial cell behavior to the specific peptides presented on the substrate. The results demonstrate that while soluble VR-BP promoted endothelial cell proliferation as demonstrated previously, covalently immobilized VR-BP inhibited endothelial cell attachment and proliferation, and modulated tubulogenesis. Covalently immobilized VR-BP also inhibited the effects of soluble VEGF protein. Taken together, these results suggest that subtle changes in the context in which a ligand is presented to cells can lead to completely different biological responses. Therefore, extending our understanding of how the context of ligand presentation effects cell responses could ultimately lead to improved strategies to induce or inhibit growth factor signaling.
Figure 1.
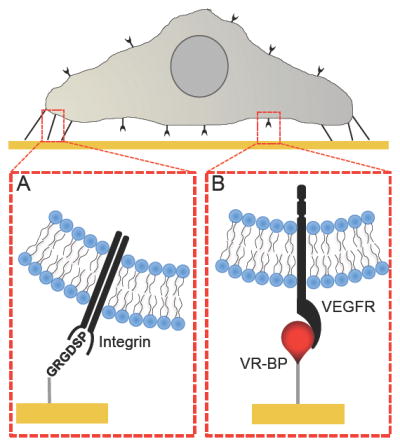
Screening HUVEC microenvironments using alkanethiolate self-assembled monolayer arrays presenting the (A) cell adhesion ligand Gly-Arg-Gly-Asp-Ser-Pro (GRGDSP) and (B) Covalently immobilized VEGF receptor-binding peptide (VR-BP).
Insight, Innovation, Integration
Here we use a chemically-defined cell culture array to demonstrate that a VEGF receptor-binding peptide (VR-BP) elicits pro-angiogenic effects in soluble form, but elicits anti-angiogenic effects when covalently immobilized, including antagonism of soluble VEGF. These results demonstrate that the context in which a growth factor is presented to cells can lead to very different cell responses, ranging from stimulation to inhibition. This novel mechanism to regulate endothelial cell behavior suggests a more broadly applicable approach for context-dependent growth factor regulation.
Experimental methods
Materials and reagents
Carboxylic acid-capped hexa(ethylene glycol) undecanethiol (HS-C11-(O-CH2-CH2)6-O-CH2-COOH) (referred to herein as “HS-C11-EG6-COOH”) was purchased from Prochimia (Sopot, Poland). 11-tri(ethylene glycol)-undecane-1-thiol (HS-C11-(O-CH2-CH2)3-OH) (referred to herein as “HS-C11-EG3-OH”) was synthesized as described elsewhere.[15] Fmoc-protected amino acids and Rink amide MBHA peptide synthesis resin were purchased from NovaBiochem (San Diego, CA). Hydroxybenzotriazol (HOBt) was purchased from Advanced Chemtech (Louisville, KY). Diisopropylcarbodiimide (DIC) was purchased from Anaspec (San Jose, CA). N-hydroxysuccinimide (NHS), n-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), sodium dodecyl sulfate (SDS), trifluoroacetic acid (TFA), diethyl ether, and deionized ultrafiltered (DIUF) H2O were purchased from Fisher Scientific (Fairlawn, NJ). Triisopropylsilane (TIPS), piperidine, dimethylformamide (DMF), acetone, hexanes, and acetonitrile were purchased from Sigma-Aldrich (St. Louis, MO). Absolute ethanol (EtOH) was purchased from AAPER Alcohol and Chemical Co. (Shelbyville, KY). All purchased items were of analytical grade and used as received. Thin films of 1000 Å Au <111>, 50 Å Ti on 1″ × 3″ X 0.040″ glass slides were purchased from Evaporated Metal Films, Inc. (Ithaca, NY. Cat. No. TA134) while thin films of 100 Å Au <111>, 20 Å Ti on 1″ × 3″ X 0.040″ glass were purchased from Platypus Technologies, LLC (Madison, WI. Cat. No. AU.0100.ALSI).
Peptide synthesis
Standard solid phase Fmoc-peptide synthesis (Fmoc SPPS) was performed using a 316c automated peptide synthesizer (C S Bio, Menlo Park, Ca). Rink amide MBHA resin was used as the solid phase, and HOBt and DIC were used for amino acid activation and coupling. After coupling the final amino acid, a 4-hour incubation in TFA, TIPS, and DIUF (95:2.5:2.5) released the peptide from resin and removed protecting groups. Released peptide was extracted from the TFA/TIPS/DIUF cocktail via precipitation in cold diethyl ether. Lyophilized peptides were analyzed using matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry with a Bruker Reflex II (Billerica, MA). The purity of synthesized peptides was verified to be greater than 90% via HPLC using a C18 analytical column (Shimadzu, Kyoto, Japan) with a gradient of 0–70% H2O + 0.1% TFA/acetonitrile and a flow rate of 0.9 mL/minute. VR-BP and a scrambled control peptide were synthesized with an N-termnal tri-glycine spacer to increase peptide spacing away from the SAM substrate. Furthermore, GWGGRGDSP and GWGGRGESP adhesion and mutant peptides were synthesized with tryptophan bearing spacers to aid in determination of peptide concentration via UV/Vis. Peptide stocks were prepared at 300 μM in PBS as pH 7.4 as determined by absorbance at 280 nm using extinction coefficients outlined by Gill and von Hippel.[16]
Circular dichroism spectroscopy
Circular dichroism (CD) was performed on AVIV Model 202SF CD spectrometer. Wavelength scans of CD signal were recorded with 1 nm intervals and 3 sec averaging times. CD spectra of 75 μM peptide dissolved in PBS (pH 7.4) were obtained using a 0.1 cm path length cuvette at 20°C.
Fabrication of elastomeric stencil
Elastomeric stencils containing arrays of wells were created using soft lithography.[17, 18] Briefly, master molds containing arrays of posts were fabricated from SU-8 (Microchem, Newton, MA) spin-coated silicon wafers using conventional photolithography techniques. Polydimethylsiloxane (PDMS) (Sylgard 184, Dow Corning, Midland, MI) was prepared by mixing a 10:1 ratio of base/curing agent (w/w) followed by degassing for ~30 mins. The degassed mixture was cast over the mold and cured for 4 hrs at 85 °C. Following curing, PDMS stencils were removed from molds and cleaned in hexanes using an overnight Soxhlet extraction.[19] After cleaning, elastomeric stencils were placed in vacuo to remove residual solvent from the Soxhlet extraction process.
Substrate preparation and array fabrication
Gold slides were cut using a diamond scribe and placed into a 150 mm glass Petri dish, covered with EtOH, and sonicated for ~1 min using an ultrasonic bath (Bransonic 1510, Branson, Danbury, CT). Sonicated gold chips were then rinsed with EtOH and blown dry with N2. Bulk chips for infrared analysis were immediately immersed into alkanethiolate solutions while chips for SAM arrays were fabricated using an elastomeric stencil approach. Briefly, an elastomeric stencil containing arrays of 1.1 mm diameter holes was placed on a bare gold substrate to form an array of wells on the gold substrate. Wells were then filled with 1 mM ethanolic alkanethiolate solution and incubated for 10 minutes in a chamber containing a laboratory wipe soaked with ethanol to prevent evaporation during local SAM formation. Alkanethiolate solutions were then aspirated and wells were rinsed with DIUF H2O. Carboxylate groups were then converted to active ester groups by adding a solution of 100 mM NHS and 250 mM EDC in DIUF H2O pH 5.5 to wells and incubated for 10 minutes. After an additional rinse with DIUF H2O, 300 μM solutions of peptide in PBS at pH 7.4 were added to each well and incubated for 1 hr in a humidity controlled chamber to covalently couple peptides to each array spot. After a final rinse with DIUF H2O, regions surrounding array spots were backfilled with HS-C11-EG3-OH. This was achieved by submerging the gold substrate and attached elastomeric stencil in an aqueous 0.1 mM HS-C11-EG3-OH solution (pH 2.0), removing the stencil, and incubating for 10 minutes. Following backfilling, the array was rinsed with 0.1 wt% SDS in DIUF H2O, DIUF H2O, and EtOH and then dried under a stream of N2. Arrays were stored in sterile DIUF H2O at 4°C and used within 48 hrs.
In this SAM array approach, each spot was designed to contain the same total molar density (mol/cm2) of peptide from spot to spot. Therefore, control over individual peptide density was achieved by mixing scrambled VR-BP and mutant GRGESP peptides with functional VR-BP or GRGDSP peptides, respectively. Therefore, in a typical SAM array, SAMs were locally formed within spots using an alkanethiolate mixture of 94% HS-C11-EG3-OH and 6% HS-C11-EG6-COOH to create substrates with a total of 6% carboxylate groups for peptide conjugation. Next, to create a spot presenting 5% GRGDSP and 1% VR-BP, a 300 μM peptide solution with 250 μM GRGDSP and 50 μM VR-BP was used during peptide conjugation. Likewise, to create a spot presenting 2.5% GRGDSP and 0.1% VR-BP, a 300 μM peptide solution with 125 μM GRGDSP, 125 μM GRGESP, 5 μM VR-BP and 45 μM scrambled peptide was used during peptide conjugation. In this manner, the amount of active peptide could be varied between spots while holding total peptide content constant. As mentioned previously, peptide concentrations were easily measured using UV/Vis since all peptides either contained residues that absorbed strongly at 280 nm, or were engineered to contain tryptophan residues in the poly-glycine tail.
Polarization modulation-infrared reflection-absorption spectroscopy
Polarization modulation-infrared reflection-absorption spectroscopy (PM-IRRAS) was used to confirm peptide conjugation, via carbodiimide chemistry, to carboxylate-terminated SAMs. To achieve this, bulk gold chips (1 cm × 3 cm) were prepared following the same steps described in the array fabrication process, but without use of the elastomeric stencil. IR spectra of alkanethiolate SAMs on 1000 Å Au films were obtained using a Nicolet Manga-IR 860 FT-IR spectrometer with a photoelastic modulator (PEM-90, Hinds Instruments, Hillsboro, OR), synchronous sampling demodulator (SSD-100, GWC Technologies, Madison, WI), and a liquid-N2-cooled mercury telluride (MCT) detector. All spectra were obtained at an incident angle of 83° with modulation centered at 1500 cm−1 and 2500 cm−1 to produce spectrum with a range of 1000–3000 cm−1. For each sample, 1000 scans were taken using a resolution of 4 cm−1 per modulation center. Data were acquired as differential reflectance (%ΔR/R) versus wavenumber, and baseline correction was performed as outlined by Skoda et al.[20]
Fluorescent peptide labeling
To visualize and evaluate VR-BP and scrambled peptide conjugation in SAM array spots, Alexa Fluor® 488 sulfodichlorophenol ester (AlexaFluor488 5-SDP ester, Invitrogen, Eugene, OR) was used to label epsilon primary amine groups present in lysine residues of immobilized peptides. To do this, a reaction solution of 100 μM AlexaFluor488 5-SDP ester in 0.15 M sodium bicarbonate solution at pH 8.3 reaction solution was prepared from a 10 mM stock of AlexaFluor488 5-SDP ester in DMSO. SAM arrays were immersed in freshly prepared reaction solution and incubated for 1 hr at RT. After labeling, SAM arrays were rinsed with 0.1% SDS, DUIF H2O, and EtOH and then dried under a stream of N2. A GE Healthcare Typhoon Trio Variable Mode Imager was used to scan SAM arrays containing fluorescently-labeled peptide. Fluorescent intensity was quantified using Image J (ImageJ, Freeware, NIH, Bethesda, MD) imaging software. Normalization was achieved by scaling fluorescent intensity to the average maximal intensity for each peptide, respectively.
Cell Culture
Passage 2 human umbilical vein endothelial cells were expanded at low density (less than 70% confluence) on tissue culture polystyrene to no more than 14 population doublings. During HUVEC expansion, cells were cultured in medium 199 (m199, Mediatech, Manassas, VA) containing 1% penicillin/streptomycin (Hyclone, Logan, UT) and supplemented with Clonetics EGM-2 BulletKit (Lonza Walkersville, Inc., Walkersville, MD) containing Hydrocortisone, hFGF-B, VEGF, R3-IGF-1, Ascorbic Acid, Heparin, FBS, hEGF, GA-1000 growth supplements. NIH 3T3 cells were expanded in Dulbecco’s modified eagles medium (DMEM, Mediatech, Manassas, VA) containing 1% penicillin/streptomycin and supplemented with 5% cosmic calf serum (Hyclone, Logan, UT).
Cell Assays on SAM Array
Before seeding on SAM arrays, HUVECs in cell culture flasks were starved for 24 hours in medium containing 2% FBS and then removed from the plate using a 0.05% trypsin solution and resuspended in m199 with 2% FBS. Cell suspension was seeded onto SAM arrays in sterile polystyrene Petri dishes and incubated in a humid environment at 37 °C and 5% CO2. After allowing cells to attach for ~ 1 hr, arrays were dipped in warm m199 with 2% FBS to remove loosely attached cells and then transferred to a rectangular multidish (Thermo Scientific/Nunc, Rochester, NY) containing warm media with 2% FBS and imaged ~2 hours later serving as “0 hr”. After imaging, media was replaced with m199 with 2% FBS containing varied concentrations soluble factors such as recombinant VEGF (VEGF-A165, R&D systems, Minneapolis, MN) or VR-BP. For inhibition using SU5416, 10 μM SU5416 (Sigma-Aldrich, St. Louis, MO) was present during cell seeding and the 2 hours before imaging but was washed out before addition of VEGF conditions[21]. NIH 3T3 experiments were performed using the same approach, in DMEM with 5% cosmic calf serum for all cases.
Influence of soluble peptide
HUVECs were seeded on SAM arrays with varied densities of GRGDSP and then cultured in m199 with 2% FBS containing no additional growth factor, 10 ng/ml VR-BP (4.8 nM), or 10 ng/mL recombinant VEGF (0.26 nM) for 24 hours. To assess proliferation, a Click-it EdU assay (Invitrogen, Carlsbad, CA) was used to label HUVECs in S-phase as indicated by the manufacturer. Briefly, 5 μL of 10 mM EdU in DMSO (5-ethynyl-2′-deoxyuridine) was added to each well (to achieve a final concentration of 10 μM) and placed in an incubator. After 6 hours, HUVECs were fixed using 4% buffered formalin for 15 minutes, permeabilized using 0.5% Triton X-100 (MP Biomedicals, Aurora, OH) in PBS, and then exposed to a reaction cocktail containing a reactive Alexa Fluor 488 azide to fluorescently label synthesized DNA containing the EdU nucleotide. HUVEC nuclei were then counterstained using 10 ng/mL Hoechst 33342 in PBS for 10 minutes and then imaged to assess the fraction of HUVEC nuclei staining positive for EdU incorporation.
Assaying for non-specific protein adsorption
To assay for non-specific protein adsorption on substrates presenting varied peptide densities, SAMs presenting varied densities of VR-BP or scrambled peptide, but no GRGDSP peptide, were seeded with HUVECs. After allowing HUVECs to attach overnight, staining for the f-actin cytoskeleton was performed as follows: Briefly, cells were fixed using formaldehyde and then permeabilized using 0.1 % Triton X-100. After blocking with 1% bovine serum albumin in PBS, substrates were exposed to a solution of TRITC-conjugated phalloidin in PBS for 60 minutes at room temperature. After a final rinse, arrays were mounted using Prolong Gold Antifade Reagent with DAPI (Invitrogen, Eugene, OR) and imaged using the 15× objective on a Nikon Eclipse Ti equipped with filters for TexasRed and DAPI. Exposure times and gain settings for each filter set were held constant across all samples. Analysis of projected cell area was achieved using NIS Elements software. Briefly, stacked images of HUVECs were thresholded and then automated measurements of area and counts were tabulated. For each spot, average cell projected area was calculated by dividing the thresholded actin staining (red channel) by the total number of nuclei (blue channel) in the same spot.
Real-Time Tubulogenesis Assay
SAM arrays seeded overnight with 50,000 HUVECs/cm2 were rinsed with warm media and then flipped over onto cold matrigel (Growth factor reduced matrigel, BD Biosciences, Bedford, MA) in a rectangular multidish well containing varied concentrations of soluble recombinant VEGF. Matrigel was allowed to gel for 5 minutes, and then the array and matrigel layer were covered with warm m199 with 2% FBS. Arrays were then placed in a TIZ Tokai Hit incubated, humidified stage at 37 °C and 5% CO2 and each array spot was imaged using phase contrast microscopy every 15 minutes for 48 hours. Image analysis was performed using NIS Elements software. The total cell area and length of individual objects were measured in each array spot at 24 hours after seeding. To measure mean length of capillary-like structures, each image was contrasted and thresholded using a standard method to isolate elongated objects for automated length measurements. All objects with a length less than 20 μm were excluded from length measurements. To measure total area occupied by cells, the images were contrasted and thresholded to isolate all cell bodies from the background for automated area measurements. Objects with a length less than 10 μm were excluded from area measurements. Tubulogenesis in each spot was quantified by normalizing the mean length of objects to total cell area in each array spot.
Array Imaging and Quantification
SAM array spots were imaged using a Nikon Eclipse Ti inverted microscope with a 10x PhL objective, equipped with Nikon’s Perfect Focus System. 10X images were stitched using Nikon NIS Elements software to capture entire array spots and cell counting was performed using the cell counting application in NIS Elements. Statistical analysis of all data sets was performed using a two-tailed student’s t-test, where p < 0.05 is used to denote statistical significance.
Results
VR-BP secondary structure and covalent immobilization on SAMs
Previous literature suggests that alpha-helical secondary structure is necessary for the bioactivity of VR-BP.[9, 10] Therefore, circular dichroism and infrared spectroscopic techniques were used to characterize soluble and immobilized peptide secondary structure (Figure 2). In solution, VR-BP (GGGKLTWQELYQLKYKGI) exhibited alpha-helical secondary structure as indicated by negative peaks in CD spectra at 208 nm and 220 nm, while the scrambled peptide sequence (GGGKTKQQKEIYLLWYLG) exhibited random coil structure as indicated by a negative peak at 200 nm (Figure 2A).[9, 12, 13] PM-IRRAS spectra of SAMs presenting VR-BP or scrambled peptide, immobilized via the N-terminus, both exhibited amide I (C=O) stretching bands centered around 1664 cm−1 and amide II (NH) stretching bands centered around 1548 cm−1, indicating the presence of peptide on the substrate (Figure 2B). However, spectra of VR-BP exhibited significantly greater signal in the carboxylic acid absorbance band around 1730 cm−1 when compared to scrambled peptide that may indicate differences in peptide secondary structure.[22, 23] Taken together, these results suggest that VR-BP efficiently conjugates to a SAM substrate and exhibits helical secondary structure, which was the structure originally associated with peptide bioactivity by D’Andrea and coworkers.[9, 11]
Figure 2.
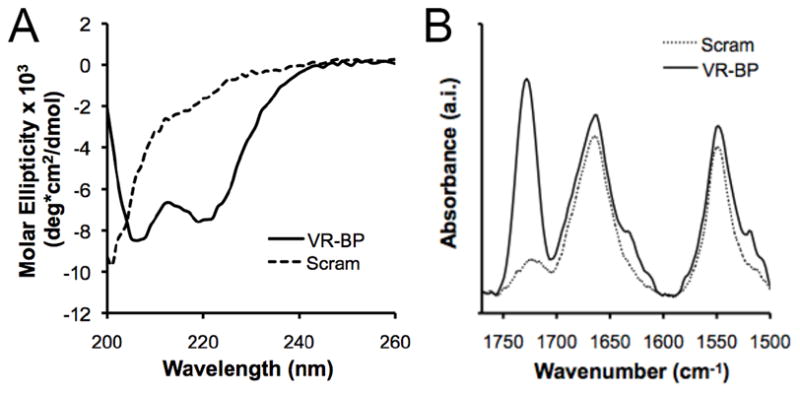
Characterization and immobilization of the vascular endothelial growth factor-receptor binding peptide (VR-BP) on self assembled monolayer substrates. Secondary structure of VR-BP (GGGKLTWQELYQLKYKGI) and the scrambled sequence (GGGKTKQQKEIYLLWYLG): (A) Circular dichroism (CD) spectra of 75 μM peptide in phosphate buffered saline at pH 7.4 at RT and (B) Polarization modulation-infrared reflection-absorption spectroscopy (PM-IRRAS) spectra of peptide conjugated to 2.5% HS-C11-EG6-COOH SAMs.
Effects of soluble VR-BP
HUVECs cultured on SAM arrays presenting the highest GRGDSP density and stimulated with soluble VR-BP exhibited significant increases in proliferation compared to cells without stimulation (Figure 3). Similarly, soluble recombinant VEGF stimulated increased HUVEC proliferation that exceeded both untreated control conditions and soluble VR-BP conditions. These results are similar to observed HUVEC proliferation in response to soluble VR-BP when cultured on gelatin-coated polystyrene plates (Supplement Figure 2) and also validate previously reported observations that VR-BP exhibits pro-angiogenic effects on endothelial cells when presented in soluble form[9, 12]. In all cases, the effects of soluble VR-BP are less substantial than the effects of recombinant VEGF protein. HUVECs also exhibited a dependence on adhesion ligand density, with increased proliferation correlating with increased GRGDSP density. Importantly, conditions presenting the mutant peptide GRGESP did not promote HUVEC attachment (data not shown), indicating that HUVEC-substrate interactions were confined to integrin-GRGDSP binding.
Figure 3.
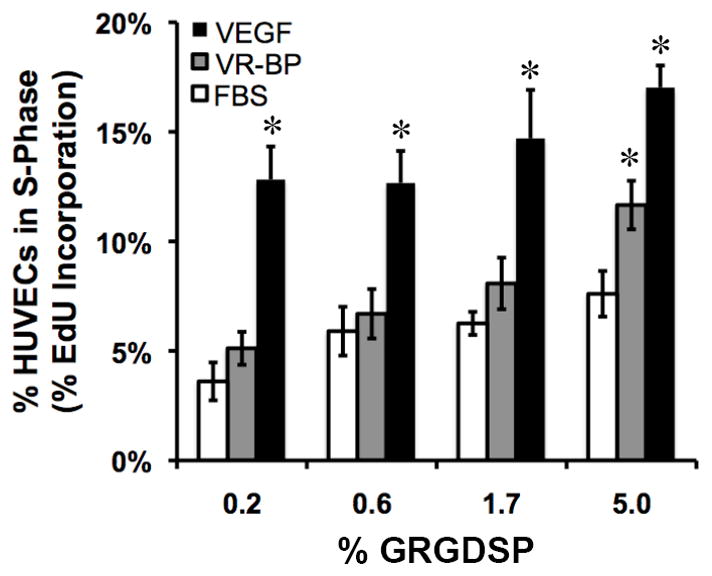
HUVEC response to soluble morphogens. HUVECs starved overnight in 2% FBS were seeded on arrays presenting varied densities of GRGDSP in the presence of 2% FBS alone or with the addition of 10 ng/mL VR-BP (4.8 nM) or 10 ng/mL VEGF (0.26 nM). (Error bars indicate standard error of the mean and asterisk indicate significance compared to FBS alone, p < 0.05)
Generating array spots with multiple peptide mixtures
Peptide immobilization in array spots was visualized using an amine reactive fluorescent molecule to label the lysine residues present in VR-BP and the scrambled peptide (Figure 4). Arrays were generated by reacting peptide solutions containing varied percentages of GRGDSP and VR-BP or scrambled peptide (at a total peptide concentration of 300 μM in PBS pH 7.4) with SAM array spots formed using 5 mole percent HS-C11-EG6-COOH and 95 mole percent HS-C11-EG3-OH. The reactions took place within an elastomeric stencil, such that each spot could incorporate a distinct peptide identity and density (Supplement Figure 1). Fluorescent signal was clearly observed in spots containing VR-BP or scrambled peptide but was not detectable in 0% conditions (containing only GRGDSP) indicating that the fluorescent labeling process was specific to the primary amine residues present in the lysine amino acids of VR-BP and scrambled peptide but not GRGDSP (Figure 4A).[24] Normalization and quantification of fluorescent intensity yielded no significant differences in VR-BP substrate incorporation compared to scrambled peptide, and both peptides exhibited 1:1 correlations between relative fluorescence intensity and the percentage of soluble peptide used during covalent immobilization (Figure 4B). Taken together, these results suggest that VR-BP and scrambled peptide react at similar efficiencies when combined with GRGDSP during conjugation and that covalently immobilized peptide density relates directly to the percentage of peptide present in solution during conjugation.
Figure 4.
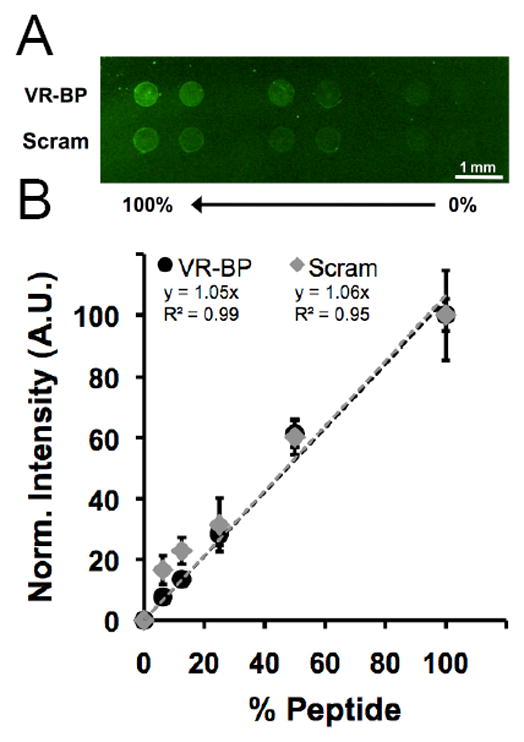
Fluorescent detection of peptide incorporation. (A) VR-BP or scrambled peptide was mixed with GRGDSP at varying percentages and coupled to SAM arrays spots with a fixed density of carboxylate. After coupling to the surface, lysine side chains were labeled using Alexa Fluor 488 sulfodichlorophenol ester and imaged using a fluorescent scanner. (B) Normalized fluorescent intensity (Error bars indicate standard deviation and % peptide refers the the percent of either VR-BP or scrambled peptide present during covalent coupling).
Substrate resistance to non-specific cell attachment
To limit confounding effects of non-specific protein adsorption, SAMs presenting varied densities of VR-BP or scrambled peptide were tested using a cell attachment assay (Figure 5). SAMs presenting a range of different total peptide densities (0–7.5%) were seeded with HUVECs overnight and then stained for f-actin (Figure 5A). Using HUVEC projected cell area as an indication of the level of non-specific cell-substrate interaction, we observed that both VR-BP and scrambled peptide exhibited similar levels of fouling with increasing peptide density (Figure 5B). Furthermore, both peptides exhibited significant changes in HUVEC spreading at peptide densities above 2.5%. Based on these results, VR-BP and scrambled peptide densities were maintained below 2.5% on SAM arrays for all cell culture assays in order to maintain chemically well-defined cell-substrate interactions in this study. Additionally, all SAM arrays presenting cell adhesion peptides and VR-BP or scrambled peptides included conditions with the mutant peptide GRGESP to identify and eliminate array spot conditions that promoted non-specific cell-substrate interactions.
Figure 5.
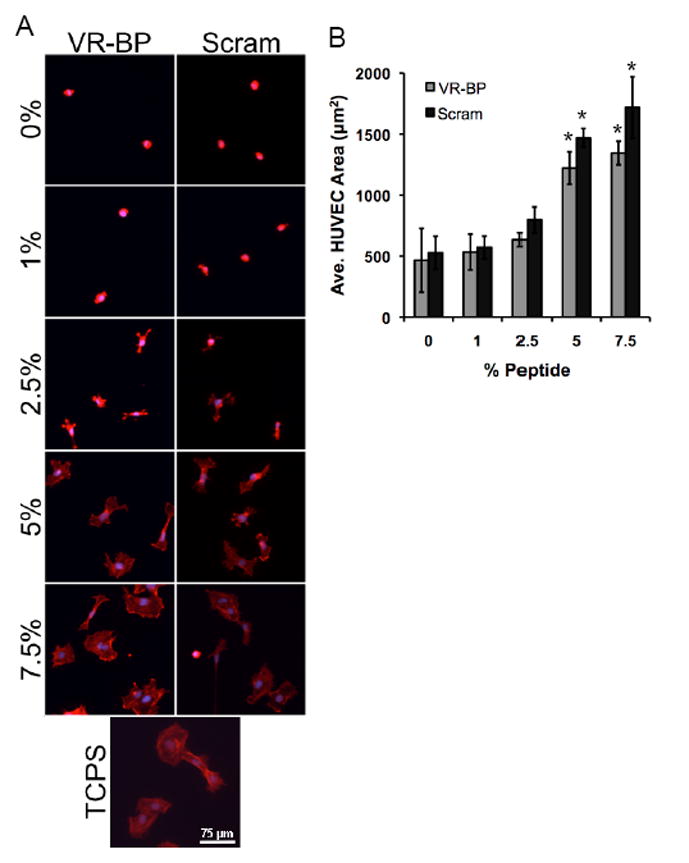
Assaying for non-specific protein adsorption and surface fouling. HUVECs were seeded on SAMs presenting varied densities of VR-BP, scrambled peptide, or tissue culture polystyrene and (A) stained for their f-actin cytoskeleton (f-actin:red, nuclei:blue). (B) HUVEC projected cell area was used as a measurement of surface fouling. (Error bars represent standard error of the mean, asterisk indicates significant increase in projected cell area compared to 0% peptide, p < 0.05) % GRGDSP % GRGDSP
Cell attachment and proliferation on substrates with covalently immobilized VR-BP
To determine whether substrates presenting VR-BP influenced cell attachment, arrays presenting varied densities of GRGDSP and 1% VR-BP or 1% scrambled peptide were seeded with HUVECs (Figure 6). NIH 3T3 fibroblasts were also seeded on similar arrays to examine how cells with no or minimal VEGF-receptor expression [25] behaved compared to HUVECs, which express both VEGFR-1 and VEGFR-2.[1] Immobilized VR-BP significantly decreased HUVEC attachment to array spots presenting the highest GRGDSP densities (Figure 6B), while VR-BP had no effect on NIH 3T3 attachment (Figure 6C), suggesting that the effects of immobilized VR-BP are specific to cells expressing VEGF receptors. Additionally, neither cell type exhibited attachment on conditions presenting 0% GRGDSP, which corresponds to 5% GRGESP, (Figure 6A) indicating that cell adhesion to SAM arrays was chemically-defined, in the sense that it was mediated by specific peptide-receptor interactions.
Figure 6.
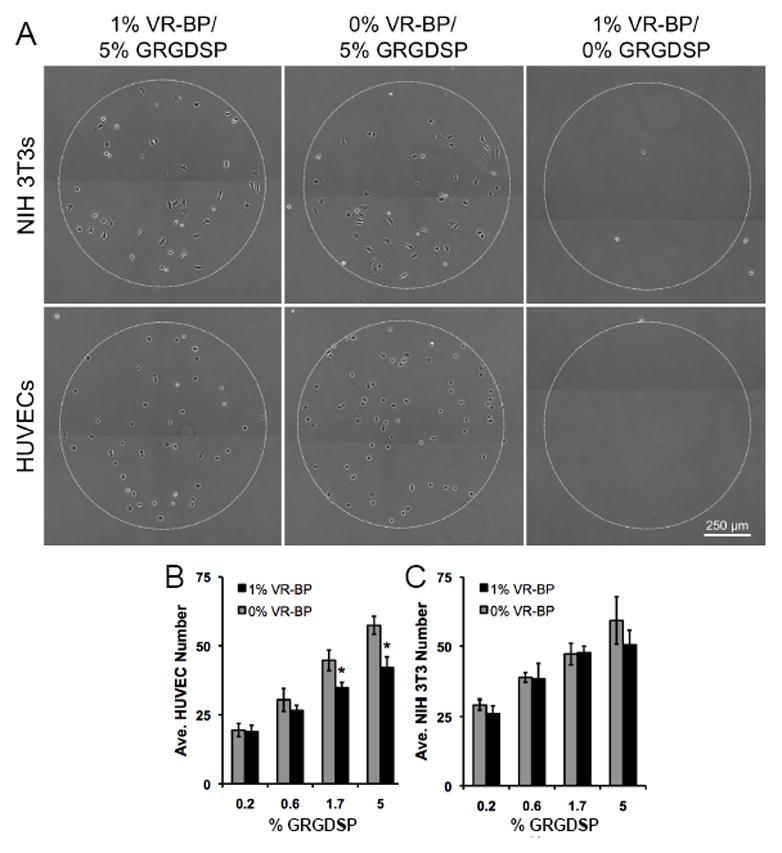
Cell attachment to surfaces presenting covalently immobilized VR-BP and varied densities of GRGDSP. (A) HUVECs and NIH 3T3 fibroblasts were seeded on SAM arrays presenting either 1% VR-BP or 0% VR-BP and varied densities of GRGDSP and allowed to attach for ~1 hr. Quantification of (B) HUVEC and (C) 3T3 fibroblast attachment. (Note: 0% VR-BP corresponds to 1% scrambled peptide and 0% GRGDSP corresponds to 5% of the mutant peptide GRGESP. Error bars represent standard error of the mean and asterisk indicates significant decrease in cell number compared to 0% VR-BP at a specific GRGDSP density, p < 0.05)
To examine the effects of immobilized VR-BP on soluble VEGF signaling, HUVECs were cultured on SAM arrays presenting varied densities of VR-BP (1% VR-BP to 0% VR-BP, where 1−X% = % scrambled peptide) and varied densities of GRGDSP in different soluble environments (Figure 7). Immobilized VR-BP at 1% density decreased HUVEC survival in conditions without soluble VEGF, or in the presence of a pharmacological inhibitor of VEGF signaling (SU5416) (Figure 7C,D). Additionally, 1% VR-BP inhibited proliferation in the presence of soluble VEGF at both high and low GRGDSP densities (Figure 7C,D). Taken together, these results indicate that, unlike the soluble form of VR-BP that exhibits mitogenic effects, substrates presenting covalently immobilized VR-BP not only block HUVEC attachment and proliferation, but also antagonize the effects of soluble VEGF signaling. It is noteworthy that the antagonist effect of immobilized VR-BP was also observed in the presence of the pharmacological inhibitor SU5416, suggesting that covalently immobilized VR-BP can antagonize VEGF signaling in concert with VEGFR tyrosine kinase inhibition.
Figure 7.
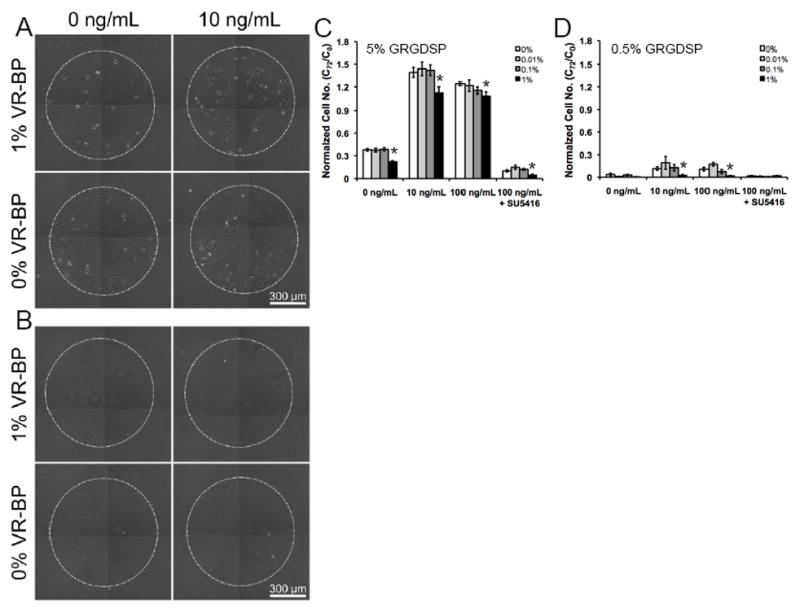
HUVEC proliferation and survival on surfaces presenting covalently immobilized VR-BP. HUVECs were seeded onto SAM arrays presenting (A) 5% or (B) 0.5% GRGDSP with varied densities of VR-BP including 1%, 0.1%, 0.01% and 0% and allowed to attach for 3 hours. After attachment, HUVECs were stimulated with VEGF concentrations of 0 ng/mL, 10ng/mL, 100 ng/mL. As indicated, 10 μM SU5416 was included during cell seeding and washed out before stimulation with VEGF. Normalized cell number after 72 hours of culture in (C) 5% or (D) 0.5% GRGDSP. (Error bars indicate standard error of the mean. Asterisk indicate significance decrease compared to 0% VR-BP at p < 0.05)
Tublulogenesis on substrates presenting VR-BP
To further characterize the effects of immobilized VR-BP on HUVEC behavior, we designed a tublulogenesis assay in which HUVECs in culture on SAM arrays presenting 5% GRGDSP and varied densities of VR-BP were placed onto a layer of matrigel and imaged continuously using time-lapse microscopy (Figure 8). Using a Nikon Eclipse Ti inverted microscope equipped with Nikon’s Perfect Focus System (which uses near-infrared light to find the gold interface of the SAM monolayer) we were able to continuously observe HUVEC behavior over time in the same z-plane. During tubule formation, HUVECs appeared to maintain contact with the SAM array substrate, as they did not migrate out of the z focal plane during the 72 hours of observation. HUVEC organization was most apparent over the first 24 hours (See Supplemental Movies S1A–D) and when quantified, soluble VEGF decreased tubulogenesis, as quantified by average tubule length per total cell area (Figure 8B). In contrast, 1% VR-BP increased tubulogenesis when compared to 0% VR-BP, both in the presence and absence of soluble VEGF. Therefore, as observed for endothelial cell attachment and proliferation, immobilized VR-BP antagonizes the effects of soluble VEGF on endothelial cell tubulogenesis.
Figure 8.
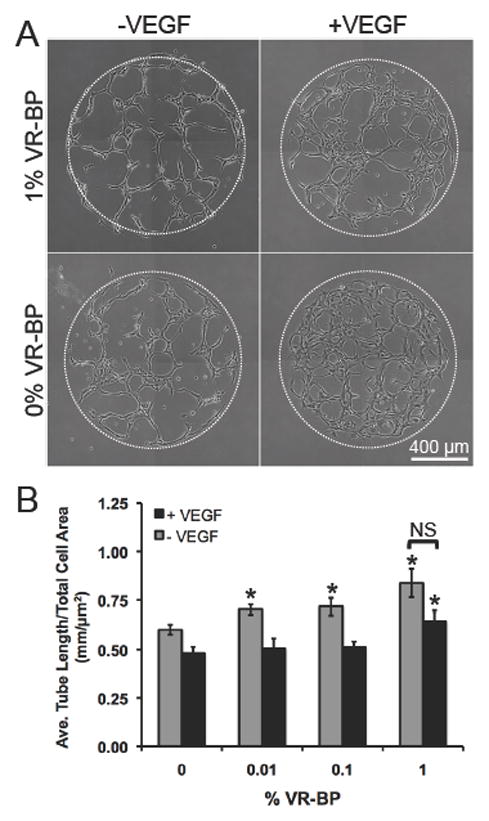
HUVEC tubulogenesis on surfaces presenting 5% GRGDSP and covalently immobilized VR-BP. (A) HUVECs on SAM arrays were placed on a thin layer of matrigel with and without 100 ng/mL VEGF and imaged for over 24 hrs (See Supplemental Movies 1A–D). (B) Tubulogenesis was quantified by averaging the length of all objects in each array spot and normalizing by total area occupied by cells. (Error bars represent standard error of the mean. Asterisk indicates significant increase compared to 0% VR-BP and “NS” indicates no significance between soluble VEGF conditions, p < 0.05)
Discussion
Here, we probed for the effects of a VEGF receptor-binding peptide, VR-BP, and discovered that the peptide stimulated endothelial cell proliferation when presented in soluble form, but antagonized HUVEC attachment and proliferation when immobilized on a chemically well-defined substrate. Importantly, this demonstrates that the same receptor-binding peptide can have opposite effects on cell behavior depending on the context in which it is presented. While previous studies using VR-BP in soluble and biomaterial-associated forms have reported increases in endothelial cell proliferation[12] and tubule formation[13, 14], our current results suggest that covalent immobilization of VR-BP alters its effect on endothelial cells. Therefore, here we focus our discussion on our results using covalently immobilized VR-BP, and we compare our results to previous studies to suggest potential mechanisms by which the context of VR-BP presentation may alter its function.
A main distinction between our current approach and previous approaches used to present VR-BP is that our substrates: (1) prevented non-specific protein interactions; (2) controlled peptide density and identity; and (3) covalently immobilized the peptide via a relatively short linker. We identified the threshold for peptide densities that led to non-specific cell-substrate attachment (Figure 5), and observed this threshold in the design of all subsequent experiments. The use of substrates that limit non-specific protein adsorption in our current work allows us to clearly define cell-substrate interactions, which is distinct when compared to substrates previously used to present VR-BP (e.g. hydroxyapatite, collagen). Combining these defined substrate properties with an elastomeric stencil approach that enabled control over spot-to-spot peptide composition allowed us to probe both a range of VR-BP conditions, and a range of cell adhesion ligand densities (Figure 4). Therefore, we were able to examine how substrates presenting covalently immobilized VR-BP influenced endothelial cell behavior in a defined manner, and also assess potential cross effects between VR-BP and cell adhesion. This experimental approach is adaptable, and can in principle be used to study the effects of a variety of other growth factor receptor-binding peptides.
The VR-BP molecule used here was similar to the VR-BP used in previous approaches, both in terms of its chemical structure and biological effects. VR-BP exhibited helical properties (Figure 2A) similar to those shown to be associated with VR-BP biological activity by D’Andrea and coworkers [9] and efficiently conjugated to SAM substrates (Figure 2B). Furthermore, soluble VR-BP promoted HUVEC proliferation, which is similar to what has been reported using bovine aortic endothelial cells[9] and mouse C166-GFP endothelial cells. [12] Interestingly, previous work by Leslie-Barbick and coworkers only observed a proliferative effect on HUVECs when VR-BP was modified with a 3.4 kDa PEG tail on the epsilon primary amine of the N-terminal lysine.[14] However, this result is perhaps not surprising since their synthetic strategy required acetylation of all other primary amines, which may have led to decreased solubility of the non-PEGylated VR-BP. Therefore, here we present evidence that when soluble VR-BP is synthesized with a poly-glycine tail and no side chain modifications it elicits a proliferative response in HUVECs. Furthermore, we also observed that soluble VR-BP has the strongest effect on endothelial cell proliferation when HUVECs are adhering to substrates with the highest density of the RGDSP cell adhesion ligand (Figure 3), suggesting that the proliferative response is dependent on both VR-BP presence and HUVEC adhesion.
The antagonistic effects of covalently immobilized VR-BP on HUVEC attachment and proliferation may be explained by considering the mechanism of soluble VEGF signaling through VEGFRs. As thoroughly characterized in previous studies, signal transduction via VEGFRs requires receptor dimerization and subsequent cross-phosphorylation to initiate intracellular signaling cascades that promote pro-angiogenic behavior, such as endothelial cell proliferation.[1–4] Although the mechanism of VEGF antagonism observed in this study is unclear, we can speculate that it may result from “pinning” of individual VEGFR molecules bound in a monomeric fashion to covalently immobilized VR-BP (Figure 9). In this potential scenario, these “pinned” receptors at the cell-substrate interface may be unable to efficiently dimerize to initiate signaling, and may also be unable to participate in synergistic interactions with integrin receptors such as αvβ3 integrin. Furthermore, VR-BP could sequester VEGFRs at the cell-substrate interface and further prevent VEGFRs from interacting with soluble VEGF (Figure 9). This proposed effect would not only decrease HUVEC proliferation in response to soluble exogenous VEGF, as observed here, but it could also decrease HUVEC proliferation in response to endogenous VEGF synthesized by HUVECs.[26, 27]
Figure 9.
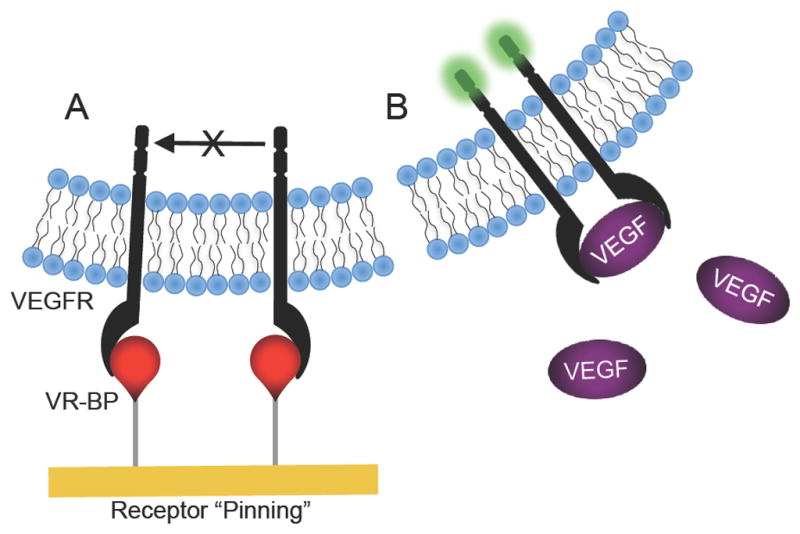
Schematic of proposed VEGFR “pinning” mechanism. We hypothesize that (A) covalently immobilized VR-BP may bind VEGFRs and prevent them from dimerizing or interacting with (B) soluble VEGF and activating intracellular signaling cascades.
It is noteworthy that previous studies using VR-BP would be less likely to observe this putative “receptor pinning” effect, as they did not covalently immobilize VR-BP via a relatively short linker. VR-BP modular peptides designed to bind to hydroxyapatite[12] or collagen[13] were immobilized non-covalently and able to release slowly over time, and VR-BP tethered to a hydrogel network may have been similar to soluble VR-BP due to the long PEG linker (3.4 kDa).[14] Presenting monomeric receptor-binding peptides on a solid substrate could provide a new, broadly applicable mechanism to inhibit activation of receptors that require dimerization. However, further studies capable of examining nanometer scale organization of VEGF receptors and peptide organization, such as fluorescence resonance energy transfer or atomic force microscopy, will be required to define the specific mechanism by which covalently immobilized VR-BP can inhibit VEGF signaling, and to address our proposed receptor pinning mechanism.
The combined effects of VR-BP and GRGDSP observed here are interesting in view of the known cross-talk between VEGFR and integrin activation. For example, previous investigations have used immunoprecipitation to identify a direct interaction between VEGFR-2 and αvβ3 integrin in the membrane of endothelial cells.[28] Furthermore, studies have demonstrated that when endothelial cells are stimulated with VEGF, activated VEGFR-2 dimers complex with αvβ3 integrins leading to increases in both integrin activation and VEGFR-2 activation.[28, 29] As a result, this synergy between VEGFRs and integrins has broad effects on endothelial cell attachment, spreading, and proliferation[30, 31]. Furthermore, if immobilized VR-BP interferes with VEGFR mobility and dimerization in the endothelial cell membrane, it may also have effects on integrin interactions with VEGFR-2 and thus significantly impact cell adhesion. Therefore, antagonism of VEGFRs via immobilized VR-BP could potentially affect both HUVEC attachment and proliferation, as observed in our results (Figure 6 and 7).
Interestingly, covalently immobilized VR-BP promoted HUVEC tubulogenesis in our study when cells were placed in contact with matrigel (Figure 8). Previous investigations into the effects of VEGF clearly demonstrate that VEGF signaling promotes endothelial cell proliferation, survival, migration, and in vivo angiogenesis. [1, 2] However, the effects of VEGF on in vitro tubulogenesis in matrigel and other three-dimensional environments are unclear.[32–34] While the proliferative and migratory effects of VEGF on endothelial cells are necessary for formation of in vivo vascular networks, some reports have suggested that the mitogenic effects of growth factor such as VEGF and bFGF can destabilize tubule formation and decrease in vitro tubulogenesis.[35] Our results corroborate these previous observations, as soluble VEGF protein was found to decrease tubulogenesis by HUVECs in our study (Figure 8). In contrast, immobilized VR-BP was found to inhibit the effects of soluble VEGF protein on tubulogenesis, and thereby increase tubulogenesis in this study (Figure 8). Therefore, we hypothesize that the enhanced tubule formation we observed in response to immobilized VR-BP represents another example of soluble VEGF inhibition. Taken together, our results indicate that soluble VEGF increases HUVEC proliferation and decreases tubulogenesis, while covalently immobilized VR-BP decreases HUVEC proliferation and favors the formation of stable tubular networks.
Conclusion
In summary, here we demonstrate that soluble VR-BP exhibits agonistic effects similar to VEGF, but covalent immobilization of VR-BP leads to antagonism of VEGF signaling. This context dependence between soluble and immobilized VR-BP was identified using a SAM array platform to rigorously control cell-substrate interactions, and systematically probe for the effects of immobilized VR-BP on endothelial cell attachment, proliferation, and tubulogenesis. Based on these findings we hypothesize that covalent immobilization of VR-BP, and potentially other ligands that bind growth factor receptors[36–45], may be capable of interfering with growth factor receptor dimerization and subsequent signal transduction. These results suggest a new mechanism for regulating growth factor signaling that may be useful to control specific biological processes, such as angiogenesis.
Supplementary Material
Supplement Figure 1. Generating defined culture substrates using alkanethiolate self-assembled monolayer arrays. (A) SAMs were generated using a simple elastomeric stencil adhered to the a gold substrate. (B) Schematic representation of SAM array fabrication: (1) Adhere elastomeric stencil to gold substrate to generate a microwell array superstructure, (2) locally form a SAM in each well with alkanethiolate mixtures containing carboxylic acid-terminated and hydroxyl-terminated oligo(ethylene-glycol) alkanethiolates, (3) covalently conjugate peptides to array spots via carbodiimide condensation of peptide n-terminal primary amine and SAM carboxylic acid terminal moities, and (4) remove mask and backfill with inert SAM.
Supplement Figure 2. HUVEC response to soluble VR-BP and VEGF using a standard tissue culture surface. HUVECs starved overnight in 2% FBS were seeded on gelatin coated 96-well plates at 3000 cells/well and cultured in the presence of 2% FBS alone or with the addition of varied VR-BP and VEGF concentrations for 24 hours. (Error bars indicate standard error of the mean and asterisk indicate significance compared to FBS alone, p < 0.05)
Acknowledgments
The authors would like to acknowledge funding from the National Institutes of Health (R01HL093282 and the Biotechnology Training Program NIGMS 5 T32GM08349) and the National Science Foundation (DMR 0906123). Fluorescent scans were obtained using a GE Healthcare Typhoon Trio Variable Mode Imager at the Scientific Instrumentation Facility of the UW Carbone Cancer Center, Madison, WI. Circular dichroism data were obtained at the University of Wisconsin - Madison Biophysics Instrumentation Facility, which was established with support from the University of Wisconsin - Madison and grants BIR-9512577 (NSF) and S10 RR13790 (NIH). We also thank Jae Sung Lee for his assistance obtaining circular dichroism data and Sam Gellman from the University of Wisconsin for helpful discussions.
References
- 1.Cross MJ, et al. VEGF-receptor signal transduction. Trends Biochem Sci. 2003;28(9):488–94. doi: 10.1016/S0968-0004(03)00193-2. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 3.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6(4):273–86. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 4.D’Andrea LD, et al. Peptide-based molecules in angiogenesis. Chem Biol Drug Des. 2006;67(2):115–26. doi: 10.1111/j.1747-0285.2006.00356.x. [DOI] [PubMed] [Google Scholar]
- 5.Soker S, et al. Inhibition of vascular endothelial growth factor (VEGF)-induced endothelial cell proliferation by a peptide corresponding to the exon 7-encoded domain of VEGF165. J Biol Chem. 1997;272(50):31582–8. doi: 10.1074/jbc.272.50.31582. [DOI] [PubMed] [Google Scholar]
- 6.Fuh G, et al. Requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J Biol Chem. 1998;273(18):11197–204. doi: 10.1074/jbc.273.18.11197. [DOI] [PubMed] [Google Scholar]
- 7.Boesen TP, et al. Single-chain vascular endothelial growth factor variant with antagonist activity. J Biol Chem. 2002;277(43):40335–41. doi: 10.1074/jbc.M204107200. [DOI] [PubMed] [Google Scholar]
- 8.Siemeister G, et al. An antagonistic vascular endothelial growth factor (VEGF) variant inhibits VEGF-stimulated receptor autophosphorylation and proliferation of human endothelial cells. Proc Natl Acad Sci U S A. 1998;95(8):4625–9. doi: 10.1073/pnas.95.8.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Andrea LD, et al. Targeting angiogenesis: structural characterization and biological properties of a de novo engineered VEGF mimicking peptide. Proc Natl Acad Sci U S A. 2005;102(40):14215–20. doi: 10.1073/pnas.0505047102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santulli G, et al. In vivo properties of the proangiogenic peptide QK. J Transl Med. 2009;7:41. doi: 10.1186/1479-5876-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diana D, et al. Structural determinants of the unusual helix stability of a de novo engineered vascular endothelial growth factor (VEGF) mimicking peptide. Chemistry. 2008;14(14):4164–6. doi: 10.1002/chem.200800180. [DOI] [PubMed] [Google Scholar]
- 12.Lee JS, Wagoner Johnson AJ, Murphy WL. A modular, hydroxyapatite-binding version of vascular endothelial growth factor. Adv Mater. 2010;22(48):5494–8. doi: 10.1002/adma.201002970. [DOI] [PubMed] [Google Scholar]
- 13.Chan TR, Stahl PJ, Yu SM. Matrix-Bound VEGF Mimetic Peptides: Design and Endothelial-Cell Activation in Collagen Scaffolds. Advanced Functional Materials. 2011;21(22):4252–4262. doi: 10.1002/adfm.201101163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leslie-Barbick JE, et al. The promotion of microvasculature formation in poly(ethylene glycol) diacrylate hydrogels by an immobilized VEGF-mimetic peptide. Biomaterials. 2011;32(25):5782–5789. doi: 10.1016/j.biomaterials.2011.04.060. [DOI] [PubMed] [Google Scholar]
- 15.Prime KL, Whitesides GM. Adsorption of Proteins onto Surfaces Containing End-Attached Oligo(Ethylene Oxide) - a Model System Using Self-Assembled Monolayers. Journal of the American Chemical Society. 1993;115(23):10714–10721. [Google Scholar]
- 16.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Analytical Biochemistry. 1989;182(2):319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 17.Walker GM, Beebe DJ. A passive pumping method for microfluidic devices. Lab on a Chip. 2002;2(3):131–134. doi: 10.1039/b204381e. [DOI] [PubMed] [Google Scholar]
- 18.Jo BH, et al. Three-dimensional micro-channel fabrication in polydimethylsiloxane (PDMS) elastomer. Journal of Microelectromechanical Systems. 2000;9(1):76–81. [Google Scholar]
- 19.Thibault C, et al. Poly(dimethylsiloxane) Contamination in Microcontact Printing and Its Influence on Patterning Oligonucleotides. Langmuir. 2007;23(21):10706–10714. doi: 10.1021/la701841j. [DOI] [PubMed] [Google Scholar]
- 20.Skoda MWA, et al. Optimizing the PMIRRAS signal from a multilayer system and application to self-assembled monolayers in contact with liquids. Journal of Electron Spectroscopy and Related Phenomena. 2009;172(1–3):21–26. [Google Scholar]
- 21.Fong TA, et al. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999;59(1):99–106. [PubMed] [Google Scholar]
- 22.Buffeteau T, et al. Anisotropic Optical Constants of Alpha-Helix and Beta-Sheet Secondary Structures in the Infrared. The Journal of Physical Chemistry B. 2000;104(18):4537–4544. [Google Scholar]
- 23.Frey BL, Corn RM. Covalent attachment and derivatization of poly(L-lysine) monolayers on gold surfaces as characterized by polarization-modulation FT-IR spectroscopy. Analytical Chemistry. 1996;68(18):3187–3193. [Google Scholar]
- 24.Fluorescent signal from spots presenting only VR-BP had significantly higher fluorescent intensity compared to spots presenting only scrambled peptide suggesting that the fluorescent probe reacts more efficiently with the lysine residues in the VR-BP compared to scrambled peptide, perhaps to the differences in peptide secondary structure observed in CD and PM-IRRAS data (Figure 2).
- 25.Millauer B, et al. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature. 1994;367(6463):576–9. doi: 10.1038/367576a0. [DOI] [PubMed] [Google Scholar]
- 26.Lee S, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130(4):691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helotera H, Alitalo K. The VEGF family, the inside story. Cell. 2007;130(4):591–2. doi: 10.1016/j.cell.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 28.Mahabeleshwar GH, et al. Mechanisms of integrin-vascular endothelial growth factor receptor cross-activation in angiogenesis. Circ Res. 2007;101(6):570–80. doi: 10.1161/CIRCRESAHA.107.155655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soldi R, et al. Role of alphavbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBO J. 1999;18(4):882–92. doi: 10.1093/emboj/18.4.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Byzova TV, et al. A mechanism for modulation of cellular responses to VEGF: activation of the integrins. Mol Cell. 2000;6(4):851–60. [PubMed] [Google Scholar]
- 31.Somanath PR, Ciocea A, Byzova TV. Integrin and growth factor receptor alliance in angiogenesis. Cell Biochem Biophys. 2009;53(2):53–64. doi: 10.1007/s12013-008-9040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xin X, et al. Hepatocyte growth factor enhances vascular endothelial growth factor-induced angiogenesis in vitro and in vivo. Am J Pathol. 2001;158(3):1111–20. doi: 10.1016/S0002-9440(10)64058-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen CJ, et al. Decreased cell adhesion promotes angiogenesis in a Pyk2-dependent manner. Exp Cell Res. 317(13):1860–71. doi: 10.1016/j.yexcr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lafleur MA, et al. Endothelial tubulogenesis within fibrin gels specifically requires the activity of membrane-type-matrix metalloproteinases (MT-MMPs) J Cell Sci. 2002;115(Pt 17):3427–38. doi: 10.1242/jcs.115.17.3427. [DOI] [PubMed] [Google Scholar]
- 35.Saunders RL, Hammer DA. Assembly of Human Umbilical Vein Endothelial Cells on Compliant Hydrogels. Cell Mol Bioeng. 3(1):60–67. doi: 10.1007/s12195-010-0112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zouani OF, et al. Differentiation of pre-osteoblast cells on poly(ethylene terephthalate) grafted with RGD and/or BMPs mimetic peptides. Biomaterials. 2010;31(32):8245–53. doi: 10.1016/j.biomaterials.2010.07.042. [DOI] [PubMed] [Google Scholar]
- 37.Briscoe H, et al. A novel tumor necrosis factor (TNF) mimetic peptide prevents recrudescence of Mycobacterium bovis bacillus Calmette-Guerin (BCG) infection in CD4+ T cell-depleted mice. J Leukoc Biol. 2000;68(4):538–44. [PubMed] [Google Scholar]
- 38.Dower WJ. Targeting growth factor and cytokine receptors with recombinant peptide libraries. Curr Opin Chem Biol. 1998;2(3):328–34. doi: 10.1016/s1367-5931(98)80005-7. [DOI] [PubMed] [Google Scholar]
- 39.Shrivastava A, Nunn AD, Tweedle MF. Designer peptides: learning from nature. Curr Pharm Des. 2009;15(6):675–81. doi: 10.2174/138161209787315620. [DOI] [PubMed] [Google Scholar]
- 40.Binetruy-Tournaire R, et al. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J. 2000;19(7):1525–33. doi: 10.1093/emboj/19.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engstrom U, et al. Identification of a peptide antagonist for platelet-derived growth factor. J Biol Chem. 1992;267(23):16581–7. [PubMed] [Google Scholar]
- 42.Li Z, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005;19(14):1978–85. doi: 10.1096/fj.05-4058com. [DOI] [PubMed] [Google Scholar]
- 43.Maruta F, et al. Identification of FGF receptor-binding peptides for cancer gene therapy. Cancer Gene Ther. 2002;9(6):543–52. doi: 10.1038/sj.cgt.7700470. [DOI] [PubMed] [Google Scholar]
- 44.Michon IN, et al. The effect of TGF-beta receptor binding peptides on smooth muscle cells. Biochem Biophys Res Commun. 2002;293(4):1279–86. doi: 10.1016/S0006-291X(02)00378-9. [DOI] [PubMed] [Google Scholar]
- 45.Qin X, et al. Identification of a novel peptide ligand of human vascular endothelia growth factor receptor 3 for targeted tumour diagnosis and therapy. J Biochem. 2007;142(1):79–85. doi: 10.1093/jb/mvm109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Figure 1. Generating defined culture substrates using alkanethiolate self-assembled monolayer arrays. (A) SAMs were generated using a simple elastomeric stencil adhered to the a gold substrate. (B) Schematic representation of SAM array fabrication: (1) Adhere elastomeric stencil to gold substrate to generate a microwell array superstructure, (2) locally form a SAM in each well with alkanethiolate mixtures containing carboxylic acid-terminated and hydroxyl-terminated oligo(ethylene-glycol) alkanethiolates, (3) covalently conjugate peptides to array spots via carbodiimide condensation of peptide n-terminal primary amine and SAM carboxylic acid terminal moities, and (4) remove mask and backfill with inert SAM.
Supplement Figure 2. HUVEC response to soluble VR-BP and VEGF using a standard tissue culture surface. HUVECs starved overnight in 2% FBS were seeded on gelatin coated 96-well plates at 3000 cells/well and cultured in the presence of 2% FBS alone or with the addition of varied VR-BP and VEGF concentrations for 24 hours. (Error bars indicate standard error of the mean and asterisk indicate significance compared to FBS alone, p < 0.05)