

Abstract
This communication describes the development of a mild method for the cross-coupling of arylboronic acids with CF3I via the merger of photoredox and Cu catalysis. This method has been applied to the trifluoromethylation of electronically diverse aromatic and heteroaromatic substrates and tolerates many common functional groups.
Trifluoromethyl substituents are widely prevalent in pharmaceuticals and agrochemicals.1 As such, the development of mild and versatile synthetic methods for generating carbon–CF3 bonds has become a field of intense research effort. Over the past 3 years, a variety of Pd2,3 and Cu4,5-based cross-coupling protocols have been developed for the trifluoromethylation of aryl halides, aryl boronic acids, and aromatic carbon– hydrogen bonds. As exemplified in Scheme 1a/b for the Cu-promoted trifluoromethylation of boronic acids, these transformations typically involve “CF3−” 4c,4e,4l,5i or “CF3+” 4f,5d,e reagents and are believed to proceed via nucleophilic or electrophilic transfer of the CF3 group to the Cu center, respectively. Many of these new methods represent significant progress in comparison to the traditional Swarts reaction6 (which requires highly reactive fluorinating reagents and harsh conditions).
Scheme 1.

Cu-Mediated/Catalyzed Trifluoromethylation of Boronic Acids
Despite these important advances, most current strategies for aryl–CF3 cross-coupling suffer from one or more limitations. In some cases, temperatures greater than 100 °C2b,3a,b,4d and/or strong acids or bases (TFA3a or tBuOK4j) are necessary. Other methods require expensive trifluoromethylating reagents (e.g., S-(trifluoromethyl)thiophenium salts,3a,4f,5d Togni’s reagent,5b,5e,5g or TESCF3).3b,5a Finally, many protocols exhibit limited substrate scope/generality.3c,5a,b
One attractive approach to begin to address these limitations would be to access alternative and potentially complementary mechanistic manifolds. We reasoned that a radical pathway (Scheme 1c) would be particularly interesting, since CF3• can be generated under mild, neutral conditions from commercially available and relatively inexpensive CF3I.7 In particular, we noted a recent report by MacMillan demonstrating the conversion of CF3I to CF3• at room temperature in the presence of a photocatalyst, visible light, and a reductant (Scheme 2).8 On the basis of this work, we hypothesized that the merger of visible light photocatalysis (to generate CF3•) with Cu catalysis (to generate reactive Cu–aryl species) (Scheme 2) might provide a mild and general method for the trifluoromethylation of boronic acid derivatives.
Scheme 2.

Proposed New Pathway for Radical Trifluoromethylation of Boronic Acids via Cu/Ru Photocatalysis
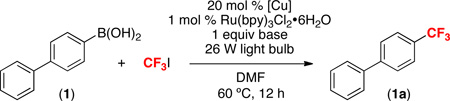
Our initial investigations focused on the Cu and Ru-photocatalyzed trifluoromethylation of 1,1’-biphenyl-4-ylboronic acid with CF3I to form 4-(trifluoromethyl)-1,1’-biphenyl (1a). We were delighted to find that Cu/Ru photocatalysis provided product 1a in modest to excellent yield under a number of conditions (Table 1). A variety of different bases (to promote transmetalation) and reaction solvents were screened for this reaction (see Table S1) and the use of K2CO3 in DMF proved optimal. Copper(I) catalysts generally performed better than CuII salts, and the highest yield of 1a (76%) was obtained with CuOAc. The optimal conditions were as follows: 1 equiv of boronic acid 1, 5 equiv of CF3I, 1 equiv of K2CO3, 20 mol % of CuOAc, and 1 mol % of Ru(bpy)3Cl2·6H2O with irradiation from two 26 W household light bulbs. The major side product was 4-iodo-1,1’-biphenyl (formed in 9% yield under the optimal conditions).
Table 1.
Optimization of Reaction Between 1 and CF3I[a]
 | |||
|---|---|---|---|
| Entry | [Cu] | Base | Yield |
| 1 | Cu(OTf)2 | K2CO3 | 14% |
| 2 | [Cu(OTf)]2•C6H6 | K2CO3 | 28% |
| 3 | Cul | K2CO3 | 34% |
| 4 | Cu | K2CO3 | 40% |
| 5 | Cu(OAc)2 | K2CO3 | 68% |
| 6 | CuOAC | K2CO3 | 76% |
| 7 | CuOAC | NaOAc | 34% |
| 8 | CuOAC | KF | 50% |
| 9 | CuOAC | none | 6% |
| 10[b] | CuOAC | K2CO3 | 1% |
| 11[c] | none | K2CO3 | 3% |
| 12[d] | CuOAC | K2CO3 | 3% |
General conditions: substrate (0.05 mmol, 1 equiv), CF3I (5 equiv), [Cu] (0.2 equiv), Ru(bpy)3Cl2•6H2O (0.01 equiv), base (1 equiv), DMF (0.17 M in substrate), 60 °C, 12 h, 26 W compact fluorescent light bulb. 19F NMR yield.
General conditions, but with no light.
General conditions, but with no CuOAc.
General conditions, but with no Ru(bpy)3Cl2•6H2O.
This Cu/Ru-catalyzed coupling between 1 and CF3I is practical and easily scalable. The reaction in Table 1, entry 6 was performed on a 0.05 mmol scale and provided 76% yield as determined by 19F NMR spectroscopy and GCMS. Nearly identical isolated yields were obtained on both 1 and 5 mmol scales (72% and 70% isolated yield, respectively).
A variety of control reactions were conducted to establish the role of each component of the reaction mixture. As shown in Table 1, entries 10–12, when light, CuOAc or Ru(bpy)3Cl2·6H2O were excluded under otherwise identical conditions, ≤3% yield of 1a was obtained.9 These results clearly indicate the necessity of all three components to achieve high yields under these conditions, consistent with the major pathway to 1a proceeding via dual Ru/Cu catalysis (vide infra). The iodinated side product 4-iodo-1,1’-biphenyl was also subjected to the reaction conditions to establish whether it is an intermediate in the boronic acid trifluoromethylation process. Less than 2% of the aryl–CF3 product was formed, strongly suggesting that the major pathway to 1a does not involve an iodinated intermediate. Finally, the reactivity of boronic acid substrate 1 was investigated under conditions reported by Baran and MacMillan to promote C–H trifluoromethylation reactions via in situ generation of CF3•.10 In both cases, <2% of 1a was observed. These results indicate that 1a is not formed by the direct reaction of CF3• with the boronic acid.
This transformation was next applied to a variety of different aryl- and heteroaryl boronic acid derivatives. Representative examples are shown in Scheme 3 and were selected to highlight not only the broad scope but also the limitations of this method.11 Aromatic boronic acids bearing both electron-donating (t-butyl, methoxy) as well as electron-withdrawing (cyano, trifluoromethyl, fluoro, methyl ester) substituents underwent trifluoromethylation in high yield. A variety of different potentially reactive functional groups (aromatic alcohols, ketones, aldehydes, esters, and amides) were quite well-tolerated. A boronic acid embedded in the estrone framework underwent trifluoromethylation to generate 22a in 80% isolated yield. Most remarkably, 4-iodo-phenylboronic acid underwent selective trifluoromethylation to form 7a, leaving the aryl iodide intact for subsequent functionalization. This demonstrates the complementarity of this method to many other Cu-catalyzed trifluoromethylation protocols.5a,5c,5f Furthermore, it provides additional evidence against the possibility of aryl iodide intermediates in this transformation.
Scheme 3.

Substrate Scope for Cu/Ru-Catalyzed Trifluoromethylation of Boronic Acids[a]
[a] General conditions: substrate (1 equiv), CF3I (5 equiv), [Cu] (0.2 equiv), Ru(bpy)3Cl2•6H2O (0.01 equiv), K2CO3 (1 equiv), DMF (0.17 M in substrate), 60 °C, 12 h, 26 W compact fluorescent light bulb; Isolated yield (≥95% purity). [b] 19F NMR yield. [c] 0.5 equiv of CuOAc. [d] Isolated as a 1:1 mixture with inseparable protodeboronated product. [e] 0.1 equiv of CuOAc. [f] Isolated as a 10:1 mixture with inseparable protodeboronated product. [g] 3 equiv of CF3I. [h] Reaction run at 70 °C. [i] Reaction run at 40 °C. [j] 0.05 equiv of CuOAc.
The use of sterically hindered substrates such as 1-naphthyl and 2,4,6-trimethylphenyl boronic acid is typically challenging for copper-mediated cross-coupling reactions.12 As shown in Scheme 2, similar effects were seen in the current transformation, with products 9a and 10a being formed in modest yields (42% and 39%, respectively). In these cases, competing protodeboronation was problematic, and the major side product was naphthalene or mesitylene, respectively.
Heteroaromatic substrates are of particular relevance to the pharmaceutical and agrochemical industries due to the prevalence of heteroarenes in biologically active compounds.13 Boronic acids derived from pyridine, quinoline, furan and thiophene all underwent trifluoromethylation in modest to good yield.10 In some of these cases, modification of the catalyst loading and/or reaction temperature was required to achieve optimal yield. Importantly, with all of these substrates, trifluoromethylation of the boronic acid moiety out-competed uncatalyzed C–H trifluoromethylation of the heterocycle with CF3•. Thus, this method provides an attractive route for the site-selective installation of CF3 substituents into these scaffolds.
Related conditions could also be applied to analogous perfluoroalkylation reactions. This is a significant advantage of the current method, since perfluoroalkyl analogues of other common trifluoromethylating reagents (e.g., R3SiCF3, S-(trifluoromethyl)thiophenium salts, or Togni’s reagent) are expensive and/or not commercially available. As shown in Scheme 4, perfluorobutyl and perfluorodecyl iodides reacted with 1 to afford products 1b and 1c in good yields under the Cu/Ru catalyzed conditions.
Scheme 4.

Perfluoroalkylation of 1 Catalyzed by Cu/Ru[a]
[a] General conditions: substrate (1 equiv), CuOAc (0.5–1 equiv) Ru(bpy)3Cl2•6H2O (0.01 equiv), K2CO3 (1 equiv), DMF (0.17 M in substrate), 60 °C, 12 h, 26 W compact fluorescent light bulb; Isolated yield. [b] 5 equiv of C4F9I, 0.5 equiv of CuOAc. [c] 1.2 equiv of C10F21I, 1 equiv of CuOAc.
While a detailed mechanistic picture of this transformation remains to be elucidated, a possible set of catalytic cycles is shown in Scheme 5. In this sequence, photoexcitation of Ru(bpy)32+ to Ru(bpy)32+* is followed by 1e− reduction by CuI to generate Ru(bpy)3+ and CuII.14 Reduction of CF3I by Ru(bpy)3+ then affords CF3• and I−. Notably, literature reduction potential data indicates that both of these reactions should be thermodynamically favorable (see Figure S1). The CF3• could then react with CuII to generate a CuIII(CF3) intermediate. Subsequent base-promoted transmetalation between CuIII and the aryl boronic acid would afford CuIII(aryl)(CF3), which could undergo aryl–CF3 bond-forming reductive elimination to release the organic product and regenerate the CuI catalyst.15
Scheme 5.

Possible Mechanism for Cu/Ru-Catalyzed Trifluoromethylation of Boronic Acids
In summary, this communication describes a mild and general approach for the Cu/Ru-catalyzed trifluoromethylation/perfluoroalkylation of aryl boronic acids. This method takes advantage of visible light photoredox catalysis to generate RF• under mild conditions and merges it with copper-catalyzed aryl boronic acid functionalization. The combination has enabled a method for the trifluoromethylation of a wide variety of aromatic and heteroaromatic substrates bearing many common functional groups. This transformation demonstrates the feasibility of achieving Cu-catalyzed trifluoromethylation via a radical pathway. Furthermore, it represents a new example of combining organometallic and photoredox catalysis to achieve synthetically useful organic transformations.16
Supplementary Material
Acknowledgements
We thank the NIH NIGMS (GM073836) for financial support. We thank Dr. Rebecca Loy and Dr. Brannon Gary for helpful discussions.
Footnotes
Supporting Information Available: Experimental details and spectroscopic and analytical data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Schlosser M. Angew. Chem. Int. Ed. 2006;45:5432. doi: 10.1002/anie.200600449. [DOI] [PubMed] [Google Scholar]; (b) Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (c) Hagmann WK. J. Med. Chem. 2008;51:4359. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]; (d) Kirk KL. Org. Process Res. Dev. 2008;12:305. [Google Scholar]; (e) Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (f) Tomashenko OA, Grushin VV. Chem. Rev. 2011;111:4475. doi: 10.1021/cr1004293. [DOI] [PubMed] [Google Scholar]; (g) Roy S, Gregg BT, Gribble GW, Le V-D. Tetrahedron. 2011;67:2161. [Google Scholar]
- 2.(a) Grushin VV, Marshall WJ. J. Am. Chem. Soc. 2006;128:4632. doi: 10.1021/ja0602389. [DOI] [PubMed] [Google Scholar]; (b) Grushin VV, Marshall WJ. J. Am. Chem. Soc. 2006;128:12644. doi: 10.1021/ja064935c. [DOI] [PubMed] [Google Scholar]; (c) Grushin VV. Acc. Chem. Res. 2010;43:160. doi: 10.1021/ar9001763. [DOI] [PubMed] [Google Scholar]; (d) Ball ND, Kampf JW, Sanford MS. J. Am. Chem. Soc. 2010;132:2878. doi: 10.1021/ja100955x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ye Y, Ball ND, Kampf JW, Sanford MS. J. Am. Chem. Soc. 2010;132:14682. doi: 10.1021/ja107780w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ball ND, Gary JB, Ye Y, Sanford MS. J. Am. Chem. Soc. 2011;133:7577. doi: 10.1021/ja201726q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Wang X, Truesdale L, Yu J-Q. J. Am. Chem. Soc. 2010;132:3648. doi: 10.1021/ja909522s. [DOI] [PubMed] [Google Scholar]; (b) Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. Science. 2010;328:1679. doi: 10.1126/science.1190524. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mu X, Chen S, Zhen X, Liu G. Chem. Eur. J. 2011;17:6039. doi: 10.1002/chem.201100283. [DOI] [PubMed] [Google Scholar]; (d) Loy RN, Sanford MS. Org. Lett. 2011;13:2548. doi: 10.1021/ol200628n. [DOI] [PubMed] [Google Scholar]; (e) Mu X, Wu T, Wang H-Y, Guo Y-L, Liu G. J. Am. Chem. Soc. 2011;134:878. doi: 10.1021/ja210614y. [DOI] [PubMed] [Google Scholar]
- 4.(a) Dubinina GG, Furutachi H, Vicic DA. J. Am. Chem. Soc. 2008;130:8600. doi: 10.1021/ja802946s. [DOI] [PubMed] [Google Scholar]; (b) Dubinina GG, Ogikubo J, Vicic DA. Organometallics. 2008;27:6233. [Google Scholar]; (c) Chu L, Qing FL. Org. Lett. 2010;12:5060. doi: 10.1021/ol1023135. [DOI] [PubMed] [Google Scholar]; (d) McReynolds KA, Lewis RS, Ackerman LKG, Dubinina GG, Brennessel WW, Vicic DA. J. Fluorine Chem. 2010;131:1108. [Google Scholar]; (e) Senecal TD, Parsons AT, Buchwald SL. J. Org. Chem. 2011;76:1174. doi: 10.1021/jo1023377. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhang CP, Wang ZL, Chen QY, Zhang CT, Gu YC, Xiao JC. Angew. Chem. Int. Ed. 2011;50:1896. doi: 10.1002/anie.201006823. [DOI] [PubMed] [Google Scholar]; (g) Tomashenko OA, Escudero EC, Belmonte MM, Grushin VV. Angew. Chem. Int. Ed. 2011;50:7655. doi: 10.1002/anie.201101577. [DOI] [PubMed] [Google Scholar]; (h) Morimoto H, Tsubogo T, Litvinas ND, Hartwig JF. Angew. Chem. Int. Ed. 2011;50:3793. doi: 10.1002/anie.201100633. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Litvinas ND, Fier PS, Hartwig JF. Angew. Chem. Int. Ed. 2012;51:536. doi: 10.1002/anie.201106668. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Zanardi A, Novikov MA, Martin E, Benet-Buchholz J, Grushin VV. J. Am. Chem. Soc. 2011;133:20901. doi: 10.1021/ja2081026. [DOI] [PubMed] [Google Scholar]; (k) Kremlev MM, Mushta AI, Tyrra W, Yagupolskii YL, Naumann D, Möller A. J. Fluorine Chem. 2012;133:67. [Google Scholar]; (l) Khan BA, Buba AE, Gooßen LJ. Chem. Eur. J. 2012;18:1577. doi: 10.1002/chem.201102652. [DOI] [PubMed] [Google Scholar]
- 5.(a) Oishi M, Kondo H, Amii H. Chem. Commun. 2009:1909. doi: 10.1039/b823249k. [DOI] [PubMed] [Google Scholar]; (b) Shimizu R, Egami H, Nagi T, Chae J, Hamashima Y, Sodeoka M. Tetrahedron Lett. 2010;51:5947. [Google Scholar]; (c) Knauber T, Arikan F, G R, Goossen LJ. Chem. Eur. J. 2011;17:2689. doi: 10.1002/chem.201002749. [DOI] [PubMed] [Google Scholar]; (d) Xu J, Luo DF, Xiao B, Liu J, Gong TJ, Fu Y, Liu L. Chem. Commun. 2011;47:4300. doi: 10.1039/c1cc10359h. [DOI] [PubMed] [Google Scholar]; (e) Liu T, Shen Q. Org. Lett. 2011;13:2342. doi: 10.1021/ol2005903. [DOI] [PubMed] [Google Scholar]; (f) Li YCT, Wang H, Zhang R, Jin K, Wang X, Duan C. Synlett. 2011;12:1713. [Google Scholar]; (g) Liu T, Shao X, Wu Y, Shen Q. Angew. Chem. Int. Ed. 2012;51:540. doi: 10.1002/anie.201106673. [DOI] [PubMed] [Google Scholar]; (h) Chu L, Qing FL. J. Am. Chem. Soc. 2011;134:1298. doi: 10.1021/ja209992w. [DOI] [PubMed] [Google Scholar]; (i) Jiang X, Chu L, Qing FL. J. Org. Chem. 2012;77:1251. doi: 10.1021/jo202566h. [DOI] [PubMed] [Google Scholar]
- 6.Swarts F. Bull. Acad. R. Belg. 1892;24:309. [Google Scholar]
- 7.(a) Strekowski L, Hojjat M, Patterson SE, Kiselyov AS. J. Heterocyclic Chem. 1994;31:1413. [Google Scholar]; (b) Sato K, Omote M, Ando A, Kumadaki I. Org. Lett. 2004;6:4359. doi: 10.1021/ol048134v. [DOI] [PubMed] [Google Scholar]; (c) Mikami K, Tomita Y, Ichikawa Y, Amikura K, Itoh Y. Org. Lett. 2006;8:4671. doi: 10.1021/ol0611301. [DOI] [PubMed] [Google Scholar]; (d) Itoh Y, Houk KN, Mikami K. J. Org. Chem. 2006;71:8918. doi: 10.1021/jo0616153. [DOI] [PubMed] [Google Scholar]; (e) Kino T, Nagase Y, Ohtsuka Y, Yamamoto K, Uraguchi D, Tokuhisa K, Yamakawa T. J. Fluorine Chem. 2010;131:98. [Google Scholar]; (f) Ye Y, Lee SH, Sanford MS. Org. Lett. 2011;13:5464. doi: 10.1021/ol202174a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Nagib DA, Scott ME, MacMillan DWC. J. Am. Chem. Soc. 2009;131:10875. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pham PV, Nagib DA, MacMillan DWC. Angew. Chem. Int. Ed. 2011;50:6119. doi: 10.1002/anie.201101861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reactions between some electron-deficient substrates and CF3I showed ~20% of the trifluoromethylated products in the absence of Ru catalyst. See Supporting Information for more details.
- 10.(a) Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS. Proc. Natl. Acad. Sci. USA. 2011;108:14411. doi: 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nagib DA, MacMillan DWC. Nature. 2011;480:224. doi: 10.1038/nature10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The trifluoromethylated products that are liquids were isolated via Kugelrohr distillation. This isolation procedure typically afforded ≥95% pure products (contaminated with traces of protodeboronated material). In many cases (eg, 6a, 9a, 18a, 20a), >98% pure products could be obtained via subsequent careful purification by column chromatography, albeit in reduced yields. See Supporting Information for full details.
- 12.Hassan J, Sévignon M, Gozzi C, Schulz E, Lemaire M. Chem. Rev. 2002;102:1359. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 13.Top Pharmaceuticals Poster Njardarson Group. [accessed Feb 14, 2012]; http://cbc.arizona.edu/njardarson/group/sites/default/files/Top 200 Brand-name Drugs by US Retail Sales in 2010sm_0.pdf. [Google Scholar]
- 14.The observation that CuI salts generally perform better than CuII salts in this transformation is consistent with this proposal.
- 15.The order of steps ii and iii in Scheme 5 could also easily be reversed. Without detailed evidence about the resting state of the Cu-catalyst, we cannot draw definitive conclusions about the species most likely to react with CF3•.
- 16.Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J. Am. Chem. Soc. 2011;133:18566. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.