

Abstract
Osteogenesis imperfecta (OI) is a heterogenous group of genetic disorders of bone fragility. OI type V is an autosomal-dominant disease characterized by calcification of the forearm interosseous membrane, radial head dislocation, a subphyseal metaphyseal radiodense line, and hyperplastic callus formation; the causative mutation involved in this disease has not been discovered yet. Using linkage analysis in a four-generation family and whole-exome sequencing, we identified a heterozygous mutation of c.−14C>T in the 5′-untranslated region of a gene encoding interferon-induced transmembrane protein 5 (IFITM5). It completely cosegregated with the disease in three families and occurred de novo in five simplex individuals. Transfection of wild-type and mutant IFITM5 constructs revealed that the mutation added five amino acids (Met-Ala-Leu-Glu-Pro) to the N terminus of IFITM5. Given that IFITM5 expression and protein localization is restricted to the skeletal tissue and IFITM5 involvement in bone formation, we conclude that this recurrent mutation would have a specific effect on IFITM5 function and thus cause OI type V.
Main Text
Osteogenesis imperfecta (OI) is a heterogeneous group of disorders characterized by intrinsic bone fragility, resulting in frequent fractures as well as in deformities of the spine and limbs. The phenotypic severity of OI ranges from perinatal lethal to subtle fracture susceptibility or generalized osteopenia. The Sillence classification system1 categorizes OI individuals into four types (type I [MIM 166200], type II [MIM 166210], type III [MIM 259420], and type IV [MIM 166220]), and types V (MIM 610967), VI (MIM 613982), and VII (MIM 610682) have subsequently been added on the basis of characteristic phenotypes and laboratory findings.2 About 90% of OI individuals fit into one of the types I through IV; their conditions are caused by dominant mutations of either COL1A1 (MIM 120150) or COL1A2 (MIM 120160). Mutations in SERPINF1 (MIM 172860) and CRTAP (MIM 605497) were found responsible for OI types VI and VII, respectively.3,4 Further mutations responsible for autosomal-recessive OI were identified in LEPRE1 (MIM 610339),5 PPIB (MIM 123841),6 SERPINH1 (MIM 600943),7 and FKBP10 (MIM 607063),8 which lead to the molecular-genetic division of OI into 11 types.9 However, given the discovery of a large number of genes responsible for recessive OI forms, the genotype-phenotype relationships are difficult to explain. It has been suggested that we retain the Sillence classification as a clinical scoring system devoid of any direct relationship to a specific gene.10
OI type V is a specific disease entity with distinguishing clinical and radiological features, and its causative mutation has not been discovered yet. It is characterized by an autosomal-dominant inheritance pattern, absence of blue sclera, absence of dentinogenesis imperfecta, propensity to hyperplastic callus formation, calcification of the forearm interosseous membrane, radial-head dislocation, and a subphyseal metaphyseal radiodense line (Figure 1).11,12 It is a dominantly inherited OI subtype not related to COL1A1 and COL1A2 mutations.13
Figure 1.
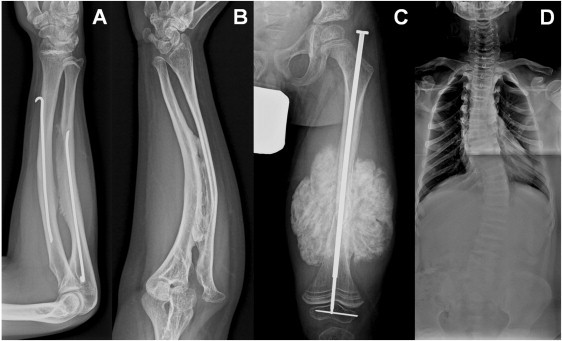
Radiologic Findings for OI Type V
(A) The forearm of individual II:1 from family 9 at 15 years of age shows calcification of the interosseous membrane without radial-head dislocation.
(B) The forearm of individual II:2 from family 3 at 27 years of age shows calcification of the interosseous membrane with radial-head dislocation.
(C) The left femur i of individual II:1 from family 4 at 7 years of age. Note hyperplastic callus after open reduction of the femur shaft fracture.
(D) Scoliosis and vertebral body collapse in individual IV:6 from family 1 at 11 years of age.
To identify the causative mutation for OI type V, we recruited 19 Korean individuals with the disease: 13 affected individuals from three families and six simplex individuals (Figure S1 in the Supplemental Data available with this article online). This study was approved by the institutional review boards of Seoul National University Hospital and Seoul National University Dental Hospital. Informed consent was obtained from all subjects. The clinical and radiographic findings of 12 affected individuals (11 familial and one simplex) were reported previously.14 An additional seven individuals (two familial and five simplex) were included in this study. Their clinical and radiographic features are summarized in Table 1. The height of 12 individuals was within normal range (−2 to +2 SD), but seven individuals were markedly short when their height was plotted against normal Korean growth curves. Most individuals had less than ten fractures; however, three individuals had 20–30 fractures. Physical impairment ranged from an individual's being wheel-chair-bound to no physical impairment. All affected individuals showed various degrees of calcification of the forearm interosseous membrane, and radial-head dislocation was observed in those individuals older than 20 years of age, except that it was also observed in an 8-year-old boy (family 5-II:1). Sanger sequencing and deletion/duplication analysis using MLPA for COL1A1 and COL1A2 failed to show pathogenic mutations in the index individuals of three familial cases or any other simplex individuals.
Table 1.
Clinical and Radiological Data of the Individuals
| Individual | Age (yr) | Sex | Height (z Valuea) | BMDb | CFIM | Radial-Head Dislocation | Number of Fracturesc | Hyperplastic Callus | Scoliosis | Disability at Other than Elbow or Forearm | Physical Activity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 1-II:1 | 82 | M | −4.09a | N.A. | + | + | 1 | N.A. | + | - | Appropriate for age |
| Family III:1 | 59 | F | −2.43a | N.A. | + | + | 4 | N.A. | + | - | No limitation in daily life |
| Family III:3 | 46 | F | −0.9a | N.A. | + | + | 1 | N.A. | + | - | No limitation in daily life |
| Family III:5 | 52 | M | −0.94a | N.A. | + | + | 4 | N.A. | + | - | No limitation in daily life |
| Family IV:2 | 34 | F | −0.14a | N.A. | ± | + | 2 | N.A. | - | - | No limitation in daily life |
| Family IV:3 | 32 | M | 0.11a | N.A. | + | + | 10 | N.A. | + | - | No limitation in daily life |
| Family IV:4 | 32 | M | −0.94a | N.A. | + | + | 5 | N.A. | N.A. | - | No limitation in daily life |
| Family IV:6 | 11 | M | 1.88 | 0.289/4.3/NA/0 | + | - | ∼10 | - | + | - | Unlimited ambulation, cannot run |
| Family V:1 | 8 | M | 0.80 | 0.584/2.6/NA/13.5 | + | - | 5 | - | - | - | Can sprint |
| Family 2-II:2 | 49 | M | −3.21a | N.A. | + | + | 7∼9 | + | + | Bony ankylosis of both hips | Independent ambulation with a cane |
| Family III:1 | 20 | F | −2.43 | 0.681/10.5/−1.27/0 | + | + | 15 | + | - | - | Can run slowly |
| Family 3-II:2 | 27 | F | −2.05 | N.A. | + | + | ∼30 | + | - | - | No limitation in daily life |
| Family III:1 | 6 | F | 0.02 | 0.64/4.8/NA/10.5 | + | - | ∼20 | + | - | Ankylosis of elbow, left | Independent but limited ambulation |
| Family 4-II:1 | 7 | M | −2.52 | 0.339/5.2/-3.70/3.5 | + | - | ∼5 | + | - | - | Can run slowly |
| Family 5-II:1 | 8 | F | −1.96 | 0.264/0.8/NA/0 | + | + | ∼20 | + | + | - | Can sprint |
| Family 6-II:1 | 4 | F | 0.03 | 0.401/3.9/NA/0 | + | - | ∼10 | + | - | - | Can run slowly |
| Family 7-II:1 | 2 | F | −1.76 | N.A. | + | - | 8 | + | - | - | Starts independent ambulation |
| Family 8-II:1 | 14 | M | −0.77 | 0.640/5.7/−0.05/12 | + | ± | ∼10 | - | + | Migrating bone pain | Wheelchair bound due to painful LOM, both knees and elbows |
| Family 9-II:1 | 16 | M | −3.84 | 0.504/7.9/−1.87/0 | + | - | ∼10 | + | + | SNHL | Can run slowly |
Abbreviations are as follows: CFIM, calcification of the forearm interosseous membrane; N.A., information not available; LOM, limit of motion; and SNHL, sensorineural hearing loss.
The z value was calculated from 2007 data collected from normal children and young adults of the Korean population. Because people from the old Korean generation tend to be shorter, the z values of those over 30 years of age should be underestimated.
Bone mineral density measured by dual-energy X-ray absorptiometry (DXA) is described as BMD at L2-4 (g/cm2)/age at measurement (year)/calculated z value/accumulated dose of pamidronate infused before the measurement (mg/kg). z value could be calculated from 5 years of age.
The number of fractures was recorded according to the affected individual's and/or parents' memory.
To identify the disease locus, we performed genome-wide linkage analysis on a four-generation family (family 1 in Figure S1). Using 407 microsatellite markers with an average interval of 10 cM, we genotyped 14 family members (nine affected and five unaffected). A maximum LOD score of 2.52 was obtained at D11S4046. Additional markers defined the locus at the 11pter-11p15.4 region spanning 9.1 Mb from D11S4149 (Figure S1 and Table S1). Sanger sequencing performed on the coding region and exon-intron boundaries of the 171 genes in this region failed to identify a causative mutation. Cytogenetic analysis revealed no significant structural change in the chromosome, and aCGH showed no pathogenic copy-number variation.
Then, we performed whole-exome sequencing in all four members of family 4 (Figure S1) by using the Illumina TruSeq DNA sample prep kit and 90 bp paired-end sequencing on an Illumina HiSeq 2000. High-quality sequencing data with an average of 95.1% coverage and 42.1× sequencing depth were obtained (Table S2). Alignment of the sequencing data to the NCBI human reference genome (NCBI build 37.2, hg19) was performed, and the sequence variations were annotated with dbSNP build 135. Sequence variations (including those of the 5′-untranslated region [5′-UTR], 3′-UTR, and intron region) unique to the proband were selected manually. Among the variations located in the linked region of family 1 (Table S3), we focused on a heterozygous change in a gene encoding interferon-induced transmembrane protein 5 (IFITM5); this change was a transition from C to T in the 5′-UTR (c.–14C>T) (Figure 2) (RefSeq accession number NM_001025295.1). Sanger sequencing confirmed that this variation completely cosegregated with the disease in family 1 (Table S4). The variation was found in all affected individuals and not in unaffected family members who participated. Furthermore, it was not found in 200 unrelated normal chromosomes from individuals with the same ethnic background. Cosegregation in the other two families (families 2 and 3, Figure S1) and de novo occurrence in the five simplex individuals confirmed that this variation is a disease-causing mutation of OI type V.
Figure 2.
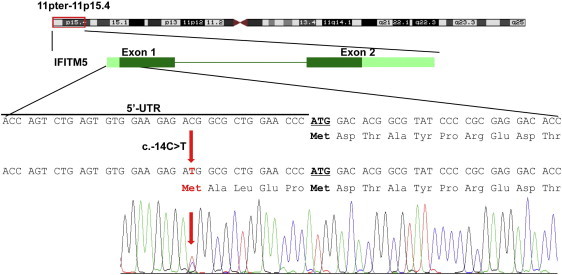
Mutation of IFITM5 in OI Type V
Schematic representation of IFITM5 is shown under the linked region (11pter-11p15.4). Dark green boxes indicate coding exons, and bright green boxes indicate UTRs. The mutation (c.−14C>T) is indicated by the red arrow, and the additional five amino acids generated by this mutation are shown under the gene sequence. The sequencing chromatogram of an affected individual is shown below.
The mutation was predicted to generate an in-frame translation start codon that would add five amino acids (Met-Ala-Leu-Glu-Pro) to the N terminus of IFITM5. Because we did not have any cultured IFITM5-expressing cells from the affected individuals at hand, we transfected wild-type and mutant IFITM5 constructs to investigate their translation. The PCR product of IFITM5 that had been amplified from cDNA from the DF/HCC DNA Resource Core (Boston, MA) with primers introducing HindIII and BamHI sequences (Table S4) was cloned. PCR mutagenesis introduced the mutations c.−14C>T and/or c.2T>C (Table S4). The wild-type and three mutated cDNAs (c.−14C>T, c.2T>C, and both) were subcloned into pEGFPN1 vector with HindIII and BamHI. HEK293 cells were transiently transfected with lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). Immunoblot analysis of the transfected cell lysates revealed that the translation started at the start codon generated by the mutation, not at the original start codon (Figure 3). It confirmed that the mutation generates a mutated form of IFITM5 having five additional amino acids at its N terminus.
Figure 3.
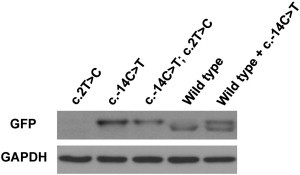
In Vitro Translation Assay
Total cell lysates of HEK293 cells transfected with three mutated cDNAs (c.2T>C, c.−14C>T, and both), wild-type, or wild-type along with c.−14C>T cDNA were immunoblotted with GFP antibody. When the original start codon was mutated (c.2T>C), no translation was observed. Translation started at the start codon generated by the mutation (c. −14C>T) regardless of the status of the original start codon. Its translaton product was slightly larger than that of the wild-type. Cotransfection with wild-type and c.−14C>T-mutated constructs produced double bands. GAPDH was used as an internal control.
We investigated postnatal spatial localization of IFITM5 by using polyclonal rabbit IFITM5 antibody (Abgent, San Diego, CA) at a 1:50 dilution to conduct an immunohistochemical study on the proximal tibia from a 3-week-old female BALB/c mouse. IFITM5 immunoreactivity was localized at the osteoblasts; periosteum; periphyseal fibrochondrosseous structure, including the groove of Ranvier and the ring of LaCroix; and the superficial chondrocytes of the articular cartilage; but not at the physeal chondrocytes (Figure S2). We also examined the mRNA expression of IFITM5 in various human tissues, including bone, cartilage, hip joint capsule, circulating leukocytes, fascia lata, ligamentum teres, muscle, and skin by RT-PCR with gene-specific primers (Table S4). IFITM5 was expressed exclusively in the bone and cartilage in humans, and IFITM5 immnunoreactivity was localized at the osteoblasts, periosteum, perichondrium, and superficial layer of articular cartilage (Figure S2).
IFITM5 is a member of the interferon-induced transmembrane gene family. It is located in chromosomal region 11p15.5 and encodes a 132 amino acid protein, which has two transmembrane domains, such that it has extracellular N and C termini and an intracellular loop.15 Although IFITM5 was named because it is clustered close to other IFITM family members and because it has an IFITM-like gene structure, it does not have interferon response elements and is not induced by interferons.15 Hence, another name, bone-restricted ifitm-like protein (BRIL), was suggested.15 The N and C termini would be the most likely domains to interact with other proteins or cells.15 The function of IFITM5 has not been well elucidated, but it was suggested to be involved in the process of bone formation. Its mRNA expression and protein localization are highly restricted to the skeletal tissue. In situ hybridization and immunohistochemistry of mouse embryos showed Ifitm5 expression and protein localization at the osteoblasts of the long bones, vertebrae, facial bones, and the periosteum, but not at the proliferating and resting zones of the growth plate or other soft-tissue organs.15,16 Our data indicate similar spatial mRNA expression and protein localization patterns in postnatal mice and humans.
Ifitm5 overexpression in UMR106 cells and primary rat osteoblasts resulted in a dose-dependent increase in mineralization, whereas knockdown of Ifitm5 by shRNA in MC3T3 osteoblasts induced reduced mineralization.15 However, Ifitm5 knockout mice showed less critical change than expected. Ifitm5−/− mice had bent and smaller long bones than Ifitm5+/− mice, but there was no significant difference in bone morphometric parameters between Ifitm5−/− and Ifitm5+/− mice.16 Hence, simple loss-of-function of IFITM5 cannot explain the complex phenotype of OI type V.
The mutation identified in this study is unique in that it occurs in a UTR. We missed this mutation in the linkage analysis and subsequent Sanger sequencing of candidate genes because we paid attention only to the coding and splicing regions. Additionally, it is unique in that all affected individuals from nine unrelated families share the same mutation in IFITM5. This suggests that the mutation might have a specific effect on the function of IFITM5. Unlike individuals with type-I-collagen-related OI, individuals with OI type V showed a relatively normal texture of soft tissues, as detected during a surgical procedure (unpublished data), absence of dentinogenesis imperfecta, and involvement of non-calcified connective tissue such as the forearm interosseous membrane. These findings indicate that OI type V might be a disorder of skeletal bone deposition rather than a generalized connective-tissue disease.
In light of the contradictory components of the OI type V phenotype, such as osteopenia versus ectopic calcification or hyperplastic callus formation, the effect of the c.−14C>T mutation might depend on the tissues where it is expressed and the events that the tissues undergo. The mutant IFITM5 seems to interfere with normal bone formation in the cortical and trabecular bone tissue, resulting in osteopenia and subsequent bone fragility. However, it develops ectopic calcification consistently at the forearm interosseous membrane and less consistently at the subperiosteal region of the long bones. Hyperplastic callus formation is an exaggerated response of bone formation during bone healing; it is possibly provoked by post-traumatic inflammatory reaction. A subphyseal metaphyseal radiodense line also develops paradoxically in the osteopenic long bones. Therefore, we postulate that the mutant IFITM5 having five additional amino acids at its N terminus dysregulates the process of bone formation—usually by suppressing bone formation but sometimes by leading to the development of ectopic calcification or exaggerated bone formation in a certain location or in a certain situation.
Given the consistent presence of the c.−14C>T mutation in IFITM5 in all affected individuals, the restricted spatial pattern of IFITM5 expression and protein localization, and its involvement in bone formation, we conclude that this specific mutation is responsible for OI type V. The mechanism by which this mutation leads to the disease is probably related to site-specific dysregulation of bone formation. Further study with a specific mutation knockin mouse would elucidate the pathogenic mechanism of this mutation and help us to better understand the physiologic function of IFITM5.
Acknowledgments
The authors sincerely thank all the family members for their participation in this study. This work was supported by a grant from the Bio & Medical Technology Development Program (2011-0027790) and by a Science Research Center grant to the Bone Metabolism Research Center (2012-0000487) of the National Research Foundation (NRF) funded by the Korean government (MEST) and by a grant from the Korea Healthcare Technology Research and Development Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea (A080588).
Contributor Information
Tae-Joon Cho, Email: tjcho@snu.ac.kr.
Jung-Wook Kim, Email: pedoman@snu.ac.kr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
dbSNP 135, http://www.ncbi.nlm.nih.gov/projects/SNP/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC database, version hg19, www.genome.ucsc.edu/
References
- 1.Sillence D.O., Senn A., Danks D.M. Genetic heterogeneity in osteogenesis imperfecta. J. Med. Genet. 1979;16:101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rauch F., Glorieux F.H. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- 3.Becker J., Semler O., Gilissen C., Li Y., Bolz H.J., Giunta C., Bergmann C., Rohrbach M., Koerber F., Zimmermann K. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2011;88:362–371. doi: 10.1016/j.ajhg.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 5.Cabral W.A., Chang W., Barnes A.M., Weis M., Scott M.A., Leikin S., Makareeva E., Kuznetsova N.V., Rosenbaum K.N., Tifft C.J. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Dijk F.S., Nesbitt I.M., Zwikstra E.H., Nikkels P.G., Piersma S.R., Fratantoni S.A., Jimenez C.R., Huizer M., Morsman A.C., Cobben J.M. PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 2009;85:521–527. doi: 10.1016/j.ajhg.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christiansen H.E., Schwarze U., Pyott S.M., AlSwaid A., Al Balwi M., Alrasheed S., Pepin M.G., Weis M.A., Eyre D.R., Byers P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:389–398. doi: 10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alanay Y., Avaygan H., Camacho N., Utine G.E., Boduroglu K., Aktas D., Alikasifoglu M., Tuncbilek E., Orhan D., Bakar F.T. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forlino A., Cabral W.A., Barnes A.M., Marini J.C. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011;7:540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Dijk F.S., Pals G., Van Rijn R.R., Nikkels P.G., Cobben J.M. Classification of Osteogenesis Imperfecta revisited. Eur. J. Med. Genet. 2010;53:1–5. doi: 10.1016/j.ejmg.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Glorieux F.H., Rauch F., Plotkin H., Ward L., Travers R., Roughley P., Lalic L., Glorieux D.F., Fassier F., Bishop N.J. Type V osteogenesis imperfecta: a new form of brittle bone disease. J. Bone Miner. Res. 2000;15:1650–1658. doi: 10.1359/jbmr.2000.15.9.1650. [DOI] [PubMed] [Google Scholar]
- 13.Cundy T. Recent advances in osteogenesis imperfecta. Calcif. Tissue Int. 2012;90:439–449. doi: 10.1007/s00223-012-9588-3. [DOI] [PubMed] [Google Scholar]
- 14.Lee D.Y., Cho T.J., Choi I.H., Chung C.Y., Yoo W.J., Kim J.H., Park Y.K. Clinical and radiological manifestations of osteogenesis imperfecta type V. J. Korean Med. Sci. 2006;21:709–714. doi: 10.3346/jkms.2006.21.4.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moffatt P., Gaumond M.H., Salois P., Sellin K., Bessette M.C., Godin E., de Oliveira P.T., Atkins G.J., Nanci A., Thomas G. Bril: a novel bone-specific modulator of mineralization. J. Bone Miner. Res. 2008;23:1497–1508. doi: 10.1359/jbmr.080412. [DOI] [PubMed] [Google Scholar]
- 16.Hanagata N., Li X., Morita H., Takemura T., Li J., Minowa T. Characterization of the osteoblast-specific transmembrane protein IFITM5 and analysis of IFITM5-deficient mice. J. Bone Miner. Metab. 2011;29:279–290. doi: 10.1007/s00774-010-0221-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.